Table of Contents
|
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
FORM 10-K
☑ | Annual Report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
| For the Fiscal Year Ended December 31, 2016 |
OR
¨ | Transition Report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
| For the transition period from to . |
Commission file number 333-199861
MYLAN N.V.
(Exact name of registrant as specified in its charter)
The Netherlands |
| 98-1189497 |
(State or other jurisdiction of incorporation or organization) |
| (I.R.S. Employer Identification No.) |
Building 4, Trident Place, Mosquito Way, Hatfield, Hertfordshire, AL10 9UL, England
(Address of principal executive offices)
+44 (0) 1707-853-000
(Registrant's telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
Title of Each Class: |
| Name of Each Exchange on Which Registered: |
Ordinary shares, nominal value €0.01 |
| The NASDAQ Stock Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☑ No ¨
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No ☑
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☑ No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☑ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☑
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act.:
Large accelerated filer | ☑ |
|
| Accelerated filer | ¨ |
Non-accelerated filer | ¨ | (Do not check if a smaller reporting company) |
| Smaller reporting company | ¨ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ¨ No ☑
The aggregate market value of the outstanding ordinary shares, nominal value €0.01 , of the registrant other than shares held by persons who may be deemed affiliates of the registrant, as of June 30, 2016 , the last business day of the registrant's most recently completed second fiscal quarter, was approximately $21,822,318,620 .
The number of ordinary shares outstanding, nominal value €0.01 , of the registrant as of February 24, 2017 was 535,496,988 .
INCORPORATED BY REFERENCE
Document | Part of Form 10-K into Which Document is Incorporated |
An amendment to this Form 10-K will be filed no later than 120 days after the close of registrant's fiscal year. | III |
|
Table of Contents
MYLAN N.V.
INDEX TO FORM 10-K
For the Year Ended December 31, 2016
|
| Page |
PART I |
| |
ITEM 1. | Business | 3 |
ITEM 1A. | Risk Factors | 25 |
ITEM 1B. | Unresolved Staff Comments | 52 |
ITEM 2. | Properties | 52 |
ITEM 3. | Legal Proceedings | 52 |
|
|
|
PART II |
| |
ITEM 5. | Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | 53 |
ITEM 6. | Selected Financial Data | 55 |
ITEM 7. | Management's Discussion and Analysis of Financial Condition and Results of Operations | 57 |
ITEM 7A. | Quantitative and Qualitative Disclosures about Market Risk | 85 |
ITEM 8. | Financial Statements and Supplementary Data | 87 |
ITEM 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure | 177 |
ITEM 9A. | Controls and Procedures | 177 |
ITEM 9B. | Other Information | 177 |
|
|
|
PART III |
| |
ITEM 10. | Directors, Executive Officers and Corporate Governance | 178 |
ITEM 11. | Executive Compensation | 178 |
ITEM 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 178 |
ITEM 13. | Certain Relationships and Related Transactions, and Director Independence | 178 |
ITEM 14. | Principal Accounting Fees and Services | 178 |
|
|
|
PART IV |
| |
ITEM 15. | Exhibits and Consolidated Financial Statement Schedules | 179 |
Signatures | 189 | |
2
Table of Contents
PART I
ITEM 1. | Business |
Mylan N.V. , along with its subsidiaries (collectively, the "Company," " Mylan ," "our" or "we"), is a leading global pharmaceutical company, which develops, licenses, manufactures, markets and distributes generic, brand name and over-the-counter ("OTC") products in a variety of dosage forms and therapeutic categories. Mylan is committed to setting new standards in healthcare by creating better health for a better world, and our mission is to provide the world's 7 billion people access to high quality medicine. To do so, we innovate to satisfy unmet needs; make reliability and service excellence a habit; do what's right, not what's easy; and impact the future through passionate global leadership.
Mylan offers one of the industry's broadest product portfolios, including approximately 7,500 marketed products around the world, to customers in more than 165 countries and territories. We operate a global, high quality, vertically-integrated manufacturing platform around the world and one of the world's largest active pharmaceutical ingredient ("API") operations. We also operate a strong and innovative research and development ("R&D") network that has consistently delivered a robust product pipeline including a variety of dosage forms, therapeutic categories and biosimilars.
Overview
Throughout its history, Mylan has been recognized as a leader in the United States ("U.S.") generic pharmaceutical industry. Our leadership position is the result of, among other factors, our ability to efficiently obtain Abbreviated New Drug Application ("ANDA") approvals and our reliable high quality supply chain. Mylan is one of the largest pharmaceutical companies in the world today in terms of revenue and is recognized as an industry leader because of our organic growth and transformative acquisitions beginning in 2007. Our most recent significant acquisitions are described below.
2016 Acquisitions
On June 15, 2016 , we completed the acquisition of the non-sterile, topicals-focused business (the " Topicals Business ") of Renaissance Acquisition Holdings, LLC (" Renaissance ") for approximately $1.0 billion in cash at closing, including amounts deposited into escrow for potential contingent payments, subject to customary adjustments. The Topicals Business provides the Company with a complementary portfolio of approximately 25 products, an active pipeline of approximately 25 products, and an established U.S. sales and marketing infrastructure targeting dermatologists. The Topicals Business also provides an integrated manufacturing and development platform.
On August 5, 2016, we acquired approximately 94% of the total number of outstanding shares of Meda AB (publ.) (" Meda "). The total purchase price for Meda was approximately $6.92 billion , net of cash acquired. Subsequent to the August 5, 2016 closing, a compulsory acquisition proceeding was initiated in accordance with the Swedish Companies Act (Sw. aktiebolagslagen (2005:551)) to acquire the remaining Meda shares. On November 1, 2016, Mylan made an offer to the remaining Meda shareholders to tender all their Meda shares to Mylan for cash consideration of 161.31kr per Meda share (the "November Offer") to provide such remaining Meda shareholders with an opportunity to sell their Meda shares to Mylan in advance of the automatic acquisition of their shares for cash in connection with the compulsory acquisition proceeding. At the end of November 2016, Mylan completed the acquisition of approximately 19 million Meda shares duly tendered into the November Offer. As of March 1, 2017, Mylan obtained full legal ownership to the remaining Meda shares pursuant to the compulsory acquisition proceeding, and now owns 100% of the total number of outstanding Meda shares. The Meda shareholders whose shares are subject to the compulsory acquisition proceeding, representing approximately 1% of the total number of outstanding Meda shares, will automatically receive cash consideration plus statutory interest for their Meda shares as determined in the compulsory acquisition proceeding.
The acquisition of Meda created a more diversified and expansive portfolio of branded and generic medicines along with a strong and growing portfolio of OTC products. Meda has a balanced global footprint with significant scale in key geographic markets, particularly the U.S. and Europe. We have significantly expanded and strengthened our presence in emerging markets including China, Southeast Asia and the Middle East. These markets provide opportunities for future growth and expansion and are complemented by Mylan's historical presence in India, Brazil and certain countries in Africa (including South Africa).
2015 Acquisitions
On February 27, 2015 , we completed the acquisition of Mylan Inc. and Abbott Laboratories ' non-U.S. developed markets specialty and branded generics business (the " EPD Business ") in an all-stock transaction. The purchase price for the
3
Table of Contents
EPD Business, which was on a debt-free basis, was $6.31 billion based on the closing price of Mylan Inc.'s stock as of the acquisition date, as reported by the NASDAQ Global Select Stock Market ("NASDAQ"). The acquired EPD Business enhanced our already expansive product portfolio by more than 100 specialty and branded generic pharmaceutical products in five major therapeutic areas and included several patent protected, novel and/or hard-to-manufacture products. Additionally, we significantly expanded and strengthened our presence in Europe, Japan, Canada, Australia and New Zealand.
On November 20, 2015, we completed the acquisition of certain female healthcare businesses from Famy Care Limited (such businesses " Jai Pharma Limited "), a specialty women's healthcare company with global leadership in generic oral contraceptive products, through our wholly owned subsidiary Mylan Laboratories Limited ("Mylan India") for a cash payment of $750 million plus additional contingent payments of up to $50 million for the filing for approval with, and receipt of approval from, the U.S. Food and Drug Administration ("FDA") of a product under development with Jai Pharma Limited .
One Mylan
Through these transactions, along with our previous transformative acquisitions of Agila Specialties (‘‘ Agila ''), Mylan India, Merck KGaA's generics and specialty pharmaceutical business, Bioniche Pharma Holdings Limited (" Bioniche Pharma ") and Pfizer Inc.'s respiratory delivery platform (the "respiratory delivery platform"), we have created a horizontally and vertically integrated platform with global scale, augmented our diversified product portfolio and further expanded our range of capabilities, all of which we believe position us well for the future.
Today, Mylan has a robust worldwide commercial presence, including leadership positions in the U.S., Australia, several key European markets, such as France and Italy, as well as other markets around the world. Mylan 's global portfolio of approximately 7,500 marketed products around the world that covers a vast array of therapeutic categories. We offer an extensive range of dosage forms and delivery systems, including oral solids, topicals, liquids and semisolids while focusing on those products that are difficult to formulate and manufacture, and typically have longer life cycles than traditional generic pharmaceuticals, including transdermal patches, high potency formulations, injectables, controlled-release and respiratory products. In addition, we offer a wide range of antiretroviral therapies ("ARVs"), upon which approximately 50% of patients being treated for HIV/AIDS in the developing world depend. Mylan also operates one of the largest API manufacturers, supplying low cost, high quality API for our own products and pipeline, as well as for a number of third parties. With the acquisition of Meda, we gained access to an extensive portfolio of OTC products. OTC products are key complements to prescribed drugs because they are easily accessible, save patients' time and reduce cost pressures on healthcare systems.
We believe that the breadth and depth of our business and platform provide certain competitive advantages in major markets in which we operate, including less dependency on any single market or product. As a result, we are better able to successfully compete on a global basis than compared to many of our competitors.
Our Operations
Mylan N.V. was originally incorporated as a private limited liability company, New Moon B.V., in the Netherlands in 2014. Mylan became a public limited liability company in the Netherlands through its acquisition of the EPD Business on February 27, 2015 . Mylan's corporate seat is located in Amsterdam, the Netherlands , its principal executive offices are located in Hatfield, Hertfordshire, England and Mylan N.V. group's global headquarters are located in Canonsburg, Pennsylvania .
The Company has made a number of significant acquisitions since 2015, and as part of the holistic, global integration of these acquisitions, the Company is focused on how to best optimize and maximize all of its assets across the organization and across all geographies. On December 5, 2016, the Company announced restructuring programs in certain locations representing initial steps in a series of actions that are anticipated to further streamline its operations globally. The Company continues to develop the details of the cost reduction initiatives, including workforce actions and other potential restructuring activities beyond the programs already announced. Refer to Item 7 and Note 16 Restructuring included in Item 8 in this Form 10-K for additional information related to our restructuring initiatives.
As a result of our acquisition of Meda on August 5, 2016 and the integration of our portfolio across our branded, generics and OTC platforms in all of our regions, effective October 1, 2016, we expanded our reportable segments. We are reporting our results in three segments on a geographic basis as follows: North America, Europe and Rest of World. Our North America segment is primarily made up of our operations in the U.S. and Canada. Also included in our North America segment are the operations of our previously reported Specialty segment. Our Europe segment is made up of our operations in 35 countries within the region. Our Rest of World segment is primarily made up of our operations in India, Australia, Japan and New Zealand. Also included in our Rest of World segment are our operations in emerging markets, which includes countries in Africa (including South Africa) as well as Brazil and other countries throughout Asia and the Middle East.
4
Table of Contents
The chart below reflects third party net sales by reportable segment for the year ended December 31, 2016 .
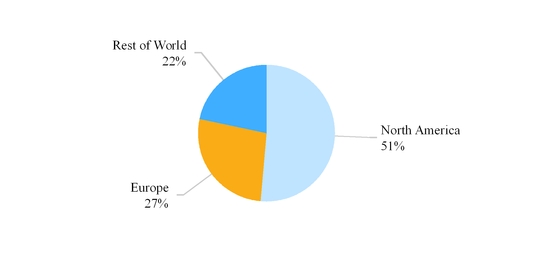
Our third party net sales are derived primarily from the sale of generic and branded generic pharmaceuticals, branded pharmaceuticals, OTC products and API. Our API business is conducted through Mylan India , which is included within our Rest of World segment. Refer to Note 13 Segment Information included in Item 8 in this Form 10-K for additional information related to our reportable segments.
Our global operational footprint, including the locations of our manufacturing and R&D facilities and capabilities, along with the individual site's primary activities, are detailed on the map below.
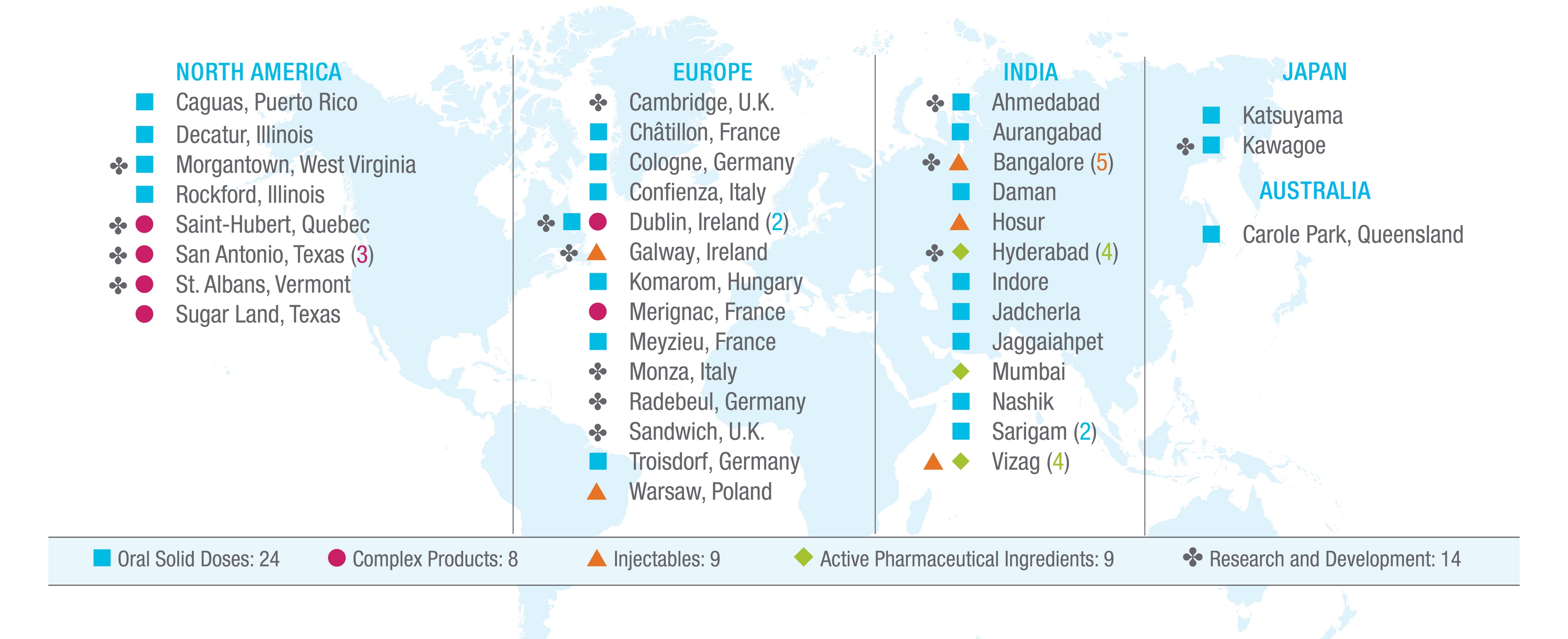
Our global manufacturing platform is an important component of our business model. We own eleven manufacturing and distribution facilities in the U.S. including Puerto Rico, with significant sites in Morgantown, West Virginia; San Antonio, Texas; St. Albans, Vermont; Caguas, Puerto Rico; and Greensboro, North Carolina. Outside the U.S. and Puerto Rico, we utilize production and distribution facilities in eleven countries, including key facilities in India, Australia, Japan, Ireland, Hungary and France. Through our manufacturing facilities, which we operate around the globe, we have a manufacturing capacity capable of producing approximately 80 billion oral solid doses, 4,800 kiloliters of APIs, 500 million injectable units, and 1.5 billion complex products (transdermals, dermals, topicals, respiratory, oral films, and other specialty items) per year.
The Company also leases manufacturing, warehousing, distribution and administrative facilities in numerous locations, within and outside of the U.S., including properties in New York, Canada, France, India, Ireland and the United Kingdom (the "U.K."). All of the facilities listed above are included in our reportable segments based on the location of the facility. Our global R&D centers of excellence are located in Morgantown, West Virginia and Hyderabad, India. We also have specific technology focused development sites in Texas, Vermont, Canada, Ireland, Germany, Italy, the U.K., India and Japan.
5
Table of Contents
In addition, under our collaboration agreements with Biocon Limited ("Biocon") for the development of generic biologic compounds and insulin analog products, certain state of the art manufacturing facilities owned by Biocon in India and Malaysia are to be used for the manufacture of products under the agreements, which are excluded from the chart above.
We believe that all of our facilities are in good operating condition, the machinery and equipment are well-maintained, the facilities are suitable for their intended purposes and they have capacities adequate for the current operations.
Unless otherwise indicated, industry data included in Item 1 is sourced from Quintiles IMS Holdings, Inc. ("IMS") and is for the twelve months ended November 2016 .
North America Segment
Our North America segment primarily develops, manufactures, sells and distributes pharmaceutical products in tablet, capsule, injectable, transdermal patch, gel, nebulized and cream or ointment form. For the year ended December 31, 2016 , North America segment third party net sales were $5.63 billion . Our North America segment includes our operations in the U.S. and Canada, each of which is discussed further below.
The U.S. generics market is the largest in the world, in terms of value, with generic prescription sales of $59.1 billion for the twelve months ended November 2016 . As of December 2016, according to the GPhA Savings Study, in terms of generic prescription volume, approximately 89% of all pharmaceutical products sold in the U.S. were generic products, which demonstrates the high level of generic penetration in this market. Mylan holds a top two ranking within the U.S. generics prescription market in terms of both sales and prescriptions dispensed. Approximately one in every 13 prescriptions dispensed in the U.S. is a Mylan product. Our sales of products in the U.S. are derived primarily from the sale of oral solid dosages, injectables, transdermal patches, gels, creams, ointments and unit dose offerings. In the U.S., we have one of the largest product portfolios among all generic pharmaceutical companies. With the acquisition of the Topicals Business, we gained a complementary portfolio of approximately 25 branded and generic topical products, including an active pipeline of approximately 25 products as well as an established U.S. sales and marketing infrastructure targeting dermatologists. The Topicals Business also brings Mylan an integrated manufacturing and development platform along with a leading topicals-focused contract development and manufacturing organization.
In addition, we manufacture and sell a diverse portfolio of injectable products across several key therapeutic areas, including respiratory and allergy, infectious disease, cardiovascular, oncology and central nervous system and anesthesia. Mylan's injectable manufacturing capabilities include vials, pre-filled syringes, ampoules and lyophilization with a focus on oncology, penems, penicillins, ophthalmics and peptides.
Our unit dose business focuses on providing one of the largest product portfolios along with innovative packaging and barcoding that supports bedside verification throughout the U.S. and Canada for hospitals, group purchasing organizations ("GPOs"), long term care facilities, wholesalers, surgical services, home infusion service providers, correctional facilities, specialty pharmacies and retail outlets.
The EpiPen® Auto-Injector , which is used in the treatment of severe allergic reactions, is an epinephrine auto-injector that has been sold in the U.S. and internationally since the mid-1980s. Mylan has worldwide rights to the EpiPen® Auto-Injector , which is supplied to Mylan by a wholly owned subsidiary of Pfizer Inc. Anaphylaxis is a severe allergic reaction that is rapid in onset and may cause death, either through swelling that shuts off airways or through a significant drop in blood pressure. In December 2010, the National Institute of Allergy and Infectious Diseases, a division of the National Institutes of Health, introduced the "Guidelines for the Diagnosis and Management of Food Allergy in the United States." These guidelines state that epinephrine is the first line treatment for anaphylaxis. The EpiPen® Auto-Injector is the number one dispensed epinephrine auto-injector. On December 16, 2016, Mylan launched the first authorized generic for the EpiPen® Auto-Injector , which has the same drug formulation and device functionality as the branded product.
Perforomist® Inhalation Solution , Mylan's Formoterol Fumarate Inhalation Solution, was launched in October 2007. Perforomist® Inhalation Solution is a long-acting beta 2 -adrenergic agonist indicated for long-term, twice-daily administration in the maintenance treatment of bronchoconstriction in chronic obstructive pulmonary disorder ("COPD") patients, including those with chronic bronchitis and emphysema. Mylan holds several U.S. and international patents protecting Perforomist® Inhalation Solution . Mylan also markets ULTIVA® , which is an analgesic agent used during the induction and maintenance of general anesthesia for inpatient and outpatient procedures and is generally administered by an infusion device.
6
Table of Contents
With the acquisition of Meda, we acquired certain key branded products, including Dymista® which is used for the treatment of seasonal allergic rhinitis and was launched in the U.S. in 2012 and in Europe in 2013, and is now being rolled out in certain emerging markets.
We believe that the breadth and quality of our product offerings help us to successfully meet our customers' needs and to better compete in the generics industry over the long-term. The future growth of our U.S. generics business is partially dependent upon continued acceptance of generic products as affordable alternatives to branded pharmaceuticals, a trend which is largely outside of our control. However, we believe that we can maximize the value of our generic product opportunities by continuing our proven track record of bringing to market high quality products that are difficult to formulate or manufacture. Throughout Mylan's history, we have successfully introduced many generic products that are difficult to formulate or manufacture and continue to be meaningful contributors to our business several years after their initial launch. Additionally, we expect to achieve growth in our U.S. business by launching new products for which we may attain FDA first-to-file status with Paragraph IV certification. As described further in the "Product Development and Government Regulation" discussion below, a first-filed ANDA with a Paragraph IV certification qualifies the product approval holder for a period of generic marketing and distribution exclusivity.
In Canada , we have successfully leveraged the acquired EPD Business allowing us to further broaden our presence in this market. We currently rank seventh in terms of market share in the generic prescription market and Mylan products are sold in eight out of ten pharmacies in Canada. As in the U.S., growth in Canada will be dependent upon acceptance of generic products as affordable alternatives to branded pharmaceuticals. Further, we plan to leverage the strength and reliability of the collective Mylan brand to foster continued brand awareness and growth throughout the region.
Europe Segment
Our European operations are conducted through our wholly owned subsidiaries in 35 countries across the region. For the year ended December 31, 2016 , Europe segment third party net sales were $2.95 billion . The types of markets within Europe vary from country to country; however, when combined, the European market is the second largest generic pharmaceutical market in the world in terms of value. Within Europe, by value, the generic prescription market in Germany is the largest , followed by the U.K., France, Spain and Italy, respectively.
In Europe, the manner in which products are marketed varies by country. In addition to selling pharmaceuticals under their International Nonproprietary Name ("INN") (i.e., API), in certain European countries, branded generic pharmaceutical products are given a unique brand name, as these markets tend to be more responsive to the promotion efforts generally used to promote brand products.
The European generic prescription market also varies significantly by country in terms of the extent of generic penetration, the key decision maker in terms of drug choice and other important aspects. Some countries, including Germany, the U.K., the Netherlands, Denmark and Poland, are characterized by relatively high generic penetration, ranging between 69% and 73% of total prescription market sales in the twelve months ended November 2016 , based on volume. Conversely, other major European markets, including France, Italy and Spain, are characterized by much lower generic penetration, ranging between 21% and 44% of total prescription sales in the twelve months ended November 2016 , based on volume. However, actions taken by governments, particularly in these latter under-penetrated countries, to reduce healthcare costs could encourage further use of generic pharmaceutical products. In some of these under-penetrated markets, in addition to growth from new product launches, we expect our future growth to be driven by increased generic utilization and penetration.
As a result of the acquisitions of Meda and the EPD Business, our product portfolio has been diversified with OTC products and additional branded and branded generic products in Europe. In addition, Mylan has significantly expanded and strengthened its presence in Europe. In particular, we have grown our presence in several markets in Central and Eastern Europe, including Poland, Greece, the Czech Republic and Slovakia and gained access into new markets, such as Romania and Bulgaria. Following these recent acquisitions, our revenues in Europe are now significantly diversified across our generics, branded and branded generic portfolios.
Of the top ten generic prescription markets in Europe, we hold leadership positions in several of the markets, including the number one market share position in France and the number two market share position in Italy. In France , we have the highest market share of approximately 26% . Our future growth in the French market is expected to come primarily from new product launches. Further growth can be possible in case of increased generic utilization and penetration through government initiatives. In addition, the acquired EPD Business and the acquisition of Meda have positioned us as a major healthcare provider in three key channels including practitioners, hospitals and pharmacies, our primary customers in this market.
7
Table of Contents
In Italy , we have the second highest market share in the generic prescription market, with a share of approximately 18% in terms of volume and value. We believe that the Italian generic market is still under-penetrated, with generics representing approximately 21% of the Italian pharmaceutical market, based on volume. The Italian government has put forth only limited measures aimed at encouraging generic use, and as a result, generic substitution is still in its early stages. As leaders of the generic market, we can benefit from increased generic utilization.
In addition to France and Italy, we have grown our presence in several European markets including the U.K., Spain and markets in Eastern Europe. In the U.K. , Mylan is ranked third in the U.K. generic prescription market, in terms of value. Mylan is well positioned in the U.K. as a preferred supplier to wholesalers and is also focused on areas such as retail pharmacy chains and hospitals. The acquisition of the EPD Business in the U.K. has provided us with an additional branded off-patent market presence, particularly in the areas of pancreatic enzyme replacement therapy and hormone replacement therapy.
In Spain , we have the ninth highest market share in the generic prescription market based on volume. The generic market comprised approximately 35% of the total Spanish pharmaceutical market by volume for the twelve months ended November 2016 . Within the overall Spanish pharmaceutical market, our position has expanded due to the acquired EPD Business. Our portfolio and depth in this market has been further expanded with the acquisition of Meda by adding OTC products. As a result of these acquisitions, we have diversified our product offerings in Spain and generic prescription products now account for approximately half of our sales in Spain. We view further generic utilization and penetration of the Spanish market to be one of the key drivers of our growth in this country.
As a result of the acquisitions of Meda and the EPD Business, we have strengthened and expanded our presence in Germany and have diversified our portfolio to reduce our reliance on the tender system. A tender system is part of the market in Germany, and as a result, health insurers play a major role. Under a tender system, health insurers invite manufacturers to submit bids that establish prices for generic pharmaceuticals.
In the Nordic region, which we define as Sweden, Norway, Denmark, Finland and Iceland, our presence has expanded significantly as a result of our recent acquisitions. For instance, we now have the fourth highest market share in Sweden, in terms of volume and value, and the fifth highest market share in Norway, based on value.
We also have a notable presence in other European generic prescription markets, including Portugal and Belgium, where we hold the third and fifth highest market share, respectively, in terms of volume. In the Netherlands, we have the third highest market share in the generic prescription market, which is characterized by relatively high generic penetration.
Rest of World Segment
We market pharmaceuticals in Rest of World primarily through our subsidiaries in India, Australia, Japan, New Zealand, and emerging markets. In addition, in certain emerging markets, we often use distributors to market our products. Our export business is primarily focused on countries in Africa, and through Mylan India , we market API to third parties and also supply other Mylan subsidiaries. For the year ended December 31, 2016 , Rest of World segment third party net sales were $2.38 billion .
The Indian generics market is the largest in the world, in terms of volume. In India , we are one of the world's largest API manufacturers as measured by the number of drug master files ("DMFs") filed with regulatory agencies. Mylan India 's manufacturing capabilities include a range of dosage forms, such as tablets, capsules and injectables, in a wide variety of therapeutic categories. Mylan India has nine API and intermediate manufacturing facilities and a total of sixteen finished dosage form ("FDF") facilities, which includes nine oral solid dose ("OSD") facilities and seven injectable facilities, all located in India. Our presence in India goes beyond manufacturing, sales and marketing. With a global R&D center of excellence in Hyderabad, India and technology driven R&D sites in Bangalore, India and Hyderabad, India, we are able to create unique and efficient R&D capabilities.
Mylan India markets high quality API to third parties around the world and ARV products for people living with HIV/AIDS. In addition, Mylan India has a growing commercial presence, with products representing approximately 20% of the Hepatitis C market share in India. Our current areas of focus include Critical Care, Hepato Care, HIV Care, Onco Care and Women's Care. We continue to expand our products in the therapeutic categories such as hepatology, oncology and critical care. In November 2015, we completed our acquisition of Jai Pharma Limited, which significantly broadened our women's care portfolio and strengthened our technical capabilities in terms of dedicated hormone manufacturing.
In Australia , we have the highest market share in the generic pharmaceutical market, with an estimated 22% market share by volume. Mylan is the number one supplier by volume to Australia's national pharmaceuticals program. The acquired
8
Table of Contents
EPD Business has enabled Mylan to broaden its product portfolio in this market. The generic pharmaceutical market in Australia had sales of approximately $1.3 billion during the twelve months ended November 2016 .
Mylan has been among the fastest growing companies in the Japan generics market for the last three years. We also maintain manufacturing capabilities in Japan, which play a key role in supplying our businesses throughout the country. Currently, the market in Japan is largely composed of hospitals and clinics, but pharmacies are playing a greater role as generic substitution, aided by recent pro-generics government action, becomes more prevalent. Japan is the second largest single pharmaceutical market in the world by value, behind the U.S., and the fourth largest generic prescription market worldwide by volume, with sales of approximately $7.0 billion during the twelve months ended November 2016 . Beginning in 2013, we established an exclusive long-term strategic collaboration with Pfizer Japan Inc. ("Pfizer Japan") to develop, manufacture, distribute and market generic drugs in Japan. Under the agreement, both parties operate separate legal entities in Japan and collaborate on current and future generic products, sharing the costs and profits resulting from such collaboration. Mylan 's responsibilities, under the agreement, primarily consist of managing operations, including R&D and manufacturing. Pfizer Japan's responsibilities primarily consist of the commercialization of the combined generics portfolio and managing the marketing and sales effort. The Japanese government has stated that it now intends to grow the generic share to at least 70% by mid-2017 and to at least 80% at the earliest possible date between 2018 and the end of 2020 . As of August 2016 , the generic share reached 66% , up from approximately 59% in August 2015 .
With the acquisition of the EPD Business, we have strengthened our position in the Japanese market as we have acquired a wide portfolio of branded products that are promoted by our own sales force. The acquired EPD Business is run independently from our strategic collaboration with Pfizer Japan.
In addition to our operations in India, Australia and Japan, we also have a notable presence in New Zealand . In New Zealand, we are the largest generics company in the country, with 33% of the market share by volume. Additionally, among all pharmaceutical companies we are the largest company in New Zealand by volume. New Zealand is generally a government tender market where pharmaceutical suppliers can gain exclusivity of up to three years . In New Zealand, we have broadened our market presence and profile with the addition of the acquired EPD Business.
In Brazil , we operate a commercial business focused on providing high quality generic injectable products to the Brazilian hospital segment. Our sales into this market segment are made through distributors as well as through tenders. Brazil is the fourth largest generic pharmaceutical market in the world, behind the U.S., the combined European market and China, in terms of value. In the coming years, the Brazilian generic pharmaceutical market is expected to continue its growth trajectory primarily because of the increase of off patent reference drugs, the growth of biological products and the growth of emerging markets. Our goal is to continue to build upon this local platform in order to further access the nearly $12 billion Brazilian generic pharmaceutical market.
With the acquisition of Meda, we have grown our presence in emerging markets, such as China which is the third largest generic market in the world by value behind the U.S. and combined Europe, with generic market sales of approximately $25.0 billion for the twelve months ended November 2016. We also gained access to other emerging markets including countries within Southeast Asia and the Middle East. Our portfolio in emerging markets includes prescription, non-prescription and OTC products, and we now have the opportunity to reach these markets through an organized sales force and direct access to the healthcare providers, as well as through distributor relationships.
Product Development and Government Regulation
North America
U.S.
Prescription pharmaceutical products in the U.S. are generally marketed as either brand or generic drugs. Brand products are usually marketed under brand names through marketing programs that are designed to generate physician and consumer loyalty. Brand products are generally patent protected, which provides a period of market exclusivity during which time they are sold with little or no competition for the compound, although there are typically other participants in the therapeutic area. Additionally, brand products may benefit from other periods of non-patent market exclusivity.
Generic pharmaceutical products are the pharmaceutical and therapeutic equivalents of an approved brand drug, known as the reference listed drug ("RLD") that is listed in the FDA publication entitled Approved Drug Products with Therapeutic Equivalence Evaluations , popularly known as the "Orange Book." The Drug Price Competition and Patent Term Restoration Act of 1984 (the "Hatch-Waxman Act") provides that generic drugs may enter the market after the approval of an
9
Table of Contents
ANDA, which generally requires that similarity to an RLD, including bioequivalence, be demonstrated, any patents on the RLD have expired or been found to be invalid or not infringed, and any market exclusivity periods related to the RLD have ended. Because approved generic drugs have been found to be the same as their respective RLDs, they can be expected to have the same safety and effectiveness profile as the RLD. Accordingly, generic products provide a safe, effective and cost-efficient alternative to users of these reference brand products. Branded generic pharmaceutical products are generic products that are more responsive to the promotion efforts generally used to promote brand products. Growth in the generic pharmaceutical industry has been, and will continue to be, driven by the increased market acceptance of generic drugs, as well as the number of brand drugs for which patent terms and/or other market exclusivities have expired.
We obtain new generic products primarily through internal product development. Additionally, we increasingly collaborate with other companies by entering into licensing or co-development agreements. All applications for FDA approval must contain information relating to product formulation, raw material suppliers, stability, manufacturing processes, packaging, labeling and quality control. Information to support the bioequivalence of generic drug products or the safety and effectiveness of new drug products for their intended use is also required to be submitted. There are generally four types of applications used for obtaining FDA approval of new products:
New Drug Application ("NDA") - An NDA is filed when approval is sought to market a newly developed branded product and, in certain instances, for a new dosage form, a new delivery system or a new indication for a previously approved drug.
ANDA - An ANDA is filed when approval is sought to market a generic equivalent of a drug product previously approved under an NDA and listed in the FDA's Orange Book or for a new dosage strength for a drug previously approved under an ANDA.
Biologics License Application ("BLA") - A BLA is similar to an NDA, but is submitted to seek approval to market a drug product that is a biologic, which generally is a product derived from a living organism.
Biosimilars Application - This is an abbreviated approval pathway for a biologic product that is "highly similar" to a product previously approved under a BLA.
The ANDA development process is generally less time-consuming and complex than the NDA development process. It typically does not require new preclinical and clinical studies, because it relies on the studies establishing safety and efficacy conducted for the RLD previously approved through the NDA process. The ANDA process, however, does typically require one or more bioequivalence studies to show that the ANDA drug is bioequivalent to the previously approved reference listed brand drug. Bioequivalence studies compare the bioavailability of the proposed drug product with that of the RLD product containing the same active ingredient. Bioavailability is a measure of the rate and extent to which the active ingredient or active moiety is absorbed from a drug product and becomes available at the site of action. Thus, a demonstration of bioequivalence confirms the absence of a significant difference between the proposed product and the reference listed brand drug in terms of the rate and extent to which the active ingredient or active moiety becomes available at the site of drug action when administered at the same molar dose under similar conditions. An ANDA also typically must show that the proposed generic product is the same as the RLD in terms of active ingredient(s), strength, dosage form, route of administration and labeling.
Generic products are generally introduced to the marketplace at the expiration of patent protection for the brand product or at the end of a period of non-patent market exclusivity. However, if an ANDA applicant files an ANDA containing a certification of invalidity, non-infringement or unenforceability related to a patent listed in the Orange Book with respect to a reference drug product, the applicant may be able to market the generic equivalent prior to the expiration of patent protection for the brand product. Such patent certification is commonly referred to as a Paragraph IV certification. Generally, if the patent owner brings an infringement action within 45 days from receiving notification by the applicant, the FDA may not approve the ANDA application until the earlier of the rendering of a court decision favorable to the ANDA applicant or the expiration of 30 months. An ANDA applicant that is first to file a substantially complete ANDA containing a Paragraph IV certification is eligible for a period of generic marketing exclusivity. This exclusivity, which under certain circumstances may be required to be shared with other applicable ANDA sponsors with Paragraph IV certifications, lasts for 180 days, during which the FDA cannot grant final approval to other ANDA sponsors holding applications for a generic equivalent to the same reference drug.
In addition to patent exclusivity, the holder of the NDA for the listed drug may be entitled to a period of non-patent market exclusivity, during which the FDA cannot approve (or in some cases, accept for review) an application for a generic version product. If the reference drug is a new chemical entity (which generally means the active moiety has not previously been approved), the FDA may not accept an ANDA for a generic product for up to five years following approval of the NDA for the new chemical entity. If it is not a new chemical entity, but the holder of the NDA conducted clinical trials essential to
10
Table of Contents
approval of the NDA or a supplement thereto, the FDA may not approve an ANDA for reference NDA product before the expiration of three years from the date of approval of the NDA or supplement. Certain other periods of exclusivity may be available if the RLD is indicated for treatment of a rare disease or the sponsor conducts pediatric studies in accordance with FDA requirements.
Supplemental ANDAs are required for approval of various types of changes to an approved application and these supplements may be under review for six months or more. In addition, certain types of changes may only be approved once new bioequivalence studies are conducted or other requirements are satisfied.
A number of branded pharmaceutical patent expirations are expected over the next several years. These patent expirations should provide additional generic product opportunities. We intend to concentrate our generic product development activities on branded products with significant sales in specialized or growing markets or in areas that offer significant opportunities and other competitive advantages. In addition, we intend to continue to focus our development efforts on technically difficult-to-formulate products or products that require advanced manufacturing technology.
The Biologics Price Competition and Innovation Act ("BPCIA") authorizes the FDA to license a biological product that is a "biosimilar" to an FDA-licensed biologic through an abbreviated pathway. The BPCIA establishes criteria for determining that a product is biosimilar to an already licensed biologic, known as the "reference product," and establishes a process by which an abbreviated BLA for a biosimilar product is submitted, reviewed and approved. This abbreviated approval pathway is intended to permit a biosimilar product to come to market more quickly and less expensively than if a full BLA were submitted, by relying to some extent on FDA's previous review and approval of the reference product. Generally, a biosimilar must be shown to be highly similar to, and have no clinically meaningful differences in safety, purity or potency from, the reference product. The BPCIA provides periods of exclusivity that protect a reference product from biosimilars competition. Under the BPCIA, the FDA may not accept a biosimilar application for review until four years after the date of first licensure of the reference product, and the biosimilar may not be licensed until twelve years after the reference product's approval. Additionally, the BPCIA establishes procedures by which the biosimilar applicant must provide information about its application and product to the reference product sponsor, and by which information about potentially relevant patents is shared and litigation over patents may proceed in advance of approval. The BPCIA also provides a period of exclusivity for the first biosimilar to be determined by the FDA to be interchangeable with the reference product.
Because the BPCIA is a relatively new law, we anticipate that its contours will be defined as the statute is implemented over a period of years. This likely will be accomplished by a variety of means, including FDA issuance of guidance documents, proposed regulations, and decisions in the course of considering specific applications. In that regard, the FDA has to date issued various guidance documents and other materials providing indications of the agency's thinking regarding any number of issues implicated by the BPCIA. Additionally, the FDA's approval in 2015 of the first biosimilar application helped define the agency's approach to certain issues.
An additional requirement for FDA approval of NDAs and ANDAs is that our manufacturing procedures and operations conform to FDA requirements and guidelines, generally referred to as current Good Manufacturing Practices ("cGMP"). The requirements for FDA approval encompass all aspects of the production process, including validation and recordkeeping, the standards around which are continuously changing and evolving.
Facilities, procedures, operations and/or testing of products are subject to periodic inspection by the FDA, the Drug Enforcement Administration ("DEA") and other authorities. In addition, the FDA conducts pre-approval and post-approval reviews and plant inspections to determine whether our systems and processes are in compliance with cGMP and other FDA regulations. Our suppliers are subject to similar regulations and periodic inspections.
In 2012, the Food and Drug Administration Safety and Innovation Act ("FDASIA") was enacted into law. FDASIA is intended to enhance the safety and security of the U.S. drug supply chain by holding all drug manufacturers supplying products to the U.S. to the same FDA inspection standards. Specifically, prior to the passage of FDASIA, U.S. law required U.S. based manufacturers to be inspected by FDA every two years but remained silent with respect to foreign manufacturers, causing some foreign manufacturers to go as many as nine years without a routine FDA cGMP inspection, according to the Government Accountability Office.
FDASIA also includes the Generic Drug User Fee Agreement ("GDUFA"), a novel user fee program to provide FDA with approximately $1.5 billion in total user fees through 2018 focused on three key aims:
Safety – Ensure that industry participants, foreign or domestic, are held to consistent quality standards and are inspected with foreign and domestic parity using a risk-based approach.
11
Table of Contents
Access – Expedite the availability of generic drugs by bringing greater predictability to the review times for abbreviated new drug applications, amendments and supplements and improving timeliness in the review process.
Transparency – Enhance FDA's visibility into the complex global supply environment by requiring the identification of facilities involved in the manufacture of drugs and associated APIs, and improve FDA's communications and feedback with industry.
Under GDUFA, 70% of the total fees are being derived from facility fees paid by FDF manufacturers and API facilities listed or referenced in pending or approved generic drug applications. The remaining 30% of the total fees are being derived from application fees, including generic drug application fees, prior approval supplement fees and DMF fees.
The process required by the FDA before a pharmaceutical product with active ingredients that have not been previously approved may be marketed in the U.S. generally involves the following:
• | laboratory and preclinical tests; |
• | submission of an Investigational New Drug ("IND") application, which must become effective before clinical studies may begin; |
• | adequate and well-controlled human clinical studies to establish the safety and efficacy of the proposed product for its intended use; |
• | submission of an NDA or BLA containing the results of the preclinical tests and clinical studies establishing the safety and efficacy of the proposed product for its intended use, as well as extensive data addressing matters such as manufacturing and quality assurance; |
• | scale-up to commercial manufacturing; and |
• | FDA approval of an NDA or BLA. |
Preclinical tests include laboratory evaluation of the product and its chemistry, formulation and stability, as well as toxicology and pharmacology studies to help define the pharmacological profile of the drug and assess the potential safety and efficacy of the product. The results of these studies are submitted to the FDA as part of the IND. They must demonstrate that the product delivers sufficient quantities of the drug to the bloodstream or intended site of action to produce the desired therapeutic results before human clinical trials may begin. These studies must also provide the appropriate supportive safety information necessary for the FDA to determine whether the clinical studies proposed to be conducted under the IND can safely proceed. The IND automatically becomes effective 30 days after receipt by the FDA unless the FDA, during that 30-day period, raises concerns or questions about the conduct of the proposed trials, as outlined in the IND. In such cases, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials may begin. In addition, an independent institutional review board must review and approve any clinical study prior to initiation.
Human clinical studies are typically conducted in three sequential phases, which may overlap:
• | Phase I – The drug is initially introduced into a relatively small number of healthy human subjects or patients and is tested for safety, dosage tolerance, mechanism of action, absorption, metabolism, distribution and excretion. |
• | Phase II – Studies are performed with a limited patient population to identify possible adverse effects and safety risks, to assess the efficacy of the product for specific targeted diseases or conditions, and to determine dosage tolerance and optimal dosage. |
• | Phase III – When Phase II evaluations demonstrate that a dosage range of the product is effective and has an acceptable safety profile, Phase III trials are undertaken to evaluate further dosage and clinical efficacy and to test further for safety in an expanded patient population at geographically dispersed clinical study sites. |
The results of the product development, preclinical studies and clinical studies are then submitted to the FDA as part of the NDA or BLA. The NDA/BLA drug development and approval process could take from three to more than ten years.
Canada
In Canada, the approval process for all generic pharmaceuticals has two tracks that may proceed in parallel. The first track involves an examination of the product by Health Canada, the agency responsible for national public health, to ensure that the quality, safety and efficacy of the product have been established. Second persons (i.e., generic companies) may seek
12
Table of Contents
approval to sell a product by submitting an abbreviated new drug submission ("ANDS") to Health Canada to demonstrate that its product is bioequivalent to the brand reference product already marketed in Canada under a Notice of Compliance ("NOC"). When Health Canada is satisfied with the quality, safety and efficacy described in the ANDS, it issues a NOC for that product, subject to any brand patents in the second track of the approval process.
The second track of the approval process is governed by the Patented Medicines NOC Regulations ("Regulations"). The owner or exclusive licensee of patents relating to the brand drug (the "originator") may list patents relating to the medicinal ingredient, formulation, dosage form or the use of the drug on the Patent Register. Where a generic applicant makes direct or indirect reference in its ANDS to a brand product for which there are patents listed on the Patent Register, the generic must make at least one of the statutory allegations with respect to each patent listed (e.g., that the generic will await patent expiry, or the patent is invalid and/or would not be infringed). If the generic challenges the listed patent, it is required to serve the originator with a Notice of Allegation ("NOA"), which gives a detailed statement of the factual and legal basis for its allegations. If the originator wishes to seek an order prohibiting the issuance of the NOC to the generic, it must commence a court application within 45 days after it has been served with the NOA. Once an application is commenced, Health Canada may not issue a NOC until the earlier of the determination of the proceeding by the court, or the expiration of 24 months. To obtain a prohibition order, the originator must satisfy the court that the generics' allegations of invalidity and/or non-infringement are not justified.
Section C.08.004.1 of the Canadian Food and Drug Regulations is the so-called data protection provision. A generic applicant does not need to perform duplicate clinical trials similar to those conducted by the first NOC holder (i.e., the brand), but is permitted to demonstrate safety and efficacy by submitting data demonstrating that its formulation is bioequivalent to the approved brand formulation. The first party to obtain an NOC for a drug will have an eight-year period of exclusivity starting from the date it received its NOC based on that clinical data. A subsequent applicant who seeks to establish safety and efficacy by comparing its product to the product that received the first NOC will not be able to file its own application until six years after the issuance of the first NOC, and cannot receive ultimate approval for an additional two years. If the first NOC holder also conducts clinical trials in pediatric populations, it will be entitled to an extra six months of data protection. A drug is only entitled to data protection so long as it is being marketed in Canada.
Facilities, procedures, operations and/or testing of products are subject to periodic inspection by Health Canada and the Health Products and Food Branch Inspectorate. In addition, Health Canada conducts reviews and plant inspections to determine whether our systems are in compliance with the good manufacturing practices in Canada, Drug Establishment Licensing ("EL") requirements and other provisions of the Regulations. Competitors are subject to similar regulations and inspections.
Europe
The EU presents complex challenges from a regulatory perspective. There is over-arching legislation which is then implemented at a local level by the 28 individual member states, Iceland, Liechtenstein and Norway. Between 1995 and 1998, the legislation was revised in an attempt to simplify and harmonize product registration. This revised legislation introduced the mutual recognition ("MR") procedure, whereby after submission and approval by the authorities of the so-called reference member state ("RMS"), further applications can be submitted into the other chosen member states (known as concerned member states ("CMS")). Theoretically, the authorization of the RMS should be mutually recognized by the CMS. More typically, however, a degree of re-evaluation is carried out by the CMS. In November 2005, this legislation was further revised. In addition to the MR procedure, the decentralized procedure ("DCP") was introduced. The DCP is also led by the RMS, but applications are simultaneously submitted to all selected countries, provided that no national marketing authorization has been granted yet for the medicinal product in question. From 2005, the centralized procedure operated by the European Medicines Agency ("EMA") became available for generic versions of innovator products approved through the centralized authorization procedure. The centralized procedure results in a single marketing authorization (in addition to separate marketing authorizations for Iceland, Lichtenstein and Norway) which, once granted, can be used by the marketing-authorization holder to file for individual country reimbursement and make the medicine available in all of the EU countries listed on the application.
In the EU, the manufacture and sale of pharmaceutical products is regulated in a manner substantially similar to that of the U.S. requirements, which generally prohibit the handling, manufacture, marketing and importation of any pharmaceutical product unless it is properly registered in accordance with applicable law. The registration file relating to any particular product must contain medical data related to product efficacy and safety, including results of clinical testing and references to medical publications, as well as detailed information regarding production methods and quality control. Health ministries are authorized to cancel the registration of a product if it is found to be harmful or ineffective or if it is manufactured or marketed other than in accordance with registration conditions.
13
Table of Contents
Pursuant to the MR procedure, a marketing authorization is first sought in one member state from the national regulatory agency (the "RMS"). The RMS makes its assessment report on the quality, efficacy and safety of the medicinal product available to the other CMSs where marketing authorizations are also sought under the MR procedure.
The DCP is based on the same fundamental idea as the MR procedure. In contrast to the MR procedure, however, the DCP requires that no national marketing authorization has yet been granted for the medicinal product. The pharmaceutical company applies for marketing authorization simultaneously in all the member states of the EU in which it wants to market the product. After consultation with the pharmaceutical company, one of the member states concerned in the DCP will become the RMS. The competent agency of the RMS undertakes the scientific evaluation of the medicinal product on behalf of the other CMSs and coordinates the procedure. If all the member states involved (RMS and CMS) agree to grant marketing authorizations, this decision forms the basis for the granting of the national marketing authorizations in the respective member states.
Neither the MR nor DCPs result in automatic approval in all member states. If any member state has objections, particularly in relation to potential serious risk to public health, which cannot be resolved within the procedure scope and timelines, they will be referred to the coordination group for MR and DCPs and reviewed in a 60 -day procedure. If this 60 -day procedure does not result in a consensus by all member states, the product can be marketed in the countries whose health authorities agree that the product can be licensed. The issue raised will then enter a second referral procedure.
As with the MR procedure, the advantage of the DCP is that the pharmaceutical company receives identical marketing authorizations for its medicinal product in all the member states of the EU in which it wants to market the product. This leads to considerable streamlining of all regulatory activities in regard to the product. Variations, line extensions, renewals, and more are also handled in a coordinated manner with the RMS leading the activity.
Once a DCP has been completed, the pharmaceutical company can subsequently apply for marketing authorizations for the medicinal product in additional EU member states by means of the MR procedure.
All products, whether centrally authorized or authorized by the MR or DCP, may only be sold in other member states if the product information is in the official language of the state in which the product will be sold, which effectively requires specific packaging and labeling of the product.
Before a generic pharmaceutical product can be marketed in the EU, a marketing authorization must be obtained. If a generic pharmaceutical product is shown to be essentially the same as, or bioequivalent to, one that is already on the market and which has been authorized in the EU for a specified number of years, as explained in the section on data exclusivity below, no further preclinical or clinical trials are required for that new generic pharmaceutical product to be authorized. The generic applicant can file an abridged application for marketing authorization, but in order to take advantage of the abridged procedure, the generic manufacturer must demonstrate specific similarities, including bioequivalence, to the already authorized product. Access to clinical data of the reference drug is governed by the European laws relating to data exclusivity, which are outlined below. Other products, such as new dosages of established products, must be subjected to further testing, and "bridging data" in respect of these further tests must be submitted along with the abridged application.
An applicant for a generic marketing authorization currently cannot avail itself of the abridged procedure in the EU by relying on the originator pharmaceutical company's data until expiry of the relevant period of exclusivity given to that data. For products first authorized prior to October 30, 2005 , this period is six or ten years (depending on the member state in question and/or the regulatory procedure used by the originator) after the grant of the first marketing authorization sought for the relevant product, due to data exclusivity provisions which have been in place. From October 30, 2005 , the implementation of a new EU directive (2004/27/EC) harmonized the data exclusivity period for originator pharmaceutical products throughout the EU member states, which were legally obliged to have implemented the directive by October 30, 2005 . The new regime for data exclusivity provides for an eight -year data exclusivity period commencing from the grant of first marketing authorization. After the eight -year period has expired, a generic applicant can refer to the data of the originator pharmaceutical company in order to file an abridged application for approval of its generic equivalent product. Yet, conducting the necessary studies and trials for an abridged application, within the data exclusivity period, is not regarded as contrary to patent rights or to supplementary protection certificates for medicinal products. However, the applicant will not be able to launch its product for an additional two years. This ten -year total period may be extended to 11 years if the original marketing authorization holder obtains, within those initial eight years, a further authorization for a new therapeutic use of the product which is shown to be of significant clinical benefit. Further, specific data exclusivity for one year may be obtained for a new indication for a well-established substance, provided that significant preclinical or clinical studies were carried out in relation to the new indication. This new regime for data exclusivity applies to products first authorized after October 30, 2005 .
14
Table of Contents
Under the national procedure, a company applies for a marketing authorization in one member state. The national procedure can now only be used if the pharmaceutical company does not seek authorization in more than one member state. If it does seek wider marketing authorizations, it must use the MR or DCP.
In addition to obtaining approval for each product, in most EU countries the pharmaceutical product manufacturer's facilities must obtain approval from the national supervisory authority. The EU has a code of good manufacturing practice, with which the marketing authorization holder must comply. Regulatory authorities in the EU may conduct inspections of the manufacturing facilities to review procedures, operating systems and personnel qualifications.
In order to control expenditures on pharmaceuticals, most member states in the EU regulate the pricing and reimbursement of products and in some cases limit the range of different forms of drugs available for prescription by national health services. These controls can result in considerable price differences between member states. In addition, in past years, as part of overall programs to reduce healthcare costs, certain European governments have prohibited price increases and have introduced various systems designed to lower prices. Some European governments have also set minimum targets for generics prescribing.
Certain European markets in which Mylan does business have recently undergone, some for the first time, or will soon undergo, government-imposed price reductions or similar pricing pressures on pharmaceutical products. In addition, a number of markets in which we operate have implemented or may implement tender, or tender-like, systems for generic pharmaceuticals in an effort to lower prices. Under tender systems, health insurers invite manufacturers to submit bids that establish prices for generic pharmaceuticals. Upon winning the tender, the winning company may receive a preferential reimbursement for a period of time. Such measures are likely to have a negative impact on sales and gross profit in these markets. However, some pro-generic government initiatives in certain markets could help to offset some of this unfavorable effect by potentially increasing generic utilization.
Rest of World
Australia
The pharmaceutical industry is one of the most highly regulated industries in Australia. The Australian government is heavily involved in the operation of the industry, through the registration of medicines and licensing of manufacturing facilities, as well as subsidizing patient cost of most prescription medicines sold in Australia. The Australian government authority, the Therapeutic Goods Administration (the "TGA"), regulates the quality, safety and efficacy of therapeutic goods and is responsible for granting authorization to market pharmaceutical products in Australia and for inspecting and approving manufacturing facilities.
The TGA operates according to the Commonwealth of Australia's Therapeutic Goods Act 1989 (Cth) (the "Act"). Specifically, the Act regulates the registration, listing, quality, safety, efficacy, promotion and sale of therapeutic goods, including pharmaceuticals, supplied in Australia. The TGA carries out a range of assessment and monitoring activities to ensure that therapeutic goods available in Australia are of an acceptable standard with a goal of ensuring that the Australian community has access within a reasonable time to therapeutic advances. Australian manufacturers of all medicines must be licensed under Part 3-3 of the Act and their manufacturing processes must comply with the principles of good manufacturing practices in Australia. Similar standards and audits apply for both domestic and foreign manufactured products.
Generic medicines are subject to an abbreviated review process by the TGA, if the product can demonstrate essential similarity to the originator brand. Essential similarity means the same active ingredient in the same dose form, delivering the active ingredient to the patient at the same rate and extent, compared to the original brand. If proven, safety and efficacy is assumed to be the same.
All therapeutic goods manufactured for supply in Australia must be listed or registered in the Australian Register of Therapeutic Goods (the "ARTG") before they can be promoted or supplied for use and/or sale in Australia. The ARTG is a database kept for the purpose of compiling information in relation to therapeutic goods for use in humans and lists therapeutic goods which are approved for supply in Australia.
Medicines assessed as having a higher level of risk must be registered, while those with a lower level of risk can be listed. The majority of listed medicines are self-selected by consumers and used for self-treatment. In assessing the level of risk, factors such as the strength of a product, side effects, potential harm through prolonged use, toxicity and the seriousness of the medical condition for which the product is intended to be used are taken into account.
15
Table of Contents
Labeling, packaging and advertising of pharmaceutical products are also regulated by the Act and other relevant statutes including fair trading laws and pharmaceutical industry codes.
Australia has a five-year data exclusivity period, whereby any data relating to a pharmaceutical product cannot be referred to or used in the examination by the TGA of another company's dossier, until five years after the original product was approved.
The Pharmaceutical Benefits Scheme (the "PBS"), which has been in place since 1948, subsidizes the cost to consumers of medicines listed on the PBS, if the medicines have demonstrated acceptable clinical need, cost and effectiveness. The goal of the PBS is to make medicines available at the lowest cost compatible with reliable supply and to base access on medical need rather than ability to pay.
The government exerts a significant degree of control over the pharmaceuticals market through the PBS. More than 80% of all prescription medicine sold in Australia is reimbursed by the PBS. The PBS is operated under the Commonwealth of Australia's National Health Act 1953. This statute governs matters such as who may sell pharmaceutical products, the prices at which pharmaceutical products may be sold to consumers and the prices government pays manufacturers, wholesalers and pharmacists for subsidized medicines.
If a new medicine is to be considered for listing on the PBS, the price is determined through a full health economic analysis submitted to the government's advisory committee, the Pharmaceutical Benefits Advisory Committee (the "PBAC"), based on incremental benefit to health outcome. If the incremental benefit justifies the price requested, the PBAC then makes a recommendation to the government to consider listing the product on the PBS. In May 2014, as part of a government reform program in Australia, the Pharmaceutical Benefits Pricing Authority was abolished and the Minister for Health ("Minister"), or delegate, considers pricing matters for approximately five to six weeks following PBAC meetings. Factors contributing to pricing decisions include items such as information on the claims made in a submission, advice from the PBAC, information about the proposed price, the price and use of comparative medicines and the cost of producing the medicine, although with additional associated costs. The Minister may recommend that the proposed price is accepted; further negotiations take place for a lower price or prices within a specific range; or for some products, risk sharing arrangements to be developed and agreed upon. The Australian government's purchasing power is used to obtain lower prices as a means of controlling the cost of the program. The PBS also stipulates the wholesaler margin for drugs listed on the PBS. Wholesalers therefore have little pricing power over the majority of their product range and as a result are unable to increase profitability by increasing prices.
Following entry of the first generic product(s) onto the market, the PBS price reimbursed to pharmacies decreases by 16% for both the originator product and generic products with a brand equivalence indicator permitting substitution at the pharmacy level. Thereafter, both the originator and generic suppliers are required to disclose pricing information relating to the sale of medicines to the Price Disclosure Data Administrator, and twelve months (up until October 2014, it was 18 months) after initial generic entry, there is a further PBS price reduction based on the weighted average disclosed price if the weighted average disclosed price is 10% or more below the existing PBS price. Ongoing price disclosure cycles and calculation of the weighted average disclosed price occur every six months, and further reductions are made to the PBS price whenever the weighted average disclosed price is 10% or more below the existing PBS price. The price disclosure system has had, and will continue to have, a negative impact on sales and gross profit in this market.
Japan
In Japan, we are governed by various laws and regulations, including the Pharmaceutical Affairs Law (Law No. 145, 1960), as amended by the Pharmaceuticals and Medical Devices Law ("PMDL"), and the Products Liability Law (Law No. 85, 1994). The PMDL was amended in November 2014 to establish a fast-track authorization process for regenerative medicine products, restructure medical device regulation and establish reporting obligations for package inserts for drugs and medical devices. Regenerative medicine products are newly defined under the amended PMDL as a product for medical use in humans to reconstruct, restore, or form the structure or function of a human body, in which cells of humans are cultured or otherwise processed.
Under the amended PMDL, there are two routes to obtain authorization to manufacture and market a medicine product. The first route is the standard authorization system for drugs in which the efficacy and safety of the product must be shown in order to obtain authorization. The standard authorization procedure may take a significant amount of time to launch a regenerative medicine product because the quality of regenerative medicine products is heterogeneous by nature and therefore it is difficult to collect the data necessary to evaluate and demonstrate the efficacy. As such, the amended PMDL instituted the second route as follows: if the regenerative medicine product is heterogeneous, the efficacy of the regenerative medicine product is assumed. Thus, if the safety of the regenerative medicine product is demonstrated through clinical trials, the Minister
16
Table of Contents
of the Ministry of Health, Labor and Welfare ("MHLW") may authorize the applicant to manufacture and market the regenerative medicine product with certain conditions for a fixed term after receiving an expert opinion from the Pharmaceutical Affairs and Food Sanitation Council.
The amended PMDL also restructured medical device regulations including expanding the scope for certification in accordance with the classifications agreed upon by the Global Harmonization Task Force, new regulations on medical device software in which software may be authorized as a medical device independent of the medical device hardware into which it is incorporated, system change for medical device manufacturing so that a company may manufacture a medical device when the company registers such medical device and streamlined Quality Management Service Inspection such that the inspection is performed for each category of medical products.
In addition, under the amended PMDL, the holder of a business license for the manufacture and marketing of regenerative medicine products or medical devices must notify the MHLW of the contents of the package insert, including any cautionary statements necessary to use and deal with the products, before it manufactures and markets them. The license holder must also publish the contents of the package inserts on the website of the Pharmaceuticals and Medical Devices Agency.
Under the amended PMDL, the retailing or supply of a pharmaceutical that a person has manufactured (including manufacturing under license) or imported is defined as "marketing," and in order to market pharmaceuticals, one has to obtain a license, which we refer to herein as a Marketing License, from the MHLW. The authority to grant the "Marketing License" is delegated to prefectural governors; therefore, the relevant application must be filed with the relevant prefectural governor. A Marketing License will not be granted if the quality control system for the pharmaceutical for which the Marketing License has been applied or the post-marketing safety management system for the relevant pharmaceutical does not comply with the standards specified by the relevant Ministerial Ordinance made under the amended PMDL.
In addition to the Marketing License, a person intending to market a pharmaceutical must, for each product, obtain marketing approval from the MHLW with respect to such marketing, which we refer to herein as "Marketing Approval." Marketing Approval is granted subject to examination of the name, ingredients, quantities, structure, administration and dosage, method of use, indications and effects, performance and adverse reactions, and the quality, efficacy and safety of the pharmaceutical. A person intending to obtain Marketing Approval must attach materials, such as data related to the results of clinical trials (including a bioequivalence study, in the case of generic pharmaceuticals) or conditions of usage in foreign countries. Japan provides for market exclusivity through a reexamination system, which prevents the entry of generic pharmaceuticals until the end of the re-examination period, which can be up to eight years, and ten years in the case of drugs used to treat rare diseases ("orphan drugs").
The authority to grant Marketing Approval in relation to pharmaceuticals for certain specified purposes (e.g., cold medicines and decongestants) is delegated to the prefectural governors by the MHLW, and applications in relation to such pharmaceuticals must be filed with the governor of the relevant prefecture where the relevant company's head office is located. Applications for pharmaceuticals for which the authority to grant the Marketing Approval remains with the MHLW must be filed with the Pharmaceuticals and Medical Devices Agency. When an application is submitted for a pharmaceutical whose active ingredients, quantities, administration and dosage, method of use, indications and effects are distinctly different from those of pharmaceuticals which have already been approved, the MHLW must seek the opinion of the Pharmaceutical Affairs and Food Sanitation Council.
The amended PMDL provides that when (a) the pharmaceutical that is the subject of an application is shown not to result in the indicated effects or performance indicated in the application, (b) the pharmaceutical is found to have no value as a pharmaceutical because it has harmful effects outweighing its indicated effects or performance, or (c) in addition to (a) and (b) above, when the pharmaceutical falls within the category designated by the relevant Ministerial Ordinance as not being appropriate as a pharmaceutical, Marketing Approval shall not be granted.
The MHLW must cancel a Marketing Approval, after hearing the opinion of the Pharmaceutical Affairs and Food Sanitation Council, when the MHLW finds that the relevant pharmaceutical falls under any of (a) through (c) above. In addition, the MHLW can order the amendment of a Marketing Approval when it is necessary to do so from the viewpoint of public health and hygiene. Moreover, the MHLW can order the cancellation or amendment of a Marketing Approval when (1) the necessary materials for re-examination or re-evaluation, which the MHLW has ordered considering the character of pharmaceuticals, have not been submitted, false materials have been submitted or the materials submitted do not comply with the criteria specified by the MHLW, (2) the relevant company's Marketing License has expired or has been canceled (a Marketing License needs to be renewed every five years), (3) the regulations regarding investigations of facilities in relation to manufacturing management standards or quality control have been violated, (4) the conditions set in relation to the Marketing
17
Table of Contents
Approval have been violated, or (5) the relevant pharmaceutical has not been marketed for three consecutive years without a due reason.
Doctors and pharmacists providing medical services pursuant to national health insurance (the "NHI") are prohibited from using pharmaceuticals other than those specified by the MHLW. The MHLW also specifies the standards of pharmaceutical prices, which we refer to herein as NHI Drug Price Standards. The NHI Drug Price Standards are used as the basis of the calculation of the price paid by medical insurance for pharmaceuticals. The governmental policy relating to medical services and the health insurance system, as well as the NHI Drug Price Standards, is revised every two years. In December 2016, the Council on Economic and Fiscal Policy, announced that it intended to revise various aspects of its drug pricing policies, including among others, a move from biannual drug pricing revisions to annual revisions. The critical implementation particulars, including timing and mechanisms, of such policy changes have yet to be fully defined.
Brazil
In Brazil, pharmaceutical manufacturers and all products and services that affect the health of the population are regulated by the National Agency of Sanitary Surveillance ("ANVISA"), created by Law No. 9,782, of January 26, 1999. ANVISA is a governmental body directly linked to the Ministry of Health and is part of the Unified Health System, responsible for the sanitary control of production, storage, distribution, importation and marketing of products and services subject to sanitary surveillance. ANVISA is also responsible for registering drugs and supervising quality control, as well as issuing licenses to companies for the manufacturing, handling, packaging, distribution, advertising, importation and exportation of pharmaceutical products. ANVISA regularly monitors the market's economic regulations and is responsible for the price control of pharmaceutical drugs.
Active Pharmaceutical Ingredients
The primary regulatory oversight of API manufacturers is through inspection of the manufacturing facility in which APIs are produced, as well as the manufacturing processes and standards employed in the facility. The regulatory process by which API manufacturers generally register their products for commercial sale in the U.S. and other similarly regulated countries is via the filing of a DMF. DMFs are confidential documents containing information on the manufacturing facility and processes used in the manufacture, characterization, quality control, packaging and storage of an API. The DMF is reviewed for completeness by the FDA, or other similar regulatory agencies in other countries, in conjunction with applications filed by FDF manufacturers, requesting approval to use the given API in the production of their drug products.
Over-the-Counter Drug Products
A nonprescription, or OTC product, is a product that is sold directly to a consumer without a prescription from a healthcare professional, as compared to a prescription product, which may be sold only to consumers possessing a valid prescription. In many countries, OTC products are generally marketed with some type of safety and effectiveness review by a regulatory agency to ensure that they contain ingredients that are safe and effective when used without a physician's care. Like prescription products, an OTC product is also subjected to other general regulatory requirements, including those applicable to manufacturing practices and product advertising and promotion.
With the acquisition of Meda, the Company significantly enhanced its OTC product portfolio. The demand for OTC products is driven in part by government and healthcare provider cost pressures. The top OTC markets include developed markets like the U.S. and Europe as well as developing markets like China, Brazil and India, with the developing markets experiencing higher growth rates. In developed markets, the switch from prescription to OTC products in categories such as respiratory and gastrointestinal health has expanded access to treatments while reducing the cost for the healthcare systems.
Research and Development
R&D efforts are conducted on a global basis, primarily to enable us to develop, manufacture and market approved pharmaceutical products in accordance with applicable government regulations. Through various acquisitions, we have significantly bolstered our global R&D capabilities over the past several years, particularly in injectables and respiratory therapies. In the U.S., our largest market, the FDA is the principal regulatory body with respect to pharmaceutical products. Each of our other markets have separate pharmaceutical regulatory bodies, including, but not limited to, the National Agency for Medicines and Health Products in France, Health Canada, the Medicines and Healthcare Products Regulatory Agency in the U.K., the EMA (a decentralized body of the EU), the Federal Institute for Drugs and Medical Devices in Germany, the Health Products Regulatory Agency in Ireland, the Italian Medicines Agency, the Spanish Agency of Medicines and Medical Devices,
18
Table of Contents
the TGA in Australia, the MHLW in Japan, Drug Controller General of India, ANVISA in Brazil and the World Health Organization ("WHO"), the regulatory body of the United Nations.
Our global R&D strategy emphasizes the following areas:
• | development of both branded and generic finished dose products for the global marketplace; |
• | development of pharmaceutical products that are technically difficult to formulate or manufacture because of either unusual factors that affect their stability or bioequivalence or unusually stringent regulatory requirements; |
• | development of novel controlled-release technologies and the application of these technologies to reference products; |
• | development of drugs that target smaller, specialized or underserved markets; |
• | development of generic drugs that represent first-to-file opportunities in the U.S. market; |
• | expansion of the existing oral solid dosage product portfolio, including with respect to additional dosage strengths; |
• | development of injectable products; |
• | development of unit dose oral inhalation products for nebulization; |
• | development of APIs; |
• | development of compounds using a dry powder inhaler and/or metered-dose inhaler for the treatment of asthma, COPD and other respiratory therapies; |
• | development of monoclonal anti-bodies (which are regulated as biologics); |
• | development of products as a result of changes in product status from prescription only to OTC; |
• | completion of additional preclinical and clinical studies for approved NDA products required by the FDA, known as post-approval (Phase IV) commitments; and |
• | conducting life-cycle management studies intended to further define the profile of products subject to pending or approved NDAs. |
The success of biosimilars in the marketplace and our ability to be successful in this emerging market will depend on the implementation of balanced scientific standards for approval, while not imposing excessive clinical testing demands or other hurdles for well-established products. Furthermore, an efficient patent resolution mechanism and a well-defined mechanism to grant interchangeability after the establishment of biosimilarity with the reference biological product will be key elements determining our future success in this area.
We have a robust pipeline. As of December 31, 2016 , we had approximately 3,396 marketing license approvals pending. During 2016 , we completed 1,085 global country level product submissions, which included 44 in North America, 621 in Europe and 420 in Rest of World. These submissions included those for existing products in new markets as well as products new to the Mylan portfolio.
During the year ended December 31, 2016 , we received 873 individual country product approvals globally, which was equal to 1,234 approved new marketing licenses. Of those total individual country product approvals globally, there were 67 approvals in North America, including 50 in the U.S.; 508 approvals in Europe; and 298 approvals in Rest of World, of which 33 approvals were for ARV products. The 50 approvals in the U.S. consisted of 42 final ANDA approvals and eight tentative ANDA approvals. The 508 approvals in Europe covered 69 different products resulting in a total of 863 product marketing licenses. The 298 approvals in Rest of World included 258 approvals from emerging markets which represented 76 products in 53 countries.
As of December 31, 2016 , in the U.S. we had 247 ANDAs pending FDA approval, representing approximately $99.3 billion in annual sales for the brand name equivalents of these products for the year ended December 31, 2016 . Of those pending product applications, 42 were first-to-file Paragraph IV ANDA patent challenges, representing approximately $35.1 billion in annual brand sales for the year ended December 31, 2016 . The historic branded drug sales are not indicative of future generic sales, but are included to illustrate the size of the branded product market. Our R&D spending totaled approximately $827 million , $672 million and $582 million for the years ended December 31, 2016 , 2015 and 2014 , respectively.
19
Table of Contents
Collaboration and Licensing Agreements
We periodically enter into collaboration and licensing agreements with other pharmaceutical companies for the development, manufacture, marketing and/or sale of pharmaceutical products. Our significant collaboration agreements are focused on the development, manufacturing, supply and commercialization of multiple, high-value generic biologic compounds, insulin analog products and respiratory products. Under these agreements, we have future potential milestone payments and co-development expenses payable to third parties as part of our licensing, development and co-development programs. Payments under these agreements generally become due and are payable upon the satisfaction or achievement of certain developmental, regulatory or commercial milestones or as development expenses are incurred on defined projects. Milestone payment obligations are uncertain, including the prediction of timing and the occurrence of events triggering a future obligation. These agreements may also include potential sales-based milestones and call for us to pay a percentage of amounts earned from the sale of the product as a royalty or a profit share. These sales-based milestones or royalty obligations may be significant depending upon the level of commercial sales for each product.
On January 8, 2016, the Company entered into an agreement with Momenta Pharmaceuticals, Inc. (" Momenta ") to develop, manufacture and commercialize up to six of Momenta 's current biosimilar candidates, including Momenta 's biosimilar candidate, ORENCIA® (abatacept). Mylan paid an up-front cash payment of $45 million to Momenta. Under the terms of the agreement, the Company and Momenta are jointly responsible for product development and equally share in the costs and profits of the products with Mylan leading the worldwide commercialization efforts. Under the terms of the agreement, Momenta is eligible to receive additional contingent milestone payments of up to $200 million .
On November 2, 2016, the Company and Momenta announced that dosing had begun in a Phase 1 study to compare the pharmacokinetics, safety and immunogenicity of M834, a proposed biosimilar of ORENCIA® (abatacept), to U.S. and European Union sourced ORENCIA® in normal healthy volunteers. Under the agreement, Mylan paid $60 million related to certain milestones in 2016.
On January 30, 2015 , the Company entered into a development and commercialization collaboration with Theravance Biopharma, Inc. (" Theravance Biopharma ") for the development and, subject to FDA approval, commercialization of Revefenacin ("TD-4208"), a novel once-daily nebulized long-acting muscarinic antagonist for chronic obstructive pulmonary disease ("COPD") and other respiratory diseases. Under the terms of the agreement, Mylan and Theravance Biopharma will co-develop nebulized TD-4208 for COPD and other respiratory diseases. In addition, under the terms of the agreement, Theravance Biopharma is eligible to receive potential development and sales milestone payments totaling $220 million in the aggregate. As of December 31, 2016, Mylan has paid a total of $15 million in milestone payments to Theravance Biopharma .
In the fourth quarter of 2013, the Company entered into a licensing agreement with Pfizer Inc. for the exclusive worldwide rights to develop, manufacture and commercialize a novel long-acting muscarinic antagonist compound. Depending on the commercialization of this novel compound and the level of future sales and profits, the Company could also be obligated to make payments upon the occurrence of certain sales milestones, along with sales royalties and profit sharing payments. The Company has also entered into exclusive collaborations with Biocon on the development, manufacturing, supply and commercialization of multiple, high value generic biologic compounds and three insulin analog products for the global marketplace. The Company plans to provide funding related to the collaborations over the next several years. As the timing of cash expenditures is dependent upon a number of factors, many of which are out of the Company's control, it is difficult to forecast the amount of payments to be made over the next few years, which could be significant.
We are actively pursuing, and are currently involved in, joint projects related to the development, distribution and marketing of both generic and branded products. Many of these arrangements provide for payments by us upon the attainment of specified milestones. While these arrangements help to reduce the financial risk for unsuccessful projects, fulfillment of specified milestones or the occurrence of other obligations may result in fluctuations in cash flows and R&D expense.
Refer to Note 17 Collaboration and Licensing Agreements included in Item 8 in this Form 10-K for additional information related to our collaborations.
Patents, Trademarks and Licenses
We own or license a number of patents in the U.S. and other countries covering certain products and have also developed brand names and trademarks for other products. Generally, the branded pharmaceutical business relies upon patent protection to ensure market exclusivity for the life of the patent. We consider the overall protection of our patents, trademarks and license rights to be of significant value and act to protect these rights from infringement.
20
Table of Contents
In the branded pharmaceutical industry, the majority of an innovative product's commercial value is usually realized during the period in which the product has market exclusivity. In the U.S. and some other countries, when market exclusivity expires and generic versions of a product are approved and marketed, there can often be very substantial and rapid declines in the branded product's sales. The rate of this decline varies by country and by therapeutic category; however, following patent expiration, branded products often continue to have market viability based upon the goodwill of the product name, which typically benefits from trademark protection.
An innovator product's market exclusivity is generally determined by two forms of intellectual property: patent rights held by the innovator company and any regulatory forms of exclusivity to which the innovator is entitled.
Patents are a key determinant of market exclusivity for most branded pharmaceuticals. Patents provide the innovator with the right to lawfully exclude others from practicing an invention related to the medicine. Patents may cover, among other things, the active ingredient(s), various uses of a drug product, pharmaceutical formulations, drug delivery mechanisms and processes for (or intermediates useful in) the manufacture of products. Protection for individual products extends for varying periods in accordance with the expiration dates of patents in the various countries. The protection afforded, which may also vary from country to country, depends upon the type of patent, its scope of coverage and the availability of meaningful legal remedies in the country.
Market exclusivity is also sometimes influenced by regulatory intellectual property rights. Many developed countries provide certain non-patent incentives for the development of medicines. For example, the U.S., the EU and Japan each provide for a minimum period of time after the approval of a new drug during which the regulatory agency may not rely upon the innovator's data to approve a competitor's generic copy. Regulatory intellectual property rights are also available in certain markets as incentives for research on new indications, on orphan drugs and on medicines useful in treating pediatric patients. Regulatory intellectual property rights are independent of any patent rights and can be particularly important when a drug lacks broad patent protection. However, most regulatory forms of exclusivity do not prevent a competitor from gaining regulatory approval prior to the expiration of regulatory data exclusivity on the basis of the competitor's own safety and efficacy data on its drug, even when that drug is identical to that marketed by the innovator.
We estimate the likely market exclusivity period for each of our branded products on a case-by-case basis. It is not possible to predict the length of market exclusivity for any of our branded products with certainty because of the complex interaction between patent and regulatory forms of exclusivity and inherent uncertainties concerning patent litigation. There can be no assurance that a particular product will enjoy market exclusivity for the full period of time that we currently estimate or that the exclusivity will be limited to the estimate.
In addition to patents and regulatory forms of exclusivity, we also market products with trademarks. Trademarks have no effect on market exclusivity for a product, but are considered to have marketing value. Trademark protection continues in some countries as long as used; in other countries, as long as registered. Registration is for fixed terms and may be renewed indefinitely.
Customers and Marketing
In North America, we market products directly to wholesalers, distributors, retail pharmacy chains, long-term care facilities and mail order pharmacies. We also market our generic products indirectly to independent pharmacies, managed care organizations, hospitals, nursing homes, pharmacy benefit managers, GPOs and government entities. These customers, called "indirect customers," purchase our products primarily through our wholesale customers. In North America, wholesalers, retail drug chains, managed care organizations and payers have undergone, and are continuing to undergo, significant consolidation, which may result in these groups gaining additional purchasing leverage. Mylan markets its branded products to a number of different customer audiences in the U.S., including healthcare practitioners, wholesalers, pharmacists and pharmacy chains, hospitals, payers, pharmacy benefit managers, health maintenance organizations ("HMOs"), home healthcare, long-term care and patients. We reach these customers through our field-based sales force and National Accounts team, to increase our customers' understanding of the unique clinical characteristics and benefits of our branded products. Additionally, Mylan supports educational programs to consumers, physicians and patients.
In Europe and Rest of World, pharmaceuticals are sold to wholesalers, distributors, independent pharmacies and, in certain countries, directly to hospitals. Through a broad network of sales representatives, we adapt our marketing strategy to the different markets as dictated by their respective regulatory and competitive landscapes. Our API is sold primarily to pharmaceutical companies throughout the world, as well as to other Mylan subsidiaries.
21
Table of Contents
With the acquisition of Meda, we significantly expanded our offering of OTC products. The market for OTC products is growing and products are primarily marketed directly to consumers through a variety of media channels with an emphasis on developing and positioning the brands in a retail environment. The percentage of OTC products is generally higher in growth markets than in mature markets, often due to the fact that consumers in those markets have less access to advanced healthcare and reimbursement systems. In these circumstances, OTC products may replace prescription drugs. In more developed markets, demand for OTC products is driven by a growing interest in self-healing, wellness and improved quality of life. OTC products are commonly sold via retail channels such as pharmacies, drugstores or supermarkets directly to consumers. This makes it comparable to regular retail business with broad advertising and trade channel promotions. Consumers are often very loyal to well-known brands and as such, recommendation and reputation are very important in this market and it takes time and promotional effort to build strong brand names.
Beginning in 2015, we launched a comprehensive advertising campaign called "Better health for a better world™." The campaign represents Mylan's promise for the future as we transform into a healthcare company by setting new standards in the industry and providing 7 billion people access to high quality medicine, one person at a time. The campaign's goals are to educate consumers and customers about Mylan and to help ensure a smooth transition as we continue integrating the products from our recent acquisitions into our portfolio. In 2015, we launched the campaign in approximately 25 non-U.S. developed markets and, in 2016, introduced the campaign in more than 40 additional countries.
Major Customers
The following table represents the percentage of consolidated third party net sales to Mylan's major customers during the years ended December 31, 2016 , 2015 and 2014 .
| Percentage of Third Party Net Sales | |||||||
| 2016 |
| 2015 |
| 2014 | |||
McKesson Corporation | 16 | % |
| 15 | % |
| 19 | % |
AmerisourceBergen Corporation | 14 | % |
| 16 | % |
| 13 | % |
Cardinal Health, Inc. | 11 | % |
| 12 | % |
| 12 | % |
Consistent with industry practice, we have a return policy that allows our customers to return product within a specified period prior to and subsequent to the expiration date. See the Application of Critical Accounting Policies section of our "Management's Discussion and Analysis of Results of Operations and Financial Condition" for a discussion of our more significant revenue recognition provisions.
Competition
Our primary competitors include other generic companies (both major multinational generic drug companies and various local generic drug companies) and branded drug companies that continue to sell or license branded pharmaceutical products after patent expirations and other statutory expirations. In the branded space, key competitors are generally other branded drug companies that compete based on their clinical characteristics and benefits. Our OTC products face competition from other major pharmaceutical companies and retailers who carry their own private label brands. Our ability to compete in the various OTC markets is affected by several factors, including customer acceptance, reputation, product quality, pricing and the effectiveness of our promotional activities. OTC markets are highly fragmented in terms of product categories and geographic market coverage.
Competitive factors in the major markets in which we participate can be summarized as follows:
North America
The U.S. pharmaceutical industry is very competitive. Our competitors vary depending upon therapeutic areas and product categories. Primary competitors include the major manufacturers of brand name and generic pharmaceuticals. The primary means of competition are innovation and development, timely FDA approval, manufacturing capabilities, product quality, marketing, portfolio size, customer service, reputation and price. The environment of the U.S. pharmaceutical marketplace is highly sensitive to price. To compete effectively, we rely on cost-effective manufacturing processes to meet the rapidly changing needs of our customers around a reliable, high quality supply of generic pharmaceutical products.
Our competitors include other generic manufacturers, as well as branded companies, including those who license their products to generic manufacturers prior to patent expiration or as relevant patents expire, or who enact pricing strategies for
22
Table of Contents
their brands in order to compete directly with generics. Further regulatory approval is not required for a branded manufacturer to sell its pharmaceutical products directly or through a third-party to the generic market, nor do such manufacturers face any other significant barriers to entry into such market. Our competitors for certain branded products include branded manufacturers who offer products for the treatment of COPD and severe allergies, as well as brand companies that license their products to generic manufacturers prior to patent expiration.
The U.S. pharmaceutical market is undergoing, and is expected to continue to undergo, rapid and significant technological changes, and we expect competition to intensify as technological advances are made. We intend to compete in this marketplace by (1) developing therapeutic equivalents to branded products that offer unique marketing opportunities, are difficult to formulate and/or have significant market size, (2) developing or licensing brand pharmaceutical products that are either patented or proprietary and (3) developing or licensing pharmaceutical products that are primarily for indications having relatively large patient populations or that have limited or inadequate treatments available, among other strategies.
Our sales can be impacted by new studies that indicate that a competitor's product has greater efficacy for treating a disease or particular form of a disease than one of our products. Sales on some of our products can also be impacted by additional labeling requirements relating to safety or convenience that may be imposed on our products by the FDA or by similar regulatory agencies. If competitors introduce new products and processes with therapeutic or cost advantages, our products can be subject to progressive price reductions and/or decreased volume of sales.
Medicaid, a U.S. federal healthcare program, requires pharmaceutical manufacturers to pay rebates to state Medicaid agencies. The rebates are based on the volume of drugs that are reimbursed by the states for Medicaid beneficiaries. Sales of Medicaid-reimbursed non-innovator products require manufacturers to rebate 13% of the average manufacturer's price and, effective 2017, adjusted by the Consumer Price Index-Urban (the "CPI-U") based on certain data. Sales of the Medicaid-reimbursed innovator or single-source products require manufacturers to rebate the greater of approximately 23% of the average manufacturer's price or the difference between the average manufacturer's price and the best price adjusted by the CPI-U based on certain data. We believe that federal or state governments will continue to enact measures aimed at reducing the cost of drugs to the public.
Under Part D of the Medicare Modernization Act, Medicare beneficiaries are eligible to obtain discounted prescription drug coverage from private sector providers. As a result, usage of pharmaceuticals has increased, which is a trend that we believe will continue to benefit the generic pharmaceutical industry. However, such potential sales increases may be offset by increased pricing pressures, due to the enhanced purchasing power of the private sector providers that are negotiating on behalf of Medicare beneficiaries.
Canada is a well-established generics market characterized by a number of local and multinational competitors. The individual Canadian provinces control pharmaceutical pricing and reimbursement. A number of Canada's provinces are moving towards a tender system, which has and may continue to negatively affect the pricing of pharmaceutical products.
Europe
In France , generic penetration is relatively low compared to other large pharmaceutical markets, with low prices resulting from government initiatives. As pharmacists are the primary customers in this market, established relationships, driven by breadth of portfolio and effective supply chain management, are key competitive advantages.
In Italy , the generic product penetration is relatively small due to few incentives for market stakeholders and in part to low prices on available brand name drugs. Also to be considered is the fact that the generic market in Italy suffered a certain delay compared to other European countries due to extended patent protection. The Italian government has put forth only limited measures aimed at increasing generic usage, and as such generic substitution is still in its early stages. Pharmacists will play a key role in future market expansion, due to higher margins provided by generic versus branded products as well as a specific legislative provision which requires them to propose generic products to patients, when available.
The U.K. is one of the most competitive off-patent markets, with low barriers to entry and a high degree of fragmentation. Competition among manufacturers, along with indirect control of pricing by the government, has led to strong downward pricing pressure. Companies in the U.K. will continue to compete on price, with consistent supply chain and breadth of product portfolio also coming into play.
Spain is a rapidly growing, highly fragmented generic market with many participants. As a result of legislative changes, all regions within Spain have moved, or will move, to INN prescribing, thus making the pharmacists the key driver of product choice. Within the last few years, the Andalusia region, representing approximately 21% of the total retail market, has
23
Table of Contents
evolved into a tendering commercial model, which favors cost competitiveness. In other regions of Spain, companies are competing based on being first to market, offering a wide portfolio, building strong relationships with customers and providing a consistent supply of quality products.
The markets in the Netherlands and Germany have become highly competitive as a result of a large number of generic participants, both having one of the highest generic penetration rates in Europe and the continued use of tender systems. Under a tender system, health insurers are entitled to issue invitations to tender products. Pricing pressures resulting from an effort to win the tender should drive near-term competition. Mylan is able to play a role in tenders but also has non-tendered sales, which provide further opportunities for growth.
Rest of World
In India , the commercial pharmaceutical market is a rapidly growing, highly fragmented generic market with a significant number of participants. Companies compete in India based on price, product portfolio and the ability to provide a consistent supply of quality products. Intense competition by other API suppliers in the Indian pharmaceuticals market has, in recent years, led to increased pressure on prices. We expect that the exports of API and generic FDF products from India to developed markets will continue to increase. The success of Indian pharmaceutical companies is attributable to established development expertise in chemical synthesis and process engineering, development of FDF, availability of highly skilled labor and the low cost manufacturing base.
In Australia , the generic market is small by international standards, in terms of volume, value and the number of active participants. Generic penetration rates, however, continue to increase as government polices continue to drive volume growth.
In Japan , government initiatives have historically kept all drug prices low, resulting in little incentive for generic usage. More recent pro-generic actions by the government have led to growth in the generics market in recent years.
The Brazilian pharmaceutical market is the largest in South America. Since the entry in force of generic drug laws in Brazil , the generic segment of the pharmaceutical market has grown rapidly. The industry is highly competitive with a broad presence of multinational and national competitors.
Many emerging markets are attractive because of the growing middle class within these countries combined with an increase in the demand for pharmaceutical products. In addition to the highly competitive environment in many emerging markets, governments in many of these markets are focused on constraining healthcare costs and have enacted price controls and other related measures. Beyond pricing and market access challenges, other conditions in emerging market countries can affect our efforts to continue to grow in these markets, including potential political instability, significant currency fluctuations and limited or changing availability of funding for healthcare.
Product Liability
Global product liability litigation represents an inherent risk to firms in the pharmaceutical industry. We utilize a combination of self-insurance (including through our wholly owned captive insurance subsidiary) and traditional third-party insurance policies with regard to our product liability claims. Such insurance coverage at any given time reflects market conditions, including cost and availability, existing at the time the policy is written and the decision to obtain commercial insurance coverage or to self-insure varies accordingly.
Raw Materials
Mylan utilizes a global approach to managing relationships with its suppliers. The APIs and other materials and supplies used in our pharmaceutical manufacturing operations are generally available and purchased from many different U.S. and non-U.S. suppliers, including Mylan India . However, in some cases, the raw materials used to manufacture pharmaceutical products are available only from a single supplier. Even when more than one supplier exists, we may choose, and in some cases have chosen, only to list one supplier in our applications submitted to the various regulatory agencies. Any change in a supplier not previously approved must then be submitted through a formal approval process.
Seasonality
Certain parts of our business are affected by seasonality, including products for asthma and allergy therapies which tend to have higher sales during the second and third quarters. In addition, the timing and severity of the cough, cold and flu
24
Table of Contents
season can cause variability in sales trends for certain of our prescription and OTC products. The seasonal impact of these particular products may affect a quarterly comparison within any fiscal year; however, this impact is generally not material to our annual consolidated results.
Environment
We strive to comply in all material respects with applicable laws and regulations concerning the environment. While it is impossible to predict accurately the future costs associated with environmental compliance and potential remediation activities, compliance with environmental laws is not expected to require significant capital expenditures and has not had, and is not expected to have, a material adverse effect on our operations or competitive position.
Employees
As of December 31, 2016 , Mylan 's global workforce included more than 35,000 employees and external contractors. Certain production and maintenance employees at our manufacturing facility in Morgantown, West Virginia, are represented by the United Steel, Paper and Forestry, Rubber, Manufacturing, Energy, Allied Industrial and Service Workers International Union and its Local Union 8-957 AFL-CIO. This contract expires on April 21, 2017 and we are currently in negotiations on a successor agreement. In addition, there are non-U.S. Mylan locations that have employees who are unionized or part of works councils or trade unions.
Securities Exchange Act Reports
Mylan maintains an Internet website at the following address: mylan.com . We make available on or through our website certain reports and amendments to those reports that Mylan files with the Securities and Exchange Commission (the "SEC") in accordance with the Securities Exchange Act of 1934 (the "Exchange Act"). These include our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K. We make this information available on our website free of charge, as soon as reasonably practicable after electronically filed with, or furnished to, the SEC. The contents of our website are not incorporated by reference in this Report on Form 10-K and shall not be deemed "filed" under the Exchange Act.
The public may also read and copy any materials that we file with the SEC at the SEC's Public Reference Room at 100 F Street NE, Washington, D.C. 20549. You may obtain information about the Public Reference Room by contacting the SEC at 1.800.SEC.0330. Reports filed with the SEC are also made available on the SEC website (www.sec.gov).
ITEM 1A. | Risk Factors |
We operate in a complex and rapidly changing environment that involves risks, many of which are beyond our control. Any of the following risks, if they occur, could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or share price. These risks should be read in conjunction with the other information in this Annual Report on Form 10-K.
ABBOTT'S SUBSIDIARIES THAT HOLD ORDINARY SHARES ARE COLLECTIVELY A SIGNIFICANT BENEFICIAL SHAREHOLDER OF OURS AND THE PRESENCE OF A SIGNIFICANT BENEFICIAL SHAREHOLDER MAY AFFECT THE ABILITY OF OUR OTHER SHAREHOLDERS TO EXERCISE INFLUENCE OVER US, ESPECIALLY IN LIGHT OF CERTAIN VOTING OBLIGATIONS UNDER OUR SHAREHOLDER AGREEMENT WITH ABBOTT AND ITS SUBSIDIARIES.
Abbott's subsidiaries currently own approximately 13.0% of our outstanding ordinary shares. The shares owned by Abbott's subsidiaries are subject to the terms of a shareholder agreement, which requires the Abbott subsidiaries to vote in favor of the director nominees recommended by our board of directors and in accordance with the recommendation of our board of directors on all other matters, subject to certain exceptions for extraordinary transactions. This voting agreement is in force with respect to ordinary shares owned by Abbott's subsidiaries so long as they collectively beneficially own at least five percent of our issued and outstanding ordinary shares. Abbott's subsidiaries that hold ordinary shares are collectively a significant beneficial shareholder of ours. Having a significant beneficial shareholder that is required in many instances to vote with the recommendation of our board of directors may make it more difficult for our other shareholders to exercise influence over most matters submitted to shareholders for approval, including the election of directors, issuances of securities for equity compensation plans, amendments to the Articles, and shareholder proposals submitted pursuant to Rule 14a-8 of the Exchange Act. Additionally, Abbott subsidiaries are obligated, pursuant to the shareholder agreement, not to tender any ordinary shares in any tender or exchange offer that our board of directors recommends that the shareholders reject and, if our board of directors
25
Table of Contents
has recommended against a transaction, Abbott subsidiaries are required to vote against such transaction, which may have the effect of making it more difficult for a third party to acquire, or discouraging a third party from seeking to acquire, a majority of our outstanding ordinary shares in a public takeover offer, or control of our board of directors through a proxy solicitation.
PROVISIONS IN OUR GOVERNANCE ARRANGEMENTS OR THAT ARE OTHERWISE AVAILABLE UNDER DUTCH LAW COULD DISCOURAGE, DELAY, OR PREVENT A CHANGE IN CONTROL OF US AND MAY AFFECT THE MARKET PRICE OF OUR ORDINARY SHARES.
Some provisions of our governance arrangements that are available under Dutch law, such as our grant to a Dutch foundation (stichting) of a call option to acquire preferred shares to safeguard the interests of the Company, its businesses and its stakeholders against threats to our strategy, mission, independence, continuity and/or identity, may discourage, delay, or prevent a change in control of us, even if such a change in control is sought by our shareholders.
WE MAY BE FORCED TO DELIST, OR OTHERWISE CHOOSE TO DELIST, FROM THE TEL AVIV STOCK EXCHANGE IN THE FUTURE AND THIS COULD HAVE A NEGATIVE IMPACT ON OUR ORDINARY SHARE PRICE AND ON THE LIQUIDITY OF OUR ORDINARY SHARES.
Our ordinary shares are listed on both NASDAQ and the Tel Aviv Stock Exchange (the "TASE"). We have undertaken with the TASE that for as long as our ordinary shares are listed for trading on the TASE, if new Mylan preferred shares are issued, in response to the Dutch foundation (stichting) described above exercising its call option to acquire preferred shares or otherwise, we will take all necessary actions, as soon as practicable and no later than three Israeli business days following the issuance of such preferred shares, to notify the TASE that we are delisting our ordinary shares from the TASE (with such delisting to take effect 90 days later). Accordingly, there can be no guarantee as to how long our ordinary shares will continue to be listed on the TASE. If we delist from the TASE, that could have a negative impact on our ordinary share price and on the liquidity of our ordinary shares for our shareholders, particularly in Israel.
WE DO NOT ANTICIPATE PAYING DIVIDENDS FOR THE FORESEEABLE FUTURE, AND OUR SHAREHOLDERS MUST RELY ON INCREASES IN THE TRADING PRICE OF THE ORDINARY SHARES TO OBTAIN A RETURN ON THEIR INVESTMENT.
Mylan N.V. does not anticipate paying dividends in the immediate future. We anticipate that we will retain all earnings, if any, to support our operations and to opportunistically pursue additional transactions to deliver additional shareholder value. Any future determination as to the payment of dividends will, subject to Dutch law requirements, be at the sole discretion of our board of directors and will depend on our financial position, results of operations, capital requirements, and other factors our board of directors deems relevant at that time. Holders of Mylan N.V.'s ordinary shares must rely on increases in the trading price of their shares to obtain a return on their investment in the foreseeable future.
THE MARKET PRICE OF THE ORDINARY SHARES MAY BE VOLATILE, AND THE VALUE OF YOUR INVESTMENT COULD MATERIALLY DECLINE.
Investors who hold Mylan N.V.'s ordinary shares may not be able to sell their shares at or above the price at which they purchased such shares. The share price of Mylan N.V.'s ordinary shares fluctuates materially from time to time, and we cannot predict the price of the ordinary shares at any given time. The risk factors described herein could cause the price of the ordinary shares to fluctuate materially. In addition, the stock market in general, including the market for generic and specialty pharmaceutical companies, has experienced price and volume fluctuations. These broad market and industry factors may materially harm the market price of the ordinary shares, regardless of our operating performance. In addition, the price of the ordinary shares may be affected by the valuations and recommendations of the analysts who cover us, and if our results do not meet the analysts' forecasts and expectations, the price of the ordinary shares could decline as a result of analysts lowering their valuations and recommendations or otherwise. In the past, following periods of volatility in the market and/or in the price of a company's stock, securities class-action litigation has been instituted against us and other companies. Such litigation could result in substantial costs and diversion of management's attention and resources, which could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price. We or our shareholders also may offer or sell our ordinary shares or securities convertible into or exchangeable or exercisable for ordinary shares. An increase in the number of the ordinary shares issued and outstanding and the possibility of sales of ordinary shares or securities convertible into or exchangeable or exercisable for ordinary shares may depress the future trading price of the ordinary shares. In addition, if additional offerings occur, the voting power of our then existing shareholders may be diluted.
OUR TRANSACTIONS, INCLUDING THE MEDA TRANSACTION AND THE EPD TRANSACTION, MAY NOT ACHIEVE ALL INTENDED BENEFITS OR MAY DISRUPT OUR PLANS AND OPERATIONS.
26
Table of Contents
There can be no assurance that we will be able to successfully complete the integration of acquired businesses or assets, including Meda and the EPD Business, with Mylan, or otherwise fully realize the expected benefits of such transactions. Our ability to fully realize the anticipated benefits of such transactions will depend, to a large extent, on our ability to integrate acquired businesses or assets, including Meda or the EPD Business, with Mylan and realize the benefits of the combined businesses in each instance. The combination of independent businesses is a complex, costly, and time-consuming process. Our business may be negatively impacted if we are unable to effectively manage the expanded operations of our acquired businesses or assets. The integrations of Meda and of the EPD Business, in particular, are ongoing and continue to require significant time and focus from management and may divert attention from the day-to-day operations of our business. Additionally, the integration of the businesses could disrupt our plans and operations, which could delay the achievement of our strategic objectives.
The expected synergies and operating efficiencies of any transaction, including the Meda transaction and the EPD Transaction, may not be fully realized, which could result in increased costs and have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price. In addition, the overall integration of a business or asset may result in material unanticipated problems, expenses, liabilities, competitive responses, loss of customer relationships, and diversion of management's attention, among other potential adverse consequences. The difficulties of combining the operations of a business or asset include, among others:
• | the diversion of management's attention to integration matters, including restructuring activities; |
• | difficulties in achieving anticipated synergies, operating efficiencies, business opportunities, and growth prospects from combining an acquired business or asset with Mylan; |
• | difficulties in the integration of operations and information technology ("IT") applications, including enterprise resource planning ("ERP") systems; |
• | difficulties in the integration of employees; |
• | difficulties in managing the expanded operations of a significantly larger and more complex company; |
• | challenges in keeping existing customers and obtaining new customers; |
• | challenges in reducing reliance of transition services prior to the expiry of any period in which such services are provided by a transaction counterparty; |
• | operational or financial difficulties that would not have occurred if acquired companies, businesses, or assets continued operating in their former structures; |
• | challenges in attracting and retaining key personnel; and |
• | with respect to the EPD Business, the complexities of managing the ongoing relationship with Abbott, and certain of its business partners, which includes agreements providing for transition and other services, development and manufacturing relationships, and license arrangements. |
Many of these factors are outside of our control and any one of them could result in increased costs, decreases in the amount of expected revenues, and diversion of management's time and energy, which could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price. In addition, the overall integration of a business or asset may result in material unanticipated problems, expenses, liabilities, competitive responses, loss of customer relationships, and diversion of management's attention, among other potential adverse consequences. Furthermore, even if a business or asset is integrated successfully, we may not realize the full benefits of such transactions, including the synergies, operating efficiencies, or sales or growth opportunities that are expected. These benefits may not be achieved within the anticipated time frame or at all. All of these factors could cause dilution to our earnings per share, decrease or delay the expected accretive effect of such transactions, and/or negatively impact the price of our ordinary shares.
WE EXPECT TO BE TREATED AS A NON-U.S. CORPORATION FOR U.S. FEDERAL INCOME TAX PURPOSES. ANY CHANGES TO THE TAX LAWS OR CHANGES IN OTHER LAWS (INCLUDING UNDER APPLICABLE INCOME TAX TREATIES), REGULATIONS, RULES, OR INTERPRETATIONS THEREOF APPLICABLE TO INVERTED COMPANIES AND THEIR AFFILIATES, WHETHER ENACTED BEFORE OR AFTER THE EPD TRANSACTION, MAY MATERIALLY ADVERSELY AFFECT US.
27
Table of Contents
Under current U.S. law, we believe that we should not be treated as a U.S. corporation for U.S. federal income tax purposes as a result of the EPD Transaction. Changes to Section 7874 of the Internal Revenue Code (the "Code") or, to the U.S. Treasury Regulations promulgated thereunder, or interpretations thereof, or to other relevant tax laws (including applicable income tax treaties), could affect our status as a non-U.S. corporation for U.S. federal income tax purposes and the tax consequences to us and our affiliates. Any such changes could have prospective or retroactive application, and may apply even if enacted or promulgated now that the EPD Transaction has closed. If we were to be treated as a U.S. corporation for U.S. federal income tax purposes, or if the relevant tax laws (including applicable income tax treaties) change, we would likely be subject to significantly greater U.S. tax liability than currently contemplated as a non-U.S. corporation or if the relevant tax laws (including applicable income tax treaties) had not changed.
On August 5, 2014, the U.S. Treasury Department announced that it is reviewing a broad range of authorities for possible administrative actions that could limit the ability of a U.S. corporation to complete a transaction in which it becomes a subsidiary of a non-U.S. corporation (commonly known as an "inversion transaction") or reduce certain tax benefits after an inversion transaction takes place. On September 22, 2014 and November 19, 2015, the U.S. Treasury Department issued notices (the "Notices") announcing its intention to promulgate certain regulations that will apply to inversion transactions completed on or after September 22, 2014. Those regulations were promulgated as temporary U.S. Treasury Regulations on April 4, 2016, some of which were adopted as final U.S. Treasury Regulations on January 18, 2017, and they do not affect our belief that we expect to be treated as a non-U.S. corporation for U.S. federal income tax purposes.
In the Notices, the U.S. Treasury Department also announced that it expected to issue additional guidance to further limit and reduce the benefits of certain inversion transactions. In particular, it stated that it was considering regulations that may limit the ability of certain foreign-owned U.S. corporations to deduct certain interest payments (so-called "earnings stripping"). On April 4, 2016, the U.S. Treasury Department issued such regulations in the form of proposed U.S. Treasury Regulations. On October 13, 2016, the U.S. Treasury Department issued final and temporary U.S. Treasury Regulations. The rules described in the final and temporary U.S. Treasury Regulations will generally apply to certain intercompany arrangements entered into on or after April 5, 2016, however, certain obligations will only apply to certain intercompany arrangements entered into on or after January 1, 2018.
Additionally, there have been recent legislative proposals intended to limit or discourage inversion transactions and on May 20, 2015, the U.S. Treasury Department announced its intention to revise certain provisions of the model income tax treaties, which, if ultimately adopted by the U.S. and relevant jurisdictions, could reduce potential tax benefits for us and our affiliates by imposing U.S. withholding taxes on particular payments from our U.S. affiliates to related and unrelated foreign persons. Any such future regulatory or legislative actions regarding inversion transactions or any other changes in relevant tax laws (including under applicable income tax treaties), if taken, could apply to us, could disadvantage us as compared to other corporations, including non-U.S. corporations that have completed inversion transactions prior to September 22, 2014, and could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
THE IRS MAY NOT AGREE THAT WE SHOULD BE TREATED AS A NON-U.S. CORPORATION FOR U.S. FEDERAL INCOME TAX PURPOSES.
The U.S. Internal Revenue Service (the "IRS") may not agree that we should be treated as a non-U.S. corporation for U.S. federal income tax purposes. Although we are not incorporated in the U.S. and expect to be treated as a non-U.S. corporation for U.S. federal income tax purposes, the IRS may assert that we should be treated as a U.S. corporation for U.S. federal income tax purposes. If we were to be treated as a U.S. corporation for U.S. federal income tax purposes, we would likely be subject to significantly greater U.S. tax liability than currently contemplated as a non-U.S. corporation.
IF THE INTERCOMPANY TERMS OF CROSS BORDER ARRANGEMENTS THAT WE HAVE AMONG OUR SUBSIDIARIES ARE DETERMINED TO BE INAPPROPRIATE OR INEFFECTIVE, OUR TAX LIABILITY MAY INCREASE.
We have potential tax exposures resulting from the varying application of statutes, regulations, and interpretations which include exposures on intercompany terms of cross-border arrangements among our subsidiaries (including intercompany loans, sales, and services agreements) in relation to various aspects of our business, including manufacturing, marketing, sales, and delivery functions. Although we believe our cross-border arrangements among our subsidiaries are based upon internationally accepted standards and applicable law, tax authorities in various jurisdictions may disagree with and subsequently challenge the amount of profits taxed in their country, which may result in increased tax liability, including accrued interest and penalties, which would cause our tax expense to increase and could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
28
Table of Contents
WE MAY NOT BE ABLE TO MAINTAIN COMPETITIVE FINANCIAL FLEXIBILITY AND OUR CORPORATE TAX RATE.
We believe that our structure and operations will give us the ability to achieve competitive financial flexibility and a competitive worldwide effective corporate tax rate. The material assumptions underlying our expected tax rates include the fact that we expect certain of our businesses will be operated outside of the U.S. and, as such, will be subject to a lower tax rate than operations in the U.S., which will result in a lower blended worldwide tax rate we were previously able to achieve. We must also make assumptions regarding the effect of certain internal reorganization transactions, including various intercompany transactions. We cannot give any assurance as to what our effective tax rate will be, however, because of, among other reasons, uncertainty regarding the tax policies of the jurisdictions where we operate, potential changes of laws and interpretations thereof, and the potential for tax audits or challenges. Our actual effective tax rate may vary from our expectation and that variance may be material. Additionally, the tax laws of the United Kingdom, the Netherlands and other jurisdictions could change in the future, and such changes could cause a material change in our effective tax rate. Such a material change could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
UNANTICIPATED CHANGES IN OUR TAX PROVISIONS OR EXPOSURE TO ADDITIONAL INCOME TAX LIABILITIES AND CHANGES IN INCOME TAX LAWS AND TAX RULINGS MAY HAVE A SIGNIFICANT ADVERSE IMPACT ON OUR EFFECTIVE TAX RATE AND INCOME TAX EXPENSE.
We are subject to income taxes in many jurisdictions. Significant analysis and judgment are required in determining our worldwide provision for income taxes. In the ordinary course of business, there are many transactions and calculations where the ultimate tax determination is uncertain. The final determination of any tax audits or related litigation could be materially different from our income tax provisions and accruals.
Additionally, changes in the effective tax rate as a result of a change in the mix of earnings in countries with differing statutory tax rates, changes in our overall profitability, changes in the valuation of deferred tax assets and liabilities, the results of audits and the examination of previously filed tax returns by taxing authorities, and continuing assessments of our tax exposures could impact our tax liabilities and affect our income tax expense, which could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
Finally, potential changes to income tax laws in the U.S. include measures which would defer the deduction of interest expense related to deferred income; determine the foreign tax credit on a pooling basis; tax currently excess returns associated with transfers of intangibles offshore; and limit earnings stripping by expatriated entities. In addition, proposals have been made to encourage manufacturing in the U.S., including reduced rates of tax and increased deductions related to manufacturing. We cannot determine whether these proposals will be modified or enacted, whether other proposals unknown at this time will be made, or the extent to which the corporate tax rate might be reduced and lessen the adverse impact of some of these proposals. If enacted, and depending on its precise terms, such legislation could materially increase our overall effective income tax rate and income tax expense and could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
WE MAY BECOME TAXABLE IN A JURISDICTION OTHER THAN THE UNITED KINGDOM AND THIS MAY INCREASE THE AGGREGATE TAX BURDEN ON US.
Based on our current management structure and current tax laws of the United States, the United Kingdom, and the Netherlands, as well as applicable income tax treaties, and current interpretations thereof, the United Kingdom and the Netherlands competent authorities have determined that we are tax resident solely in the United Kingdom for the purposes of the Netherlands-United Kingdom tax treaty. We have received a binding ruling from the competent authorities in the United Kingdom and in the Netherlands confirming this treatment. We will therefore be tax resident solely in the United Kingdom so long as the facts and circumstances set forth in the relevant application letters sent to those authorities remain accurate. Even though we received a binding ruling, the applicable tax laws or interpretations thereof may change, or the assumptions on which such rulings were based may differ from the facts. As a consequence, we may become a tax resident of a jurisdiction other than the United Kingdom. As a consequence, our overall effective income tax rate and income tax expense could materially increase, which could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
WE INCUR DIRECT AND INDIRECT COSTS AS A RESULT OF OUR CORPORATE STRUCTURE.
29
Table of Contents
We have incurred costs in connection with, and will incur further costs as a result of, being a Dutch company that is a tax resident of the United Kingdom. Certain costs are not readily ascertainable and are difficult to predict, quantify, and determine. These costs include professional fees associated with complying with Dutch corporate law and financial reporting requirements, professional fees associated with complying with the tax laws of the United Kingdom, and costs and expenses incurred in connection with holding a majority of the meetings of our board of directors and certain executive management meetings in the United Kingdom, as well as any additional costs we may incur going forward as a result of our corporate structure.
WE HAVE GROWN AT A VERY RAPID PACE AND MAY OPPORTUNISTICALLY PURSUE ADDITIONAL ACQUISITION OPPORTUNITIES THAT MAKE FINANCIAL AND STRATEGIC SENSE FOR US. OUR INABILITY TO EFFECTIVELY MANAGE OR SUPPORT THIS GROWTH MAY HAVE A MATERIAL ADVERSE EFFECT ON OUR BUSINESS, FINANCIAL CONDITION, RESULTS OF OPERATIONS, CASH FLOWS, AND/OR ORDINARY SHARE PRICE.
We have grown very rapidly over the past several years as a result of increasing sales and several acquisitions and other transactions, and may opportunistically pursue additional acquisition opportunities that make financial and strategic sense for us. We evaluate various strategic transactions and business arrangements, including acquisitions, asset purchases, partnerships, joint ventures, restructurings, divestitures and investments, on an ongoing basis. These transactions and arrangements may be material both from a strategic and financial perspective.
We are currently in the process of evaluating certain potential strategic transactions, including acquisitions, and we may opportunistically pursue one or more of these transactions at any time. Some of these opportunities would be material if pursued and consummated. Our growth has, and will continue to, put significant demands on our processes, systems, and employees. We have made and expect to make further investments in additional personnel, systems, and internal control processes to help manage our growth. Attracting, retaining and motivating key employees in various departments and locations to support our growth are critical to our business, and competition for these people can be significant. If we are unable to hire and/or retain qualified employees and/or if we do not effectively invest in systems and processes to manage and support our rapid growth and the challenges and difficulties associated with managing a larger, more complex business, and/or if we cannot effectively manage and integrate our increasingly diverse and global platform, there could be a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
WE MAY BE ADVERSELY AFFECTED BY INCREASED SCRUTINY FROM THIRD PARTIES, INCLUDING GOVERNMENTS, OR NEGATIVE PUBLICITY WITH RESPECT TO MATTERS RELATING TO OUR PRODUCTS AND PRICING PRACTICES, AND OTHER MATTERS RELATED TO THE COMPANY, AND WE HAVE AND MAY CONTINUE TO EXPERIENCE PRICING PRESSURE ON THE PRICE OF CERTAIN OF OUR PRODUCTS DUE TO SOCIAL OR POLITICAL PRESSURE TO LOWER THE COST OF DRUGS, WHICH COULD REDUCE OUR REVENUE AND FUTURE PROFITABILITY.
There has been increased press coverage and increased scrutiny from third parties, including regulators, legislative bodies and enforcement agencies, with respect to matters relating to the Company's business and pricing practices, and other matters related to the Company. This increased press coverage, public scrutiny and protests by some consumers have included assertions of wrongdoing by the Company which, regardless of the factual or legal basis for such assertions, have resulted in, and may continue to result in, investigations, and calls for investigations, by governmental agencies at both the federal and state level and have resulted in, and may continue to result in, claims brought against the Company by governmental agencies or by private parties or by regulators taking other measures that could have a negative effect on the Company's business. It is not possible at this time to predict the ultimate outcome of any such investigations or claims or what other investigations or lawsuits or regulatory responses may result from such assertions, or their impact on the Company's business, financial condition, results of operations, cash flows, and/or ordinary share price. Any such investigation or claim could also result in reputational harm and reduced market acceptance and demand for our products, could harm our ability to market our products in the future, could cause us to incur significant expense, could cause our senior management to be distracted from execution of our business strategy, and could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
There has also recently been intense publicity regarding the pricing of pharmaceuticals more generally, including publicity and pressure resulting from prices charged by competitors and peer companies for new products as well as price increases by competitors and peer companies on older products that the public has deemed excessive. We have experienced and may continue to experience downward pricing pressure on the price of certain of our products due to social or political pressure to lower the cost of drugs, which could reduce our revenue and future profitability.
30
Table of Contents
Accompanying the press and media coverage of pharmaceutical pricing practices and public complaints about the same, there has been increasing U.S. federal and state legislative and enforcement interest with respect to drug pricing. In particular, U.S. federal prosecutors recently issued subpoenas to pharmaceutical companies, including Mylan, seeking information about their drug pricing practices, among other issues, and members of the Congress have sought information from certain pharmaceutical companies, including Mylan, relating to drug-price increases. Additionally, there have been several recent Congressional inquiries and proposed bills designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs. For example, in late 2015 the U.S. House of Representatives formed an Affordable Drug Pricing Task Force to advance legislation intended to control pharmaceutical drug costs and investigate pharmaceutical drug pricing. Since then, both the U.S. House of Representatives and the U.S. Senate have conducted numerous hearings with respect to pharmaceutical drug pricing practices, including in connection with the investigation of specific price increases by several pharmaceutical companies such as Mylan. In addition to the effects of any investigations or claims brought against the Company described above, our revenue and future profitability could also be negatively affected if any such inquiries, of us or of other pharmaceutical companies or the industry more generally, were to result in legislative or regulatory proposals that limit our ability to increase the prices of our products.
Any of the events or developments described above could have a material adverse impact on our business, financial condition or results of operations, as well as on our reputation.
CURRENT AND CHANGING ECONOMIC CONDITIONS MAY ADVERSELY AFFECT OUR INDUSTRY, BUSINESS, PARTNERS AND SUPPLIERS, FINANCIAL CONDITION, RESULTS OF OPERATIONS, CASH FLOWS, AND/OR ORDINARY SHARE PRICE.
The global economy continues to experience significant volatility, and the economic environment may continue to be, or become, less favorable than that of past years. Among other matters, the continued risk of a default on sovereign debt by one or more European countries, related financial restructuring efforts in Europe, and/or evolving deficit and spending reduction programs instituted by the U.S. and other governments could negatively impact the global economy and/or the pharmaceutical industry. This has led, and/or could lead, to reduced consumer and customer spending and/or reduced or eliminated governmental or third party payor coverage or reimbursement in the foreseeable future, and this may include reduced spending on healthcare, including but not limited to pharmaceutical products. While generic drugs present an alternative to higher-priced branded products, our sales could be negatively impacted if patients forego obtaining healthcare, patients and customers reduce spending or purchases, and/or if governments and/or third-party payors reduce or eliminate coverage or reimbursement amounts for pharmaceuticals and/or impose price or other controls adversely impacting the price or availability of pharmaceuticals. In addition, reduced consumer and customer spending, and/or reduced government and/or third-party payor coverage or reimbursement, and/or new government controls, may drive us and our competitors to decrease prices and/or may reduce the ability of customers to pay and/or may result in reduced demand for our products. The occurrence of any of these risks could have a material adverse effect on our industry, business, financial condition, results of operations, cash flows, and/or ordinary share price.
OUR BUSINESS, FINANCIAL CONDITION, AND RESULTS OF OPERATIONS ARE SUBJECT TO RISKS ARISING FROM THE INTERNATIONAL SCOPE OF OUR OPERATIONS.
Our operations extend to numerous countries outside the U.S. and are subject to the risks inherent in conducting business globally and under the laws, regulations, and customs of various jurisdictions. These risks include, but are not limited to:
• | compliance with a variety of national and local laws of countries in which we do business, including, but not limited to, anti-bribery and anti-corruption laws, data privacy and security and restrictions on the import and export of certain intermediates, drugs, and technologies, as well as compliance with multiple regulatory regimes; |
• | less established legal and regulatory regimes in certain jurisdictions; |
• | compliance with a variety of U.S. laws including, but not limited to, the Iran Threat Reduction and Syria Human Rights Act of 2012; and rules relating to the use of certain "conflict minerals" under Section 1502 of the Dodd-Frank Wall Street Reform and the Consumer Protection Act; |
• | changes in laws, regulations, and practices affecting the pharmaceutical industry and the healthcare system, including but not limited to imports, exports, manufacturing, quality, cost, pricing, reimbursement, approval, inspection, and delivery of healthcare; |
31
Table of Contents
• | fluctuations in exchange rates for transactions conducted in currencies other than the functional currency; |
• | differing local product preferences and product requirements; |
• | differing degrees of protection for intellectual property; |
• | adverse changes in the economies in which we or our partners and suppliers operate as a result of a slowdown in overall growth, a change in government or economic policies, or financial, political, or social change or instability in such countries that affects the markets in which we operate, particularly emerging markets; |
• | changes in employment laws, wage increases, or rising inflation in the countries in which we or our partners and suppliers operate; |
• | supply disruptions, and increases in energy and transportation costs; |
• | natural disasters, including droughts, floods, and earthquakes in the countries in which we operate; |
• | local disturbances, terrorist attacks, riots, social disruption, or regional hostilities in the countries in which we or our partners and suppliers operate; and |
• | government uncertainty, including as a result of new or changed laws and regulations. |
We also face the risk that some of our competitors have more experience with operations in such countries or with international operations generally and may be able to manage unexpected crises more easily. Furthermore, whether due to language, cultural or other differences, public and other statements that we make may be misinterpreted, misconstrued, or taken out of context in different jurisdictions. Moreover, the internal political stability of, or the relationship between, any country or countries where we conduct business operations may deteriorate. Changes in a country's political stability or the state of relations between any such countries are difficult to predict and could adversely affect our operations. Any such changes could lead to a decline in our profitability and/or adversely impact our ability to do business. Any meaningful deterioration of the political or social stability in and/or diplomatic relations between any countries in which we or our partners and suppliers do business could have a material adverse effect on our operations. The occurrence of any of the above risks could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
WE ARE SUBJECT TO THE U.S. FOREIGN CORRUPT PRACTICES ACT, U.K. BRIBERY ACT, AND SIMILAR WORLDWIDE ANTI-CORRUPTION LAWS, WHICH IMPOSE RESTRICTIONS ON CERTAIN CONDUCT AND MAY CARRY SUBSTANTIAL FINES AND PENALTIES.
We are subject to the U.S. Foreign Corrupt Practices Act, the U.K. Bribery Act and similar anti-corruption laws in other jurisdictions. These laws generally prohibit companies and their intermediaries from engaging in bribery or making other prohibited payments to government officials for the purpose of obtaining or retaining business, and some have record keeping requirements. The failure to comply with these laws could result in substantial criminal and/or monetary penalties. We operate in jurisdictions that have experienced corruption, bribery, pay-offs and other similar practices from time-to-time and, in certain circumstances, such practices may be local custom. We have implemented internal control policies and procedures that mandate compliance with these anti-corruption laws. However, we cannot be certain that these policies and procedures will protect us against liability. There can be no assurance that our employees or other agents will not engage in such conduct for which we might be held responsible. If our employees or agents are found to have engaged in such practices, we could suffer severe criminal or civil penalties and other consequences that could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
OUR FAILURE TO COMPLY WITH APPLICABLE ENVIRONMENTAL AND OCCUPATIONAL HEALTH AND SAFETY LAWS AND REGULATIONS WORLDWIDE COULD ADVERSELY IMPACT OUR BUSINESS, FINANCIAL CONDITION, RESULTS OF OPERATIONS, CASH FLOWS, AND/OR ORDINARY SHARE PRICE.
We are subject to various U.S. federal, state, and local and non-U.S. laws and regulations concerning, among other things, the environment, climate change, regulation of chemicals, employee safety and product safety. These requirements include regulation of the handling, manufacture, transportation, storage, use and disposal of materials, including the discharge of hazardous materials and pollutants into the environment. In the normal course of our business, we are exposed to risks relating to possible releases of hazardous substances into the environment, which could cause environmental or property damage or personal injuries, and which could result in (i) our noncompliance with such environmental and occupational health
32
Table of Contents
and safety laws and regulations and (ii) regulatory enforcement actions or claims for personal injury and property damage against us. If an unapproved or illegal environmental discharge occurs, or if we discover contamination caused by prior operations, including by prior owners and operators of properties we acquire, we could be liable for cleanup obligations, damages and fines. The substantial unexpected costs we may incur could have a material and adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price. In addition, our environmental capital expenditures and costs for environmental compliance may increase substantially in the future as a result of changes in environmental laws and regulations, the development and manufacturing of a new product or increased development or manufacturing activities at any of our facilities. We may be required to expend significant funds and our manufacturing activities could be delayed or suspended, which could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
CURRENCY FLUCTUATIONS AND CHANGES IN EXCHANGE RATES COULD ADVERSELY AFFECT OUR BUSINESS, FINANCIAL CONDITION, RESULTS OF OPERATIONS, CASH FLOWS, AND/OR ORDINARY SHARE PRICE.
Although we report our financial results in U.S. Dollars, a significant portion of our revenues, indebtedness and other liabilities and our costs are denominated in non-U.S. currencies, including among others the Euro, Swedish Krona, Indian Rupee, Japanese Yen, Australian Dollar, Canadian Dollar, Pound Sterling and Brazilian Real . Our results of operations and, in some cases, cash flows, have in the past been and may in the future be adversely affected by certain movements in currency exchange rates. In particular, the risk of a debt default by one or more European countries and related European or national financial restructuring efforts may cause volatility in the value of the Euro. Defaults or restructurings in other countries could have a similar adverse impact. From time to time, we may implement currency hedges intended to reduce our exposure to changes in foreign currency exchange rates. However, our hedging strategies may not be successful, and any of our unhedged foreign exchange exposures will continue to be subject to market fluctuations. The occurrence of any of the above risks could cause a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
OUR SIGNIFICANT OPERATIONS IN INDIA MAY BE ADVERSELY AFFECTED BY REGULATORY, ECONOMIC, SOCIAL, AND POLITICAL UNCERTAINTIES OR CHANGE, MAJOR HOSTILITIES, MILITARY ACTIVITY, AND/OR ACTS OF TERRORISM IN SOUTHERN ASIA.
In recent years, our Indian subsidiaries have benefited from many policies of the Government of India and the Indian state governments in which they operate, which are designed to promote foreign investment generally, including significant tax incentives, liberalized import and export duties, and preferential rules on foreign investment and repatriation. There is no assurance that such policies will continue. Various factors, such as changes in the current federal government, could trigger significant changes in India's economic liberalization and deregulation policies and disrupt business and economic conditions in India generally and our business in particular.
In addition, our financial performance may be adversely affected by general economic conditions; economic, fiscal and social policy in India, including changes in exchange rates and controls, interest rates and taxation policies; and social instability and political, economic, or diplomatic developments affecting India in the future. In particular, India has experienced significant economic growth over the last several years, but faces major challenges in sustaining that growth in the years ahead. These challenges include the need for substantial infrastructure development and improving access to healthcare and education. Our ability to recruit, train, and retain qualified employees and develop and operate our manufacturing facilities in India could be adversely affected if India does not successfully meet these challenges.
Southern Asia has, from time to time, experienced instances of civil unrest and hostilities among neighboring countries, including India and Pakistan, and within the countries themselves. Terrorist attacks, military activity, rioting, or civil or political unrest in the future could influence the Indian economy and our operations and employees by disrupting operations and communications and making travel and the conduct of our business more difficult. Resulting political or social tensions could create a greater perception that investments in companies with Indian operations involve a high degree of risk, and that there is a risk of disruption of services provided by companies with Indian operations, which could impact our customers' willingness to do business with us and have a material adverse effect on the market for our products. Furthermore, if India were to become engaged in armed hostilities, including but not limited to hostilities that were protracted or involved the threat or use of nuclear or other weapons of mass destruction, our India operations might not be able to continue. We generally do not have insurance for losses and interruptions caused by terrorist attacks, military conflicts and wars. The occurrence of any of these risks could cause a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
33
Table of Contents
AN INABILITY TO IDENTIFY OR SUCCESSFULLY BID FOR SUITABLE ACQUISITION TARGETS, OR CONSUMMATE AND EFFECTIVELY INTEGRATE RECENT AND FUTURE POTENTIAL ACQUISITIONS, OR TO EFFECTIVELY DEAL WITH AND RESPOND TO UNSOLICITED BUSINESS PROPOSALS COULD LIMIT OUR FUTURE GROWTH AND HAVE A MATERIAL ADVERSE EFFECT ON OUR BUSINESS, FINANCIAL CONDITION, RESULTS OF OPERATIONS, CASH FLOWS, AND/OR ORDINARY SHARE PRICE.
We may continue to seek to expand our product line and/or business platform organically as well as through complementary or strategic acquisitions of other companies, products, or assets or through joint ventures, licensing agreements, or other arrangements. Acquisitions or similar arrangements may prove to be complex and time consuming and require substantial resources and effort. We may compete for certain acquisition targets with companies having greater financial resources than us or other advantages over us that may hinder or prevent us from acquiring a target company or completing another transaction, which could also result in significant diversion of management time, as well as substantial out-of-pocket costs.
If an acquisition is consummated, the integration of such acquired business, product, or other assets into us may also be complex, time consuming, and result in substantial costs and risks. The integration process may distract management and/or disrupt our ongoing businesses, which may adversely affect our relationships with customers, employees, partners, suppliers, regulators, and others with whom we have business or other dealings. In addition, there are operational risks associated with the integration of acquired businesses. These risks include, but are not limited to, difficulties in achieving or inability to achieve identified or anticipated financial and operating synergies, cost savings, revenue synergies, and growth opportunities; difficulties in consolidating or inability to effectively consolidate information technology and manufacturing platforms, business applications, and corporate infrastructure; the impact of pre-existing legal and/or regulatory issues, such as quality and manufacturing concerns, among others; the risks that acquired companies or businesses do not operate to the same quality, manufacturing, or other standards as us; the impacts of substantial indebtedness and assumed liabilities; challenges associated with operating in new markets; and the unanticipated effects of export controls, exchange rate fluctuations, domestic and foreign political conditions, and/or domestic and foreign economic conditions.
In addition, in April 2015 we received an unsolicited and subsequently withdrawn non-binding expression of interest from Teva Pharmaceutical Industries Ltd. to acquire all of our outstanding shares and may receive similar proposals in the future. Such unsolicited business proposals may not be consistent with or enhancing to our financial, operational, or market strategies (which we believe have proven to be successful) and may not further the interests of our shareholders and other stakeholders, including employees, creditors, customers, suppliers, relevant patient populations and communities in which Mylan operates and may jeopardize the sustainable success of Mylan's business. However, the evaluation of and response to such unsolicited business proposals may nevertheless distract management and/or disrupt our ongoing businesses, which may adversely affect our relationships with customers, employees, partners, suppliers, regulators, and others with whom we have business or other dealings.
We may be unable to realize synergies or other benefits, including tax savings, expected to result from acquisitions, joint ventures, or other transactions or investments we may undertake, or we may be unable to generate additional revenue to offset any unanticipated inability to realize these expected synergies or benefits. Realization of the anticipated benefits of acquisitions or other transactions could take longer than expected, and implementation difficulties, unforeseen expenses, complications and delays, market factors, or deterioration in domestic and global economic conditions could reduce the anticipated benefits of any such transactions. We also may inherit legal, regulatory, and other risks that occurred prior to the acquisition, whether known or unknown to us.
Any one of these challenges or risks could impair our growth and ability to compete, require us to focus additional resources on integration of operations rather than more profitable activities, require us to reexamine our business strategy, or otherwise cause a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
WE MAY DECIDE TO SELL ASSETS, WHICH COULD ADVERSELY AFFECT OUR PROSPECTS AND OPPORTUNITIES FOR GROWTH.
We may from time to time consider selling certain assets if (i) we determine that such assets are not critical to our strategy or (ii) we believe the opportunity to monetize the asset is attractive or for various other reasons, including for the reduction of indebtedness. We have explored and may continue to explore the sale of certain non-core assets. Although our expectation is to engage in asset sales only if they advance or otherwise support our overall strategy, any such sale could reduce the size or scope of our business, our market share in particular markets or our opportunities with respect to certain markets,
34
Table of Contents
products or therapeutic categories. As a result, any such sale could have an adverse effect on our business, prospects and opportunities for growth, financial condition, results of operations, cash flows, and/or ordinary share price.
CHARGES TO EARNINGS RESULTING FROM ACQUISITIONS COULD HAVE A MATERIAL ADVERSE EFFECT ON OUR BUSINESS, FINANCIAL CONDITION, RESULTS OF OPERATIONS, CASH FLOWS AND/OR ORDINARY SHARE PRICE.
Under U.S. GAAP business acquisition accounting standards, we recognize the identifiable assets acquired, the liabilities assumed, and any noncontrolling interests in acquired companies generally at their acquisition date fair values and, in each case, separately from goodwill. Goodwill as of the acquisition date is measured as the excess amount of consideration transferred, which is also generally measured at fair value, and the net of the acquisition date amounts of the identifiable assets acquired and the liabilities assumed. Our estimates of fair value are based upon assumptions believed to be reasonable but which are inherently uncertain. After we complete an acquisition, the following factors could result in material charges and adversely affect our operating results and may adversely affect our cash flows:
• | costs incurred to combine the operations of companies we acquire, such as transitional employee expenses and employee retention, redeployment or relocation expenses; |
• | impairment of goodwill or intangible assets, including acquired in-process research and development; |
• | amortization of intangible assets acquired; |
• | a reduction in the useful lives of intangible assets acquired; |
• | identification of or changes to assumed contingent liabilities, including, but not limited to, contingent purchase price consideration, income tax contingencies and other non-income tax contingencies, after our final determination of the amounts for these contingencies or the conclusion of the measurement period (generally up to one year from the acquisition date), whichever comes first; |
• | charges to our operating results to eliminate certain duplicative pre-acquisition activities, to restructure our operations or to reduce our cost structure; |
• | charges to our operating results resulting from expenses incurred to effect the acquisition; and |
• | changes to contingent consideration liabilities, including accretion and fair value adjustments. |
A significant portion of these adjustments could be accounted for as expenses that will decrease our net income and earnings per share for the periods in which those costs are incurred. Such charges could cause a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
THE SIGNIFICANT AND INCREASING AMOUNT OF INTANGIBLE ASSETS AND GOODWILL RECORDED ON OUR BALANCE SHEET, MAINLY RELATED TO ACQUISITIONS, MAY LEAD TO SIGNIFICANT IMPAIRMENT CHARGES IN THE FUTURE WHICH COULD LEAD US TO HAVE TO TAKE SIGNIFICANT CHARGES AGAINST EARNINGS.
We regularly review our long-lived assets, including identifiable intangible assets and goodwill, for impairment. Goodwill and indefinite-lived intangible assets are subject to impairment assessment at least annually. Other long-lived assets are reviewed when there is an indication that an impairment may have occurred. The amount of goodwill and identifiable intangible assets on our consolidated balance sheet has increased significantly as a result of our acquisitions and other transactions, including Meda, and may increase further following future potential acquisitions. In addition, we may from time to time sell assets that we determine are not critical to our strategy or execution. Future events or decisions may lead to asset impairments and/or related charges. Certain non-cash impairments may result from a change in our strategic goals, business direction or other factors relating to the overall business environment. Any impairment of the value of goodwill or other intangible assets will result in a charge against earnings, which could have a material adverse effect on our business, financial condition, results of operations, shareholder's equity, and/or ordinary share price.
THE PHARMACEUTICAL INDUSTRY IS HEAVILY REGULATED AND WE FACE SIGNIFICANT COSTS AND UNCERTAINTIES ASSOCIATED WITH OUR EFFORTS TO COMPLY WITH APPLICABLE LAWS AND REGULATIONS.
35
Table of Contents
The pharmaceutical industry is subject to regulation by various governmental authorities. For instance, we must comply with applicable laws and requirements of the FDA and comparable regulatory agencies, including foreign authorities, in our other markets with respect to the research, development, manufacture, quality, safety, effectiveness, approval, labeling, storage, record-keeping, reporting, pharmacovigilance, sale, distribution, import, export, marketing, advertising, and promotion of pharmaceutical products. Failure to comply with regulations of the FDA and other foreign regulators could result in a range of consequences, including, but not limited to, fines, penalties, disgorgement, unanticipated compliance expenditures, suspension of review of applications or other submissions, rejection or delay in approval of applications, recall or seizure of products, total or partial suspension of production and/or distribution, our inability to sell products, the return by customers of our products, injunctions, and/or criminal prosecution. Under certain circumstances, a regulator may also have the authority to revoke or vary previously granted drug approvals.
The safety profile of any product will continue to be closely monitored by the FDA and comparable foreign regulatory authorities after approval. If the FDA or comparable foreign regulatory authorities become aware of new safety information about any of our marketed or investigational products, those authorities may require labeling changes, establishment of a risk evaluation and mitigation strategy or similar strategy, restrictions on a product's indicated uses or marketing, or post-approval studies or post-market surveillance.
The FDA and comparable regulatory authorities also regulate the facilities and operational procedures that we use to manufacture our products. We must register our facilities with the FDA and similar regulators in other countries. Products must be manufactured in our facilities in accordance with current good manufacturing practices ("cGMP") or similar standards in each territory in which we manufacture. Compliance with such regulations requires substantial expenditures of time, money, and effort in multiple areas, including training of personnel, record-keeping, production, and quality control and quality assurance. The FDA and other regulatory authorities, including foreign authorities, periodically inspect our manufacturing facilities for compliance with cGMP or similar standards in the applicable territory. Regulatory approval to manufacture a drug is granted on a site-specific basis. Failure to comply with cGMP and other regulatory standards at one of our or our partners' or suppliers' manufacturing facilities could result in an adverse action brought by the FDA or other regulatory authorities, which could result in a receipt of an untitled or warning letter, fines, penalties, disgorgement, unanticipated compliance expenditures, rejection or delay in approval of applications, suspension of review of applications or other submissions, suspension of ongoing clinical trials, recall or seizure of products, total or partial suspension of production and/or distribution, our inability to sell products, the return by customers of our products, orders to suspend, vary, or withdraw marketing authorizations, injunctions, consent decrees, requirements to modify promotional materials or issue corrective information to healthcare practitioners, refusal to permit import or export, criminal prosecution and/or other adverse actions.
If any regulatory body were to delay, withhold, or withdraw approval of an application; require a recall or other adverse product action; require one of our manufacturing facilities to cease or limit production; or suspend, vary, or withdraw related marketing authorization, our business could be adversely affected. Delay and cost in obtaining FDA or other regulatory approval to manufacture at a different facility also could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
Although we have established internal regulatory compliance programs and policies, there is no guarantee that these programs and policies, as currently designed, will meet regulatory agency standards in the future or will prevent instances of non-compliance with applicable laws and regulations. Additionally, despite our efforts at compliance, from time to time we receive notices of manufacturing and quality-related observations following inspections by regulatory authorities around the world, as well as official agency correspondence regarding compliance. We may receive similar observations and correspondence in the future. If we are unable to resolve these observations and address regulator's concerns in a timely fashion, our business, financial condition, results of operations, cash flows, and/or ordinary share price could be materially affected.
On September 9, 2013, prior to our completion of the Agila acquisition, the FDA issued a warning letter to Strides Arcolab for its Agila Sterile Manufacturing Facility 2 in Bangalore, India ("SFF"). On August 6, 2015, the FDA issued a second warning letter regarding this facility, the Agila Onco Therapies Limited ("OTL") facility and the Agila Sterile Product Division facility ("SPD"). On July 12, 2016, the FDA notified us that, based on its evaluation, it appeared we had addressed the issues related to SPD. On September 12, 2016, the FDA notified us that, based on its evaluation, it appeared we had addressed the issues related to SFF. We continue to work with the FDA to resolve the issues related to OTL.
We are subject to various federal, state and local laws regulating working conditions, as well as environmental protection laws and regulations, including those governing the discharge of materials into the environment and those related to climate change. If changes to such environmental laws and regulations are made in the future that require significant changes in our operations, or if we engage in the development and manufacturing of new products requiring new or different environmental or
36
Table of Contents
other controls, or if we are found to have violated any applicable rules, we may be required to expend significant funds. Such changes, delays, and/or suspensions of activities or the occurrence of any of the above risks, could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
THE USE OF LEGAL, REGULATORY, AND LEGISLATIVE STRATEGIES BY BOTH BRAND AND GENERIC COMPETITORS, INCLUDING BUT NOT LIMITED TO "AUTHORIZED GENERICS" AND REGULATORY PETITIONS, AS WELL AS THE POTENTIAL IMPACT OF PROPOSED AND NEWLY ENACTED LEGISLATION, MAY INCREASE COSTS ASSOCIATED WITH THE INTRODUCTION OR MARKETING OF OUR GENERIC PRODUCTS, COULD DELAY OR PREVENT SUCH INTRODUCTION, AND COULD SIGNIFICANTLY REDUCE OUR REVENUE AND PROFIT.
Our competitors, both branded and generic, often pursue strategies to prevent, delay, or eliminate competition from generic alternatives to branded products. These strategies include, but are not limited to:
• | entering into agreements whereby other generic companies will begin to market an authorized generic, a generic equivalent of a branded product, at the same time or after generic competition initially enters the market; |
• | launching a generic version of their own branded product prior to or at the same time or after generic competition initially enters the market; |
• | filing petitions with the FDA or other regulatory bodies seeking to prevent or delay approvals, including timing the filings so as to thwart generic competition by causing delays of our product approvals; |
• | seeking to establish regulatory and legal obstacles that would make it more difficult to demonstrate bioequivalence or to meet other requirements for approval, and/or to prevent regulatory agency review of applications, such as through the establishment of patent linkage (laws and regulations barring the issuance of regulatory approvals prior to patent expiration); |
• | initiating legislative or other efforts to limit the substitution of generic versions of brand pharmaceuticals; |
• | filing suits for patent infringement and other claims that may delay or prevent regulatory approval, manufacture, and/or scale of generic products; |
• | introducing "next-generation" products prior to the expiration of market exclusivity for the reference product, which often materially reduces the demand for the generic or the reference product for which we seek regulatory approval; |
• | persuading regulatory bodies to withdraw the approval of brand name drugs for which the patents are about to expire and converting the market to another product of the brand company on which longer patent protection exists; |
• | obtaining extensions of market exclusivity by conducting clinical trials of brand drugs in pediatric populations or by other methods; and |
• | seeking to obtain new patents on drugs for which patent protection is about to expire. |
In the U.S., some companies have lobbied Congress for amendments to the Hatch-Waxman Act that would give them additional advantages over generic competitors. For example, although the term of a company's drug patent can be extended to reflect a portion of the time an NDA is under regulatory review, some companies have proposed extending the patent term by a full year for each year spent in clinical trials rather than the one-half year that is currently permitted.
If proposals like these in the U.S., Europe, or in other countries where we or our partners and suppliers operate were to become effective, or if any other actions by our competitors and other third parties to prevent or delay activities necessary to the approval, manufacture, or distribution of our products are successful, our entry into the market and our ability to generate revenues associated with new products may be delayed, reduced, or eliminated, which could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
IF WE ARE UNABLE TO SUCCESSFULLY INTRODUCE NEW PRODUCTS IN A TIMELY MANNER, OUR FUTURE REVENUE AND PROFIT MAY BE ADVERSELY AFFECTED.
37
Table of Contents
Our future revenues and profitability will depend, in part, upon our ability to successfully and timely develop, license, or otherwise acquire and commercialize new generic products as well as branded pharmaceutical products protected by patent or statutory authority. Product development is inherently risky, especially for new drugs for which safety and efficacy have not been established and/or the market is not yet proven as well as for complex generic drugs and biosimilars. Likewise, product licensing involves inherent risks, including among others uncertainties due to matters that may affect the achievement of milestones, as well as the possibility of contractual disagreements with regard to whether the supply of product meets certain specifications or terms such as license scope or termination rights. The development and commercialization process, particularly with regard to new and complex drugs, also requires substantial time, effort and financial resources. We, or a partner, may not be successful in commercializing any of such products on a timely basis, if at all, which could adversely affect our business, financial condition, results of operations, cash flows, and/or ordinary share price.
Before any prescription drug product, including generic drug products, can be marketed, marketing authorization approval is required by the relevant regulatory authorities and/or national regulatory agencies (for example the FDA in the U.S. and the EMA in the EU). The process of obtaining regulatory approval to manufacture and market new branded and generic pharmaceutical products is rigorous, time consuming, costly, and inherently unpredictable.
Outside the U.S., the approval process may be more or less rigorous, depending on the country, and the time required for approval may be longer or shorter than that required in the U.S. Bioequivalence, clinical, or other studies conducted in one country may not be accepted in other countries, the requirements for approval may differ among countries, and the approval of a pharmaceutical product in one country does not necessarily mean that the product will be approved in another country. We, or a partner or supplier, may be unable to obtain requisite approvals on a timely basis, or at all, for new generic or branded products that we may develop, license or otherwise acquire. Moreover, if we obtain regulatory approval for a drug, it may be limited, for example, with respect to the indicated uses and delivery methods for which the drug may be marketed, or may include warnings, precautions or contraindications in the labeling, which could restrict our potential market for the drug. A regulatory approval may also include post-approval study or risk management requirements that may substantially increase the resources required to market the drug. Also, for products pending approval, we may obtain raw materials or produce batches of inventory to be used in efficacy and bioequivalence testing, as well as in anticipation of the product's launch. In the event that regulatory approval is denied or delayed, we could be exposed to the risk of this inventory becoming obsolete.
The approval process for generic pharmaceutical products often results in the relevant regulatory agency granting final approval to a number of generic pharmaceutical products at the time a patent claim for a corresponding branded product or other market exclusivity expires. This often forces us to face immediate competition when we introduce a generic product into the market. Additionally, further generic approvals often continue to be granted for a given product subsequent to the initial launch of the generic product. These circumstances generally result in significantly lower prices, as well as reduced margins, for generic products compared to branded products. New generic market entrants generally cause continued price, margin, and sales erosion over the generic product life cycle.
In the U.S., the Hatch-Waxman Act provides for a period of 180 days of generic marketing exclusivity for a "first applicant," that is the first submitted ANDA containing a certification of invalidity, non-infringement or unenforceability related to a patent listed with the ANDA's reference drug product, commonly referred to as a Paragraph IV certification. During this exclusivity period, which under certain circumstances may be shared with other ANDAs filed on the same day, the FDA cannot grant final approval to later-submitted ANDAs for the same generic equivalent. If an ANDA is awarded 180-day exclusivity, the applicant generally enjoys higher market share, net revenues, and gross margin for that generic product. However, our ability to obtain 180 days of generic marketing exclusivity may be dependent upon our ability to obtain FDA approval or tentative approval within an applicable time period of the FDA's acceptance of our ANDA. If we are unable to obtain approval or tentative approval within that time period, we may risk forfeiture of such marketing exclusivity. By contrast, if we are not a "first applicant" to challenge a listed patent for such a product, we may lose significant advantages to a competitor with 180-day exclusivity, even if we obtain FDA approval for our generic drug product. The same would be true in situations where we are required to share our exclusivity period with other ANDA sponsors with Paragraph IV certifications.
In the EU and other countries and regions, there is no exclusivity period for the first generic product. The European Commission or national regulatory agencies may grant marketing authorizations to any number of generics.
If we are unable to navigate our products through the approval process in a timely manner, there could be an adverse effect on our product introduction plans, business, financial condition, results of operations, cash flows, and/or ordinary share price.
38
Table of Contents
WE EXPEND A SIGNIFICANT AMOUNT OF RESOURCES ON RESEARCH AND DEVELOPMENT EFFORTS THAT MAY NOT LEAD TO SUCCESSFUL PRODUCT INTRODUCTIONS.
Much of our development effort is focused on technically difficult-to-formulate products and/or products that require advanced manufacturing technology, including our generic biologics program and respiratory platform. We conduct R&D primarily to enable us to gain approval for, manufacture, and market pharmaceuticals in accordance with applicable laws and regulations. We also partner with third parties to develop products. Typically, research expenses related to the development of innovative or complex compounds and the filing of marketing authorization applications for innovative and complex compounds (such as NDAs and biosimilar applications in the U.S.) are significantly greater than those expenses associated with the development of and filing of marketing authorization applications for most generic products (such as ANDAs in the U.S. and abridged applications in Europe). As we and our partners continue to develop new and/or complex products, our research expenses will likely increase. Because of the inherent risk associated with R&D efforts in our industry, including the high cost and uncertainty of conducting clinical trials (where required) particularly with respect to new and/or complex drugs, our, or a partner's, research and development expenditures may not result in the successful introduction of new pharmaceutical products approved by the relevant regulatory bodies. Also, after we submit a marketing authorization application for a new compound or generic product, the relevant regulatory authority may change standards and/or request that we conduct additional studies or evaluations and, as a result, we may incur approval delays as well as R&D costs in excess of what we anticipated.
Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. We or our partners may experience delays in our ongoing or future clinical trials, and we do not know whether planned clinical trials will begin or enroll subjects on time, need to be redesigned, or be completed on schedule, if at all.
Clinical trials may be delayed, suspended or prematurely terminated for a variety of reasons. If we experience delays in the completion of, or the termination of, any clinical trial of our product candidates, the commercial prospects of our product candidates will be harmed, and our ability to generate product revenues from any of these product candidates will be delayed. In addition, any delays in completing our clinical trials will increase our costs, slow down our product candidate development and approval process, and jeopardize our ability to commence product sales and generate revenues. Any of these occurrences may harm our business, financial condition and prospects significantly. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
Finally, we cannot be certain that any investment made in developing products will be recovered, even if we are successful in commercialization. To the extent that we expend significant resources on R&D efforts and are not able, ultimately, to introduce successful new and/or complex products as a result of those efforts, there could be a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
EVEN IF OUR PRODUCTS IN DEVELOPMENT RECEIVE REGULATORY APPROVAL, SUCH PRODUCTS MAY NOT ACHIEVE EXPECTED LEVELS OF MARKET ACCEPTANCE.
Even if we are able to obtain regulatory approvals for our new generic or branded pharmaceutical products, the success of those products is dependent upon market acceptance. Levels of market acceptance for our products could be impacted by several factors, including but not limited to:
• | the availability, perceived advantages, and relative safety and efficacy of alternative products from our competitors; |
• | the degree to which the approved labeling supports promotional initiatives for commercial success; |
• | the prices of our products relative to those of our competitors; |
• | the timing of our market entry; |
• | the effectiveness of our marketing, sales, and distribution strategy and operations; |
• | other competitor actions; and |
• | the continued acceptance of and/or reimbursement for our products by government and private formularies and/or third party payors, as well as the willingness and ability of patients to pay for our products. |
39
Table of Contents
Additionally, studies of the proper utilization, safety, and efficacy of pharmaceutical products are being conducted by the industry, government agencies, and others. Such studies, which increasingly employ sophisticated methods and techniques, can call into question the utilization, safety, and efficacy of previously marketed as well as future products. In some cases, such studies have resulted, and may in the future result, in the discontinuation or variation of product marketing authorizations or requirements for risk management programs, such as a patient registry. Any of these events could adversely affect our profitability, business, financial condition, results of operations, cash flows, and/or ordinary share price.
THE DEVELOPMENT, APPROVAL PROCESS, MANUFACTURE AND COMMERCIALIZATION OF BIOSIMILAR PRODUCTS INVOLVE UNIQUE CHALLENGES AND UNCERTAINTIES, AND OUR FAILURE TO SUCCESSFULLY INTRODUCE BIOSIMILAR PRODUCTS COULD HAVE A NEGATIVE IMPACT ON OUR BUSINESS AND FUTURE OPERATING RESULTS.
We and our partners and suppliers are actively working to develop and commercialize biosimilar products - that is, a biological product that is highly similar to an already approved reference biological product, and for which there are no clinically meaningful differences between the biosimilar and the reference biological product in terms of safety, purity and potency. Although the Biologics Price Competition and Innovation Act of 2009 established a framework for the review and approval of biosimilar products and the FDA has begun to review and approve biosimilar product applications, there continues to be significant uncertainty regarding the regulatory pathway in the U.S. and in other countries to obtain approval for biosimilar products. There is also uncertainty regarding the commercial pathway to successfully market and sell such products.
Moreover, biosimilar products will likely be subject to extensive patent clearances and patent infringement litigation, which could delay or prevent the commercial launch of a biosimilar product for many years. If we are unable to obtain FDA or other non-U.S. regulatory authority approval for our products, we will be unable to market them. Even if our biosimilar products are approved for marketing, the products may not be commercially successful and may not generate profits in amounts that are sufficient to offset the amount invested to obtain such approvals. Market success of biosimilar products will depend on demonstrating to regulators, patients, physicians and payors (such as insurance companies) that such products are safe and effective yet offer a more competitive price or other benefit over existing therapies. In addition, the development and manufacture of biosimilars pose unique challenges related to the supply of the materials needed to manufacture biosimilars. Access to and the supply of necessary biological materials may be limited, and government regulations restrict access to and regulate the transport and use of such materials. We may not be able to generate future sales of biosimilar products in certain jurisdictions and may not realize the anticipated benefits of our investments in the development, manufacture and sale of such products. If our development efforts do not result in the development and timely approval of biosimilar products or if such products, once developed and approved, are not commercially successful, or upon the occurrence of any of the above risks, our business, financial condition, results of operations, cash flows, and/or ordinary share price could be materially adversely affected.
OUR BUSINESS IS HIGHLY DEPENDENT UPON MARKET PERCEPTIONS OF US, OUR BRANDS, AND THE SAFETY AND QUALITY OF OUR PRODUCTS, AND MAY BE ADVERSELY IMPACTED BY NEGATIVE PUBLICITY OR FINDINGS.
Market perceptions of us are very important to our business, especially market perceptions of our company and brands and the safety and quality of our products. If we, our partners and suppliers, or our brands suffer from negative publicity, or if any of our products or similar products which other companies distribute are subject to market withdrawal or recall or are proven to be, or are claimed to be, ineffective or harmful to consumers, then this could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price. Also, because we are dependent on market perceptions, negative publicity associated with product quality, patient illness, or other adverse effects resulting from, or perceived to be resulting from, our products, or our partners' and suppliers' manufacturing facilities, could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
THE ILLEGAL DISTRIBUTION AND SALE BY THIRD PARTIES OF COUNTERFEIT VERSIONS OF OUR PRODUCTS OR OF DIVERTED OR STOLEN PRODUCTS COULD HAVE A NEGATIVE IMPACT ON OUR REPUTATION AND OUR BUSINESS.
The pharmaceutical drug supply has been increasingly challenged by the vulnerability of distribution channels to illegal counterfeiting and the presence of counterfeit products in a growing number of markets and over the Internet.
Third parties may illegally distribute and sell counterfeit versions of our products that do not meet the rigorous manufacturing and testing standards that our products undergo. Counterfeit products are frequently unsafe or ineffective, and
40
Table of Contents
can be potentially life-threatening. Counterfeit medicines may contain harmful substances, the wrong dose of API or no API at all. However, to distributors and users, counterfeit products may be visually indistinguishable from the authentic version.
Reports of adverse reactions to counterfeit drugs or increased levels of counterfeiting could materially affect patient confidence in the authentic product. It is possible that adverse events caused by unsafe counterfeit products will mistakenly be attributed to the authentic product. In addition, unauthorized diversions of products or thefts of inventory at warehouses, plants, or while in-transit, which are not properly stored and which are sold through unauthorized channels, could adversely impact patient safety, our reputation, and our business.
Public loss of confidence in the integrity of pharmaceutical products as a result of counterfeiting, diversion, or theft could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
OUR COMPETITORS, INCLUDING BRANDED PHARMACEUTICAL COMPANIES, AND/OR OTHER THIRD PARTIES, MAY ALLEGE THAT WE AND/OR OUR SUPPLIERS ARE INFRINGING UPON THEIR INTELLECTUAL PROPERTY, INCLUDING IN AN "AT RISK LAUNCH" SITUATION, IMPACTING OUR ABILITY TO LAUNCH A PRODUCT, AND/OR OUR ABILITY TO CONTINUE MARKETING A PRODUCT, AND/OR FORCING US TO EXPEND SUBSTANTIAL RESOURCES IN RESULTING LITIGATION, THE OUTCOME OF WHICH IS UNCERTAIN.
Companies that produce branded pharmaceutical products and other patent holders routinely bring litigation against entities selling or seeking regulatory approval to manufacture and market generic forms of their branded products, as well as other entities involved in the manufacture, supply, testing, marketing, and other aspects relating to active pharmaceutical ingredients and finished pharmaceutical products. These companies and other patent holders allege patent infringement or other violations of intellectual property rights as the basis for filing suit against an applicant for a generic product license as well as others who may be involved in some aspect of the research, production, distribution, or testing process. Litigation often involves significant expense and can delay or prevent introduction or sale of our generic products. If patents are held valid and infringed by our products in a particular jurisdiction, we and/or our supplier(s) or partner(s) may, unless we or the supplier(s) or partner(s) could obtain a license from the patent holder, need to cease manufacturing and other activities, including but not limited to selling in that jurisdiction, pay damages, and may need to surrender or withdraw the product, or destroy existing stock in that jurisdiction.
There also may be situations where we use our business judgment and decide to manufacture, market, and/or sell products, directly or through third parties, notwithstanding the fact that allegations of patent infringement(s) have not been finally resolved by the courts (i.e., an "at-risk launch"). The risk involved in doing so can be substantial because the remedies available to the owner of a patent for infringement may include, among other things, damages measured by the profits lost by the patent holder and not necessarily by the profits earned by the infringer. In the case of a finding by a court of willful infringement, the definition of which is subjective, such damages may be increased by an additional 200% in certain jurisdictions, including the U.S. Moreover, because of the discount pricing typically involved with bioequivalent (generic) products, patented branded products generally realize a substantially higher profit margin than bioequivalent products. An adverse decision in a case such as this or in other similar litigation, or a judicial order preventing us or our suppliers and partners from manufacturing, marketing, selling, and/or other activities necessary to the manufacture and distribution of our products, could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
IF WE OR ANY PARTNER OR SUPPLIER FAIL TO OBTAIN OR ADEQUATELY PROTECT OR ENFORCE OUR INTELLECTUAL PROPERTY RIGHTS, THEN WE COULD LOSE REVENUE UNDER OUR LICENSING AGREEMENTS OR LOSE SALES TO GENERIC COPIES OF OUR BRANDED PRODUCTS.
Our success depends in part on our or any partner's or supplier's ability to obtain, maintain and enforce patents, and protect trademarks, trade secrets, know-how, and other intellectual property and proprietary information. Our ability to commercialize any branded product successfully will largely depend upon our or any partner's or supplier's ability to obtain and maintain patents and trademarks of sufficient scope to lawfully prevent third-parties from developing and/or marketing infringing products. In the absence of intellectual property or other protection, competitors may adversely affect our branded products business by independently developing and/or marketing substantially equivalent products. It is also possible that we could incur substantial costs if we are required to initiate litigation against others to protect or enforce our intellectual property rights.
We have filed patent applications covering the composition of, methods of making, and/or methods of using, our branded products and branded product candidates. We may not be issued patents based on patent applications already filed or
41
Table of Contents
that we file in the future. Further, due to other factors that affect patentability, and if patents are issued, they may be insufficient in scope to cover or otherwise protect our branded products. Patents are national in scope and therefore the issuance of a patent in one country does not ensure the issuance of a patent in any other country. Furthermore, the patent position of companies in the pharmaceutical industry generally involves complex legal and factual questions and has been and remains the subject of significant litigation. Legal standards relating to scope and validity of patent claims are evolving and may differ in various countries. Any patents we have obtained, or obtain in the future, may be challenged, invalidated or circumvented. Moreover, the U.S. Patent and Trademark Office or any other governmental agency may commence opposition or interference proceedings involving, or consider other challenges to, our patents or patent applications. In addition, branded products often have market viability based upon the goodwill of the product name, which typically benefits from trademark protection. Our branded products may therefore also be subject to risks related to the loss of trademark or patent protection or to competition from generic or other branded products. Challenges can come from other businesses or governments, and governments could require compulsory licensing of this intellectual property.
Any challenge to, or invalidation or circumvention of, our intellectual property (including patents or patent applications and trademark protection) would be costly, would require significant time and attention of our management, and could cause a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
WE DEVELOP, FORMULATE, MANUFACTURE, OR IN-LICENSE AND MARKET PRODUCTS THAT ARE SUBJECT TO ECONOMIC RISKS RELATING TO INTELLECTUAL PROPERTY RIGHTS, COMPETITION, AND MARKET UNPREDICTABILITY.
Our products may be subject to the following risks, among others:
• | limited patent life, or the loss of patent protection; |
• | competition from generic or other branded products; |
• | reductions in reimbursement rates by government and other third-party payors; |
• | importation by consumers; |
• | product liability; |
• | drug research and development risks; and |
• | unpredictability with regard to establishing a market. |
In addition, developing and commercializing branded products is generally more costly than generic products. If such business expenditures do not ultimately result in the launch of commercially successful brand products, or if any of the risks above were to occur, there could be a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
WE FACE VIGOROUS COMPETITION FROM OTHER PHARMACEUTICAL MANUFACTURERS THAT THREATENS THE COMMERCIAL ACCEPTANCE AND PRICING OF OUR PRODUCTS.
The pharmaceutical industry is highly competitive. We face competition from many U.S. and non-U.S. manufacturers, some of whom are significantly larger than we are. Our competitors may be able to develop products and processes competitive with or superior to our own for many reasons, including but not limited to the possibility that they may have:
• | proprietary processes or delivery systems; |
• | larger or more productive research and development and marketing staffs; |
• | larger or more efficient production capabilities in a particular therapeutic area; |
• | more experience in preclinical testing and human clinical trials; |
• | more products; or |
42
Table of Contents
• | more experience in developing new drugs and greater financial resources, particularly with regard to manufacturers of branded products. |
The occurrence of any of the above risks could have an adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
We also face increasing competition from lower-cost generic products and other branded products. Certain of our products are not protected by patent rights or have limited patent life and will soon lose patent protection. Loss of patent protection for a product typically is followed promptly by generic substitutes. As a result, sales of many of these products may decline or stop growing over time. Various factors may result in the sales of certain of our products, particularly those acquired in the Meda transaction and the EPD Transaction, declining faster than has been projected, which could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price. In addition, legislative proposals emerge from time to time in various jurisdictions to further encourage the early and rapid approval of generic drugs. Any such proposal that is enacted into law could increase competition and worsen this negative effect on our sales and, potentially, our business, financial condition, results of operations, cash flows and/or ordinary share price.
Competitors' products may also be safer, more effective, more effectively marketed or sold, or have lower prices or better performance features than ours. We cannot predict with certainty the timing or impact of competitors' products. In addition, our sales may suffer as a result of changes in consumer demand for our products, including those related to fluctuations in consumer buying patterns tied to seasonality or the introduction of new products by competitors, which could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
A RELATIVELY SMALL GROUP OF PRODUCTS MAY REPRESENT A SIGNIFICANT PORTION OF OUR REVENUES, GROSS PROFIT, NET SALES, OR NET EARNINGS FROM TIME TO TIME.
Sales of a limited number of our products from time to time represent a significant portion of our revenues, gross profit, and net earnings. For the years ended December 31, 2016 and 2015, Mylan's top ten products in terms of sales, in the aggregate, represented approximately 27% and 28% , respectively, of the Company's third party net sales. If the volume or pricing of our largest selling products declines in the future, our business, financial condition, results of operations, cash flows, and/or ordinary share price could be materially adversely affected.
A SIGNIFICANT PORTION OF OUR REVENUES IS DERIVED FROM SALES TO A LIMITED NUMBER OF CUSTOMERS.
A significant portion of our revenues are derived from sales to a limited number of customers. If we were to experience a significant reduction in or loss of business with one or more such customers, or if one or more such customers were to experience difficulty in paying us on a timely basis, our business, financial condition, results of operations, cash flows, and/or ordinary share price could be materially adversely affected.
During the years ended December 31, 2016, 2015 and 2014, Mylan's consolidated third party net sales to Cardinal Health, Inc. were approximately 11% , 12% and 12% , respectively; Mylan's consolidated third party net sales to McKesson Corporation were approximately 16% , 15% and 19% , respectively; and Mylan's consolidated third party net sales to AmeriSourceBergen Corporation were approximately 14% , 16% and 13% , respectively, of consolidated third party net sales.
OUR BUSINESS COULD BE NEGATIVELY AFFECTED BY THE PERFORMANCE OF OUR COLLABORATION PARTNERS AND SUPPLIERS.
We have entered into strategic alliances with partners and suppliers to develop, manufacture, market and/or distribute certain products, and/or certain components of our products, in various markets. We commit substantial effort, funds and other resources to these various collaborations. There is a risk that the investments made by us in these collaborative arrangements will not generate financial returns. While we believe our relationships with our partners and suppliers generally are successful, disputes or conflicting priorities and regulatory or legal intervention could be a source of delay or uncertainty as to the expected benefits of the collaboration. A failure or inability of our partners or suppliers to fulfill their collaboration obligations, or the occurrence of any of the risks above, could have an adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
WE MAY EXPERIENCE DECLINES IN THE SALES VOLUME AND PRICES OF OUR PRODUCTS AS THE RESULT OF THE CONTINUING TREND TOWARD CONSOLIDATION OF CERTAIN CUSTOMER GROUPS, SUCH AS THE
43
Table of Contents
WHOLESALE DRUG DISTRIBUTION AND RETAIL PHARMACY INDUSTRIES, AS WELL AS THE EMERGENCE OF LARGE BUYING GROUPS.
A significant amount of our sales are to a relatively small number of drug wholesalers and retail drug chains. These customers represent an essential part of the distribution chain of generic pharmaceutical products. Drug wholesalers and retail drug chains have undergone, and are continuing to undergo, significant consolidation. This consolidation may result in these groups gaining additional purchasing leverage and, consequently, increasing the product pricing pressures facing our business. Additionally, the emergence of large buying groups representing independent retail pharmacies and the prevalence and influence of managed care organizations and similar institutions increases the negotiating power of these groups, potentially enabling them to attempt to extract price discounts, rebates, and other restrictive pricing terms on our products. The occurrence of any of the above risks could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
WE DEPEND TO A LARGE EXTENT ON THIRD-PARTY SUPPLIERS AND DISTRIBUTORS FOR RAW MATERIALS, PARTICULARLY THE CHEMICAL COMPOUND(S) THAT CONSTITUTE THE ACTIVE PHARMACEUTICAL INGREDIENTS THAT WE USE TO MANUFACTURE OUR PRODUCTS, AS WELL AS CERTAIN FINISHED GOODS, INCLUDING CERTAIN CONTROLLED SUBSTANCES. THESE THIRD-PARTY SUPPLIERS AND DISTRIBUTORS MAY EXPERIENCE DELAYS IN OR INABILITY TO SUPPLY US WITH RAW MATERIALS NECESSARY TO THE DEVELOPMENT AND/OR MANUFACTURE OF OUR PRODUCTS.
We purchase certain API (i.e., the chemical compounds that produce the desired therapeutic effect in our products) and other materials and supplies that we use in our manufacturing operations, as well as certain finished products, from many different foreign and domestic suppliers.
In certain cases, we have listed only one supplier in our applications with regulatory agencies, and there is no guarantee that we will always have timely and sufficient access to a critical raw material or finished product supplied by third parties, even when we have more than one supplier. An interruption in the supply of a single-sourced or any other raw material, including the relevant API, or in the supply of finished product, could cause our business, financial condition, results of operations, cash flows, and/or ordinary share price to be materially adversely affected. In addition, our manufacturing and supply capabilities could be adversely impacted by quality deficiencies in the products which our suppliers provide, or at their manufacturing facilities, which could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
We utilize controlled substances in certain of our current products and products in development, and therefore must meet the requirements of the Controlled Substances Act of 1970 and the related regulations administered by the DEA in the U.S., as well as similar laws in other countries where we operate. These laws relate to the manufacture, shipment, storage, sale, and use of controlled substances. The DEA and other regulatory agencies limit the availability of the controlled substances used in certain of our current products and products in development and, as a result, our procurement quota of these active ingredients may not be sufficient to meet commercial demand or complete clinical trials. We must annually apply to the DEA and similar regulatory agencies for procurement quotas in order to obtain these substances. Any delay or refusal by the DEA or such similar agencies in establishing our procurement quota for controlled substances could delay or stop our clinical trials or product launches, or could cause trade inventory disruptions for those products that have already been launched, which could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
THE SUPPLY OF API INTO EUROPE MAY BE NEGATIVELY AFFECTED BY RECENT REGULATIONS PROMULGATED BY THE EUROPEAN UNION.
All API imported into the EU has needed to be certified as complying with the good manufacturing practice standards established by the EU laws and guidance, as stipulated by the International Conference for Harmonization. These regulations place the certification requirement on the regulatory bodies of the exporting countries. Accordingly, the national regulatory authorities of each exporting country must: (i) ensure that all manufacturing plants within their borders that export API into the EU comply with EU manufacturing standards and (ii) for each API exported, present a written document confirming that the exporting plant conforms to EU manufacturing standards. The imposition of this responsibility on the governments of the nations exporting an API may cause delays in delivery or shortages of an API necessary to manufacture our products, as certain governments may not be willing or able to comply with the regulation in a timely fashion, or at all. A shortage in API may prevent us from manufacturing, or cause us to have to cease manufacture of, certain products, or to incur costs and delays to qualify other suppliers to substitute for those API manufacturers unable to export. The occurrence of any of the above risks could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
44
Table of Contents
WE HAVE A LIMITED NUMBER OF MANUFACTURING FACILITIES AND CERTAIN THIRD PARTY SUPPLIERS PRODUCING A SUBSTANTIAL PORTION OF OUR PRODUCTS, SOME OF WHICH REQUIRE A HIGHLY EXACTING AND COMPLEX MANUFACTURING PROCESS.
A substantial portion of our capacity, as well as our current production, is attributable to a limited number of manufacturing facilities and certain third party suppliers. A significant disruption at any one of such facilities within our internal or third party supply chain, even on a short-term basis, whether due to a labor strike, failure to reach acceptable agreement with labor and unions, adverse quality or compliance observation, other regulatory action, infringement of intellectual property rights, act of God, civil or political unrest, export or import restrictions, or other events could impair our ability to produce and ship products to the market on a timely basis and could, among other consequences, subject us to exposure to claims from customers. Any of these events could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
In addition, the manufacture of some of our products is a highly exacting and complex process, due in part to strict regulatory requirements. Problems may arise during manufacturing for a variety of reasons, including among others equipment malfunction, failure to follow specific protocols and procedures, problems with raw materials, natural disasters, power outages, labor unrest, and environmental factors. If problems arise during the production of a batch of product, that batch of product may have to be discarded. This could, among other things, lead to increased costs, lost revenue, damage to customer relations, time and expense spent investigating the cause, and, depending on the cause, similar losses with respect to other batches or products. If problems are not discovered before the product is released to the market, recall and product liability costs may also be incurred. If we or one of our suppliers experiences significant manufacturing problems, such problems could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
OUR REPORTING AND PAYMENT OBLIGATIONS RELATED TO OUR PARTICIPATION IN U.S. FEDERAL HEALTHCARE PROGRAMS, INCLUDING MEDICARE AND MEDICAID, ARE COMPLEX AND OFTEN INVOLVE SUBJECTIVE DECISIONS THAT COULD CHANGE AS A RESULT OF NEW BUSINESS CIRCUMSTANCES, NEW REGULATIONS OR AGENCY GUIDANCE, OR ADVICE OF LEGAL COUNSEL. ANY FAILURE TO COMPLY WITH THOSE OBLIGATIONS COULD SUBJECT US TO INVESTIGATION, PENALTIES, AND SANCTIONS.
Federal laws regarding reporting and payment obligations with respect to a pharmaceutical company's participation in federal healthcare programs, including Medicare and Medicaid, are complex. Because our processes for calculating applicable government prices and the judgments involved in making these calculations involve subjective decisions and complex methodologies, these calculations are subject to risk of errors and differing interpretations. In addition, they are subject to review and challenge by the applicable governmental agencies, and it is possible that such reviews could result in changes that may have material adverse legal, regulatory, or economic consequences.
Pharmaceutical manufacturers that participate in the Medicaid Drug Rebate Program, such as Mylan, are required to report certain pricing data to the Centers for Medicare & Medicaid Services ("CMS"), the federal agency that administers the Medicare and Medicaid programs. This data includes the Average Manufacturer Price ("AMP") for each of the manufacturer's covered outpatient drugs. CMS calculates a type of U.S. federal ceiling on reimbursement rates to pharmacies for multiple source drugs under the Medicaid program, known as the federal upper limit ("FUL"). The PPACA includes a provision requiring CMS to use the weighted average AMP for pharmaceutically and therapeutically equivalent multiple source drugs to calculate FULs, instead of the other pricing data CMS previously used. The provision was effective October 1, 2010; however, AMP-based FULs have not yet been implemented to set the federal ceiling on reimbursement rates for multiple source drugs. On January 21, 2016, CMS issued final regulations to implement the changes to the Medicaid Drug Rebate program under the Health Reform Laws (as defined below), including AMP-based FULs. These regulations became effective April 1, 2016. Although weighted average AMP-based FULs do not reveal Mylan's individual AMP, publishing a weighted average AMP available to customers and the public at large could negatively affect our commercial price negotiations.
In addition, a number of state and federal government agencies are conducting investigations of manufacturers' reporting practices with respect to Average Wholesale Prices ("AWP"). The government has alleged that reporting of inflated AWP has led to excessive payments for prescription drugs, and we may be named as a defendant in actions relating to pharmaceutical pricing issues and whether allegedly improper actions by pharmaceutical manufacturers led to excessive payments by Medicare and/or Medicaid.
Any governmental agencies or authorities that have commenced, or may commence, an investigation of us relating to the sales, marketing, pricing, quality, or manufacturing of pharmaceutical products could seek to impose, based on a claim of violation of anti-fraud and false claims laws or otherwise, civil and/or criminal sanctions, including fines, penalties, and possible exclusion from federal healthcare programs, including Medicare and Medicaid. Some of the applicable laws may
45
Table of Contents
impose liability even in the absence of specific intent to defraud. Furthermore, should there be ambiguity with regard to how to properly calculate and report payments - and even in the absence of any such ambiguity - a governmental authority may take a position contrary to a position we have taken, and may impose or pursue civil and/or criminal sanctions. Governmental agencies may also make changes in program interpretations, requirements or conditions of participation, some of which may have implications for amounts previously estimated or paid. We cannot assure you that our submissions will not be found by CMS or the U.S. Department of Veterans Affairs to be incomplete or incorrect. Any failure to comply with the above laws and regulations, and any such penalties or sanctions could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
WE MAY EXPERIENCE REDUCTIONS IN THE LEVELS OF REIMBURSEMENT FOR PHARMACEUTICAL PRODUCTS BY GOVERNMENTAL AUTHORITIES, HMOS, OR OTHER THIRD-PARTY PAYORS. IN ADDITION, THE USE OF TENDER SYSTEMS AND OTHER FORMS OF PRICE CONTROL COULD REDUCE PRICES FOR OUR PRODUCTS OR REDUCE OUR MARKET OPPORTUNITIES.
Various governmental authorities (including, among others, the United Kingdom National Health Service and the German statutory health insurance scheme) and private health insurers and other organizations, such as HMOs in the U.S., provide reimbursements or subsidies to consumers for the cost of certain pharmaceutical products. Demand for our products depends in part on the extent to which such reimbursement is available. In the U.S., third-party payors increasingly challenge the pricing of pharmaceutical products. This trend and other trends toward the growth of HMOs, managed healthcare, and legislative healthcare reform create significant uncertainties regarding the future levels of reimbursement for pharmaceutical products. Further, any reimbursement may be reduced in the future to the point that market demand for our products and/or our profitability declines. Such a decline could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
In addition, a number of markets in which we operate have implemented or may implement tender systems or other forms of price controls for generic pharmaceuticals in an effort to lower prices. Under such tender systems, manufacturers submit bids which establish prices for generic pharmaceutical products. Upon winning the tender, the winning company will receive a preferential reimbursement for a period of time. The tender system often results in companies underbidding one another by proposing low pricing in order to win the tender.
Certain other countries may consider the implementation of a tender system or other forms of price controls. Even if a tender system is ultimately not implemented, the anticipation of such could result in price reductions. Failing to win tenders, or the implementation of similar systems or other forms of price controls in other markets leading to further price declines, could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
LEGISLATIVE OR REGULATORY PROGRAMS THAT MAY INFLUENCE PRICES OF PHARMACEUTICAL PRODUCTS COULD HAVE A MATERIAL ADVERSE EFFECT ON OUR BUSINESS.
Current or future U.S. federal, U.S. state or other countries' laws and regulations may influence the prices of drugs and, therefore, could adversely affect the payment that we receive for our products. For example, programs in existence in certain states in the U.S. seek to broadly set prices, within those states, through the regulation and administration of the sale of prescription drugs. Expansion of these programs, in particular state Medicare and/or Medicaid programs, or changes required in the way in which Medicare payment rates are set and/or the way Medicaid rebates are calculated, could adversely affect the payment we receive for our products and could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
In order to control expenditure on pharmaceuticals, most member states in the EU regulate the pricing of products and, in some cases, limit the range of different forms of pharmaceuticals available for prescription by national health services. These controls can result in considerable price differences between member states.
Several countries in which we operate have implemented, or plan to or may implement, government mandated price reductions and/or other controls. When such price cuts occur, pharmaceutical companies have generally experienced significant declines in revenues and profitability and uncertainties continue to exist within the market after the price decrease. Such price reductions or controls could have an adverse effect on our business, and as uncertainties are resolved or if other countries in which we operate enact similar measures, they could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
HEALTHCARE REFORM LEGISLATION COULD HAVE A MATERIAL ADVERSE EFFECT ON OUR BUSINESS.
46
Table of Contents
In recent years, there have been numerous initiatives on the federal and state levels for comprehensive reforms affecting the payment for, the availability of and reimbursement for, healthcare services in the U.S., and it is likely that Congress and state legislatures and health agencies will continue to focus on healthcare reform in the future. The PPACA and The Health Care and Education and Reconciliation Act of 2010 (H.R. 4872), which amends the PPACA (collectively, the "Health Reform Laws"), were signed into law in March 2010. While the Health Reform Laws may increase the number of patients who have insurance coverage for our products, they also include provisions such as the assessment of a pharmaceutical manufacturer fee and an increase in the amount of rebates that manufacturers pay for coverage of their drugs by Medicaid programs.
We are unable to predict the future course of federal or state healthcare legislation. The Health Reform Laws and further changes in the law or regulatory framework that reduce our revenues or increase our costs could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
Additionally, we encounter similar regulatory and legislative issues in most other countries. In the EU and some other international markets, the government provides healthcare at low cost to consumers and regulates pharmaceutical prices, patient eligibility and/or reimbursement levels to control costs for the government-sponsored healthcare system. These systems of price regulations may lead to inconsistent and lower prices. Within the EU and in other countries, the availability of our products in some markets at lower prices undermines our sales in other markets with higher prices. Additionally, certain countries set prices by reference to the prices in other countries where our products are marketed. Thus, our inability to secure adequate prices in a particular country may also impair our ability to obtain acceptable prices in existing and potential new markets, and may create the opportunity for third party cross border trade.
If significant additional reforms are made to the U.S. healthcare system, or to the healthcare systems of other markets in which we operate, those reforms could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
WE ARE INVOLVED IN VARIOUS LEGAL PROCEEDINGS AND CERTAIN GOVERNMENT INQUIRIES AND MAY EXPERIENCE UNFAVORABLE OUTCOMES OF SUCH PROCEEDINGS OR INQUIRIES.
We are or may be involved in various legal proceedings and certain government inquiries or investigations, including, but not limited to, patent infringement, product liability, antitrust matters, breach of contract, and claims involving Medicare and/or Medicaid reimbursements, or laws relating to sales, marketing, and pricing practices, some of which are described in our periodic reports, that involve claims for, or the possibility of, fines and penalties involving substantial amounts of money or other relief, including but not limited to civil or criminal fines and penalties and exclusion from participation in various government health-care-related programs. With respect to government antitrust enforcement and private plaintiff litigation of so-called "pay for delay" patent settlements, large verdicts, settlements or government fines are possible, especially in the U.S. and EU. If any of these legal proceedings or inquiries were to result in an adverse outcome, the impact could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
With respect to product liability, we maintain a combination of self-insurance (including through our wholly owned captive insurance subsidiary) and commercial insurance to protect against and manage a portion of the risks involved in conducting our business. Although we carry insurance, we believe that no reasonable amount of insurance can fully protect against all such risks because of the potential liability inherent in the business of producing pharmaceuticals for human consumption. Emerging developments in the U.S. legal landscape relative to the liability of generic pharmaceutical manufacturers for certain product liabilities claims could increase our exposure litigation costs and damages. To the extent that a loss occurs, depending on the nature of the loss and the level of insurance coverage maintained, it could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
In addition, in limited circumstances, entities that we acquired are party to litigation in matters under which we are, or may be, entitled to indemnification by the previous owners. Even in the case of indemnification, there are risks inherent in such indemnities and, accordingly, there can be no assurance that we will receive the full benefits of such indemnification, or that we will not experience an adverse result in a matter that is not indemnified, which could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
WE HAVE A NUMBER OF CLEAN ENERGY INVESTMENTS WHICH ARE SUBJECT TO VARIOUS RISKS AND UNCERTAINTIES.
We have invested in clean energy operations capable of producing refined coal that we believe qualify for tax credits under Section 45 of the Code. Our ability to claim tax credits under Section 45 of the Code depends upon the operations in which we have invested satisfying certain ongoing conditions set forth in Section 45 of the Code. These include, among others,
47
Table of Contents
the emissions reduction, "qualifying technology", and "placed-in-service" requirements of Section 45 of the Code, as well as the requirement that at least one of the operations' owners qualifies as a "producer" of refined coal. While we have received some degree of confirmation from the IRS relating to our ability to claim these tax credits, the IRS could ultimately determine that the operations have not satisfied, or have not continued to satisfy, the conditions set forth in Section 45 of the Code. Additionally, Congress could modify or repeal Section 45 of the Code and remove the tax credits retroactively.
In addition, Section 45 of the Code contains phase out provisions based upon the market price of coal, such that, if the price of coal rises to specified levels, we could lose some or all of the tax credits we expect to receive from these investments.
Finally, when the price of natural gas or oil declines relative to that of coal, some utilities may choose to burn natural gas or oil instead of coal. Market demand for coal may also decline as a result of an economic slowdown and a corresponding decline in the use of electricity. If utilities burn less coal, eliminate coal in the production of electricity or are otherwise unable to operate for an extended period of time, the availability of the tax credits would also be reduced. The occurrence of any of the above risks could adversely affect our business, financial condition, results of operations, cash flows, and/or ordinary share price.
WE HAVE SIGNIFICANT INDEBTEDNESS WHICH COULD ADVERSELY AFFECT OUR FINANCIAL POSITION AND PREVENT US FROM FULFILLING OUR OBLIGATIONS UNDER SUCH INDEBTEDNESS. ANY REFINANCING OF THIS DEBT COULD BE AT SIGNIFICANTLY HIGHER INTEREST RATES. OUR SUBSTANTIAL INDEBTEDNESS COULD LEAD TO ADVERSE CONSEQUENCES.
Our level of indebtedness could have important consequences, including but not limited to:
• | increasing our vulnerability to general adverse economic and industry conditions; |
• | requiring us to dedicate a substantial portion of our cash flow from operations to make debt service payments, thereby reducing the availability of cash flow to fund working capital, capital expenditures, acquisitions and investments and other general corporate purposes; |
• | limiting our flexibility in planning for, or reacting to, challenges and opportunities, and changes in our businesses and the markets in which we operate; |
• | limiting our ability to obtain additional financing to fund our working capital, capital expenditures, acquisitions and debt service requirements and other financing needs; |
• | increasing our vulnerability to increases in interest rates in general because a substantial portion of our indebtedness bears interest at floating rates; and |
• | placing us at a competitive disadvantage to our competitors that have less debt. |
Our ability to service our indebtedness will depend on our future operating performance and financial results, which will be subject, in part, to factors beyond our control, including interest rates and general economic, financial and business conditions. If we do not have sufficient cash flow to service our indebtedness, we may need to refinance all or part of our existing indebtedness, borrow more money or sell securities or assets, some or all of which may not be available to us at acceptable terms or at all. In addition, we may need to incur additional indebtedness in the future in the ordinary course of business. Although the terms of our senior credit agreements and our bond indentures allow us to incur additional debt, this is subject to certain limitations which may preclude us from incurring the amount of indebtedness we otherwise desire.
In addition, although Mylan expects to maintain an investment grade credit rating, our increased indebtedness following the completion of the Meda acquisition could result in a downgrade in the credit rating of Mylan or any indebtedness of Mylan or its subsidiaries. A downgrade in the credit rating of Mylan or any indebtedness of Mylan or its subsidiaries could increase the cost of further borrowings or refinancings of such indebtedness, limit access to sources of financing in the future or lead to other adverse consequences.
In addition, if we incur additional debt, the risks described above could intensify. If global credit markets return to their recent levels of contraction, future debt financing may not be available to us when required or may not be available on acceptable terms, and as a result we may be unable to grow our business, take advantage of business opportunities, respond to competitive pressures or satisfy our obligations under our indebtedness. Any of the foregoing could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
48
Table of Contents
Our credit facilities, senior unsecured notes, accounts receivable securitization facility, other outstanding indebtedness and any additional indebtedness we incur in the future impose, or may impose, significant operating and financial restrictions on us. These restrictions limit our ability to, among other things, incur additional indebtedness, make investments, pay certain dividends, prepay other indebtedness, sell assets, incur certain liens, enter into agreements with our affiliates or restricting our subsidiaries' ability to pay dividends, merge or consolidate. In addition, our 2016 Senior Revolving Credit Agreement, 2016 Senior Term Credit Agreement, and accounts receivable securitization facility require us to maintain specified financial ratios. A breach of any of these covenants or our inability to maintain the required financial ratios could result in a default under the related indebtedness. If a default occurs, the relevant lenders could elect to declare our indebtedness, together with accrued interest and other fees, to be immediately due and payable. These factors could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
WE ENTER INTO VARIOUS AGREEMENTS IN THE NORMAL COURSE OF BUSINESS WHICH PERIODICALLY INCORPORATE PROVISIONS WHEREBY WE INDEMNIFY THE OTHER PARTY TO THE AGREEMENT.
In the normal course of business, we periodically enter into commercial, employment, legal settlement, and other agreements which incorporate indemnification provisions. In some but not all cases, we maintain insurance coverage which we believe will effectively mitigate our obligations under certain of these indemnification provisions. However, should our obligation under an indemnification provision exceed any applicable coverage or should coverage be denied, our business, financial condition, results of operations, cash flows, and/or ordinary share price could be materially adversely affected.
THERE ARE INHERENT UNCERTAINTIES INVOLVED IN ESTIMATES, JUDGMENTS AND ASSUMPTIONS USED IN THE PREPARATION OF FINANCIAL STATEMENTS IN ACCORDANCE WITH U.S. GAAP. ANY FUTURE CHANGES IN ESTIMATES, JUDGMENTS AND ASSUMPTIONS USED OR NECESSARY REVISIONS TO PRIOR ESTIMATES, JUDGMENTS OR ASSUMPTIONS OR CHANGES IN ACCOUNTING STANDARDS COULD LEAD TO A RESTATEMENT OR REVISION TO PREVIOUSLY ISSUED FINANCIAL STATEMENTS.
The Consolidated and Condensed Consolidated Financial Statements included in the periodic reports we file with the SEC are prepared in accordance with U.S. GAAP. The preparation of financial statements in accordance with U.S. GAAP involves making estimates, judgments and assumptions that affect reported amounts of assets, liabilities, revenues, expenses and income. Estimates, judgments and assumptions are inherently subject to change in the future and any necessary revisions to prior estimates, judgments or assumptions could lead to a restatement. Furthermore, although we have recorded reserves for litigation related contingencies based on estimates of probable future costs, such litigation related contingencies could result in substantial further costs. Also, any new or revised accounting standards may require adjustments to previously issued financial statements. Any such changes could result in corresponding changes to the amounts of liabilities, revenues, expenses and income. Any such changes could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
WE MUST MAINTAIN ADEQUATE INTERNAL CONTROLS AND BE ABLE ON AN ANNUAL BASIS, TO PROVIDE AN ASSERTION AS TO THE EFFECTIVENESS OF SUCH CONTROLS.
Effective internal controls are necessary for us to provide reasonable assurance with respect to our financial reports. We spend a substantial amount of management and other employee time and resources to comply with laws, regulations and standards relating to corporate governance and public disclosure. In the U.S., such regulations include the Sarbanes-Oxley Act of 2002, SEC regulations and the NASDAQ listing standards. In particular, Section 404 of the Sarbanes-Oxley Act of 2002 ("Section 404") requires management's annual review and evaluation of our internal control over financial reporting and attestation as to the effectiveness of these controls by our independent registered public accounting firm. If we fail to maintain the adequacy of our internal controls, we may not be able to ensure that we can conclude on an ongoing basis that we have effective internal control over financial reporting. Additionally, internal control over financial reporting may not prevent or detect misstatements because of its inherent limitations, including the possibility of human error, the circumvention or overriding of controls, or fraud. Therefore, even effective internal controls can provide only reasonable assurance with respect to the preparation and fair presentation of financial statements. In addition, projections of any evaluation of effectiveness of internal control over financial reporting to future periods are subject to the risk that the control may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. If we fail to maintain the adequacy of our internal controls, including any failure to implement required new or improved controls, this could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
There is a limited carveout offered by the SEC staff in its published Frequently Asked Questions on Management's Report on Internal Control Over Financial Reporting and Certification of Disclosure in Exchange Act Periodic Reports (revised September 24, 2007) which allows an acquired business to be excluded from a company's assessment of its internal controls in
49
Table of Contents
circumstances where it is not possible to conduct an assessment of the acquired business's internal controls and less than a year has passed since an acquisition. Management did not include Meda in its evaluation of the Company's internal control over financial reporting at December 31, 2016 but otherwise concluded that our internal controls were effective as of December 31, 2016. Refer to Management's Report on Internal Control over Financial Reporting included in Item 8 in this Form 10-K. There can be no assurance that our exclusion of internal controls at Meda from our assessment will not be met with negative market reaction and will not have an adverse effect on our ordinary share price.
We intend, to the extent necessary, to take appropriate measures to establish or enhance internal controls at Meda so that we meet the requirements of Section 404 and are in position to include Meda in our annual assessment of the effectiveness of internal controls as of December 31, 2017. However, it is possible that we may experience delays in implementing or be unable to implement necessary internal controls and procedures with respect to Meda. In addition, in connection with the attestation process required of our independent registered public accounting firm pursuant to Section 404, we may encounter problems or delays in completing the implementation of any requested improvements. Accordingly, either we or our independent registered public accounting firm (or both) may conclude that our internal controls are ineffective because of a material weakness in internal controls at Meda, which could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
OUR FUTURE SUCCESS IS HIGHLY DEPENDENT ON OUR CONTINUED ABILITY TO ATTRACT AND RETAIN KEY PERSONNEL. LOSS OF KEY PERSONNEL COULD LEAD TO LOSS OF CUSTOMERS, BUSINESS DISRUPTION, AND A DECLINE IN REVENUES, ADVERSELY AFFECT THE PROGRESS OF PIPELINE PRODUCTS, OR OTHERWISE ADVERSELY AFFECT OUR OPERATIONS.
It is important that we attract and retain qualified personnel in order to develop and commercialize new products, manage our business, and compete effectively. Competition for qualified personnel in the pharmaceutical industry is very intense. If we fail to attract and retain key scientific, technical, commercial, or management personnel, our business could be affected adversely. Additionally, while we have employment agreements with certain key employees in place, their employment for the duration of the agreement is not guaranteed. Current and prospective employees might also experience uncertainty about their future roles with us following the consummation and integration of our recent transactions, including the EPD Transaction and the Meda transaction, and potential future transactions, which might adversely affect our ability to retain key managers and other employees. If we are unsuccessful in retaining our key employees or enforcing certain post-employment contractual provisions such as confidentiality or non-competition, it could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
OUR ACTUAL FINANCIAL POSITION AND RESULTS OF OPERATIONS MAY DIFFER MATERIALLY FROM THE UNAUDITED PRO FORMA FINANCIAL INFORMATION INCLUDED IN THIS ANNUAL REPORT.
The unaudited pro forma financial information contained in the Form 10-K may not be an indication of what our financial position or results of operations would have been had the Meda transaction and the EPD Transaction been completed on the dates indicated nor are they indicative of the future operating results of Mylan N.V. The unaudited pro forma financial information has been derived from the historical consolidated financial statements of Mylan N.V., Mylan Inc., Meda, and the combined financial statements of the EPD Business and reflect certain adjustments related to past operating performance and acquisition accounting adjustments, such as increased amortization expense based on the fair value of assets acquired, the impact of transaction costs, and the related income tax effects. The information upon which these adjustments have been made is subjective, and these types of adjustments are difficult to make with complete accuracy. Accordingly, the actual financial position and results of our operations following the Meda transaction and the EPD Transaction may not be consistent with, or evident from, this unaudited pro forma financial information and other factors may affect our business, financial condition, results of operations, cash flows, and/or ordinary share price, including, among others, those described herein.
THE EPD BUSINESS HAS AN ONGOING RELATIONSHIP WITH ABBOTT AS A BUSINESS PARTNER, INCLUDING WITH RESPECT TO THE MANUFACTURING AND SUPPLY OF CERTAIN PRODUCTS, SHARING OF CERTAIN INTELLECTUAL PROPERTY AND PROVISION OF CERTAIN TRANSITION SERVICES.
Abbott or one of its affiliates is required to manufacture products for the EPD Business, pursuant to certain agreements providing for, among other things, manufacturing and supply services. Disruptions or disagreements related to the third-party manufacturing relationship with Abbott could impair our ability to ship products to the market on a timely basis and could, among other consequences, subject us to exposure to claims from customers.
50
Table of Contents
Mylan has certain obligations to manufacture for and supply products to Abbott. Accordingly, we may need to allocate resources to provide manufacturing capacity to Abbott in lieu of supplying products for the EPD Business, which could have a negative impact on us.
In addition, Abbott or one of its affiliates owns registrations, including marketing authorizations, for certain products of the EPD Business in certain jurisdictions, and disagreements could arise regarding Abbott's or our use of such registrations in the territory allocated to each party.
Prior to the EPD Transaction, Abbott or one of its affiliates performed various corporate functions for the EPD Business, such as accounting, information technology, and finance, among others. Abbott was required to provide some of these functions to the EPD Business for a period of time pursuant to the transition services agreement, which expired with respect to the majority of services at the end of February 2017. Pursuant to a limited extension of the transition services agreement, Abbott continues to be obligated to provide certain transition services in certain specified countries during 2017. The EPD Business may incur temporary interruptions in business operations if it cannot complete the transition effectively from Abbott's existing operational systems and transition services.
The risks related to the relationships between us and Abbott could be exacerbated if Abbott fails to perform under the agreements between Mylan and Abbott or the EPD Business fails to have necessary systems and services in place when the obligations under the agreements between Mylan and Abbott expire, and such risks could have a negative impact on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
OUR BUSINESS RELATIONSHIPS, INCLUDING CUSTOMER RELATIONSHIPS, MAY BE SUBJECT TO DISRUPTION DUE TO OUR RECENT TRANSACTIONS, INCLUDING THE MEDA TRANSACTION AND THE EPD TRANSACTION.
Parties with which we currently do business or may do business in the future, including customers and suppliers, may experience ongoing uncertainty associated with our recent transactions, including the Meda transaction and the EPD Transaction, including with respect to current or future business relationships with us. As a result, our business relationships may be subject to disruptions if customers, suppliers, and others attempt to negotiate changes in existing business relationships or consider entering into business relationships with parties other than us. For example, certain customers and collaborators have contractual consent rights or termination rights that may have been triggered by a change of control or assignment of the rights and obligations of contracts that were transferred in our recent transactions, including the Meda transaction and the EPD Transaction. In addition, our contract manufacturing business could be impaired if existing or potential customers determine not to continue or initiate contract manufacturing relationships with us. These disruptions could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
WE ARE IN THE PROCESS OF ENHANCING AND FURTHER DEVELOPING OUR GLOBAL ERP SYSTEMS AND ASSOCIATED BUSINESS APPLICATIONS, WHICH COULD RESULT IN BUSINESS INTERRUPTIONS IF WE ENCOUNTER DIFFICULTIES.
We are enhancing and further developing our global ERP and other business critical IT infrastructure systems and associated applications to provide more operating efficiencies and effective management of our business and financial operations. Such changes to ERP systems and related software, and other IT infrastructure carry risks such as cost overruns, project delays and business interruptions and delays. If we experience a material business interruption as a result of our ERP enhancements, it could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price. Refer to Management's Report on Internal Control over Financial Reporting included in Item 8 in this Form 10-K.
WE ARE INCREASINGLY DEPENDENT ON INFORMATION TECHNOLOGY AND OUR SYSTEMS AND INFRASTRUCTURE FACE CERTAIN RISKS, INCLUDING CYBERSECURITY AND DATA LEAKAGE RISKS.
Significant disruptions to our information technology systems or breaches of information security could adversely affect our business. We are increasingly dependent on sophisticated information technology systems and infrastructure to operate our business. In the ordinary course of business, we collect, store and transmit large amounts of confidential information (including trade secrets or other intellectual property, proprietary business information and personal information), and it is critical that we do so in a secure manner to maintain the confidentiality and integrity of such confidential information. We also have outsourced significant elements of our operations to third parties, some of which are outside the U.S., including significant elements of our information technology infrastructure, and as a result we are managing many independent vendor relationships with third parties who may or could have access to our confidential information. The size and complexity of our information technology
51
Table of Contents
systems, and those of our third party vendors with whom we contract, make such systems potentially vulnerable to service interruptions. The size and complexity of our and our vendors' systems and the large amounts of confidential information that is present on them also makes them potentially vulnerable to security breaches from inadvertent or intentional actions by our employees, partners or vendors, or from attacks by malicious third parties. We and our vendors could be susceptible to third party attacks on our information technology systems, which attacks are of ever increasing levels of sophistication and are made by groups and individuals with a wide range of motives and expertise, including state and quasi-state actors, criminal groups, "hackers" and others. Maintaining the security, confidentiality and integrity of this confidential information (including trade secrets or other intellectual property, proprietary, business information and personal information) is important to our competitive business position. However, such information can be difficult to protect. While we have taken steps to protect such information and invested heavily in information technology, there can be no assurance that our efforts will prevent service interruptions or security breaches in our systems or the unauthorized or inadvertent wrongful use or disclosure of confidential information that could adversely affect our business operations or result in the loss, misappropriation, and/or unauthorized access, use or disclosure of, or the prevention of access to, confidential information. A breach of our security measures or the accidental loss, inadvertent disclosure, unapproved dissemination, misappropriation or misuse of trade secrets, proprietary information, or other confidential information, whether as a result of theft, hacking, fraud, trickery or other forms of deception, or for any other cause, could enable others to produce competing products, use our proprietary technology or information, and/or adversely affect our business position. Further, any such interruption, security breach, or loss, misappropriation, and/or unauthorized access, use or disclosure of confidential information, including personal information regarding our patients and employees, could result in financial, legal, business, and reputational harm to us and could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
THE EXPANSION OF SOCIAL MEDIA PLATFORMS PRESENT NEW RISKS AND CHALLENGES.
The inappropriate use of certain social media vehicles could cause brand damage or information leakage or could lead to legal implications from the improper collection and/or dissemination of personally identifiable information or the improper dissemination of material non-public information. In addition, negative posts or comments about us on any social networking web site could seriously damage our reputation. Further, the disclosure of non-public company sensitive information through external media channels could lead to information loss as there might not be structured processes in place to secure and protect information. If our non-public sensitive information is disclosed or if our reputation is seriously damaged through social media, it could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or ordinary share price.
ITEM 1B. | Unresolved Staff Comments |
None.
ITEM 2. | Properties |
For information regarding properties, refer to Item 1 "Business" in Part I of this Annual Report.
ITEM 3. | Legal Proceedings |
For information regarding legal proceedings, refer to Note 18 Litigation , in the accompanying Notes to Consolidated Financial Statements in this Annual Report.
52
Table of Contents
PART II
ITEM 5. | Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
On February 27, 2015 , in conjunction with the acquisition of the EPD Business, Mylan Inc. became an indirect wholly owned subsidiary of Mylan N.V. , and Mylan Inc.'s common stock ceased trading on the NASDAQ. Mylan N.V. 's ordinary shares began trading on NASDAQ under the symbol " MYL " on March 2, 2015. On November 4, 2015, Mylan N.V. 's ordinary shares began trading on the Tel Aviv Stock Exchange under the symbol " MYL ."
The following table sets forth the quarterly high and low sales prices for Mylan N.V. 's ordinary shares (Mylan Inc.'s common stock prior to February 27, 2015) for the quarterly periods of 2016 and 2015 :
Year Ended December 31, 2016 | High |
| Low | ||||
Three months ended March 31, 2016 | $ | 54.44 | |
| $ | 40.04 | |
Three months ended June 30, 2016 | 49.42 | |
| 38.01 | | ||
Three months ended September 30, 2016 | 50.40 | |
| 37.65 | | ||
Three months ended December 31, 2016 | 40.50 | |
| 33.60 | | ||
Year Ended December 31, 2015 | High |
| Low | ||||
Three months ended March 31, 2015 | $ | 65.63 | |
| $ | 52.21 | |
Three months ended June 30, 2015 | 76.69 | |
| 57.46 | | ||
Three months ended September 30, 2015 | 73.91 | |
| 39.16 | | ||
Three months ended December 31, 2015 | 55.51 | |
| 37.59 | | ||
As of February 22, 2017 , there were approximately 186,000 holders of Mylan N.V. ordinary shares, including those held in street or nominee name.
The Company did not pay dividends in 2016 or 2015 and does not intend to pay dividends on its ordinary shares in the near future.
ISSUER PURCHASES OF EQUITY SECURITIES
On November 16, 2015, the Company announced that its Board of Directors had approved the repurchase of up to $1 billion of the Company's ordinary shares either in the open market through privately-negotiated transactions or in one or more self tender offers (the "Share Repurchase Program"). At December 31, 2016 , the Share Repurchase Program has approximately $932.5 million remaining for ordinary share repurchases, as the Company did not repurchase any ordinary shares during 2016 . The Share Repurchase Program does not obligate the Company to acquire any particular amount of ordinary shares.
UNREGISTERED SALES OF DEBT SECURITIES
In the past three years, we have issued unregistered securities in connection with the following transactions:
In November 2016, Mylan N.V. issued € 3.0 billion aggregate principal amount of senior unsecured debt securities, comprised of floating rate Senior Notes due 2018, 1.250% Senior Notes due 2020, 2.250% Senior Notes due 2024 and 3.125% Senior Notes due 2028. These notes were issued in a private offering exempt from the registration requirements of the Securities Act of 1933, as amended (the "Securities Act"), to persons outside of the U.S. pursuant to Regulation S under the Securities Act.
In June 2016, Mylan N.V. issued $6.5 billion aggregate principal amount of senior unsecured debt securities, comprised of 2.500% Senior Notes due 2019, 3.150% Senior Notes due 2021, 3.950% Senior Notes due 2026 and 5.250% Senior Notes due 2046. These notes were issued in a private offering exempt from the registration requirements of the Securities Act, to qualified institutional buyers in accordance with Rule 144A and to persons outside of the U.S. pursuant to Regulation S under the Securities Act. In December 2016, Mylan N.V. and Mylan Inc. filed a registration statement with the SEC with respect to an offer to exchange these notes for registered notes with the same aggregate principal amount and terms
53
Table of Contents
substantially identical in all material respects, which was declared effective on January 3, 2017. The exchange offer expired on January 31, 2017 and settled on February 3, 2017.
In December 2015, Mylan N.V. issued $1.0 billion aggregate principal amount of senior unsecured debt securities, comprised of 3.000% Senior Notes due 2018 and 3.750% Senior Notes due 2020. These notes were issued in a private offering exempt from the registration requirements of the Securities Act to qualified institutional buyers in accordance with Rule 144A and to persons outside of the United States pursuant to Regulation S under the Securities Act. In December 2016, Mylan N.V. and Mylan Inc. filed a registration statement with the SEC with respect to an offer to exchange these notes for registered notes with the same aggregate principal amount and terms substantially identical in all material respects, which was declared effective on January 3, 2017. The exchange offer expired on January 31, 2017 and settled on February 3, 2017.
STOCK PERFORMANCE GRAPH
Set forth below is a performance graph comparing the cumulative total return (assuming reinvestment of dividends), in U.S. Dollars, for the calendar years ended December 31, 2012 , 2013 , 2014 , 2015 and 2016 of $100 invested on December 31, 2011 in the Company's ordinary shares, the Standard & Poor's 500 Index and the Dow Jones U.S. Pharmaceuticals Index.
| 12/31/11 |
| 12/31/12 |
| 12/31/13 |
| 12/31/14 |
| 12/31/15 |
| 12/31/16 | ||||||
Mylan N.V. (1) | 100.00 | |
| 127.91 | |
| 202.24 | |
| 262.67 | |
| 251.96 | |
| 177.77 | |
S&P 500 | 100.00 | |
| 116.00 | |
| 153.58 | |
| 174.60 | |
| 177.01 | |
| 198.18 | |
Dow Jones U.S. Pharmaceuticals | 100.00 | |
| 113.90 | |
| 152.54 | |
| 185.19 | |
| 196.69 | |
| 192.41 | |
____________
(1) | Mylan Inc. prior to February 27, 2015 . |
54
Table of Contents
ITEM 6. | Selected Financial Data |
The selected consolidated financial data set forth below should be read in conjunction with "Management's Discussion and Analysis of Results of Operations and Financial Condition" included in Item 7 and the Consolidated Financial Statements and related Notes to Consolidated Financial Statements included in Item 8 in this Form 10-K. The functional currency of the primary economic environment in which the operations of Mylan and its subsidiaries in the U.S. are conducted is the U.S. Dollar. The functional currency of non-U.S. subsidiaries is generally the local currency in the country in which each subsidiary operates.
Mylan N.V. is the successor to Mylan Inc., the information set forth below refers to Mylan Inc. for periods prior to February 27, 2015, and to Mylan N.V. on and after February 27, 2015.
| Year Ended December 31, | ||||||||||||||||||
(In millions, except per share amounts) | 2016 |
| 2015 |
| 2014 |
| 2013 |
| 2012 | ||||||||||
Statements of Operations: |
|
|
|
|
|
|
|
|
| ||||||||||
Total revenues | $ | 11,076.9 | |
| $ | 9,429.3 | |
| $ | 7,719.6 | |
| $ | 6,909.1 | |
| $ | 6,796.1 | |
Cost of sales (1) | 6,379.9 | |
| 5,213.2 | |
| 4,191.6 | |
| 3,868.8 | |
| 3,887.8 | | |||||
Gross profit | 4,697.0 | |
| 4,216.1 | |
| 3,528.0 | |
| 3,040.3 | |
| 2,908.3 | | |||||
Operating expenses: |
|
|
|
|
|
|
|
|
| ||||||||||
Research and development | 826.8 | |
| 671.9 | |
| 581.8 | |
| 507.8 | |
| 401.3 | | |||||
Selling, general and administrative | 2,496.1 | |
| 2,180.7 | |
| 1,625.7 | |
| 1,408.5 | |
| 1,392.4 | | |||||
Litigation settlements and other contingencies, net | 672.5 | |
| (97.4 | ) |
| (32.1 | ) |
| (11.5 | ) |
| 5.2 | | |||||
Total operating expenses | 3,995.4 | |
| 2,755.2 | |
| 2,175.4 | |
| 1,904.8 | |
| 1,798.9 | | |||||
Earnings from operations | 701.6 | |
| 1,460.9 | |
| 1,352.6 | |
| 1,135.5 | |
| 1,109.4 | | |||||
Interest expense | 454.8 | |
| 339.4 | |
| 333.2 | |
| 313.3 | |
| 308.7 | | |||||
Other expense (income), net | 125.1 | |
| 206.1 | |
| 44.9 | |
| 74.9 | |
| (3.5 | ) | |||||
Earnings before income taxes and noncontrolling interest | 121.7 | |
| 915.4 | |
| 974.5 | |
| 747.3 | |
| 804.2 | | |||||
Income tax (benefit) provision | (358.3 | ) |
| 67.7 | |
| 41.4 | |
| 120.8 | |
| 161.2 | | |||||
Net earnings attributable to the noncontrolling interest | - | |
| (0.1 | ) |
| (3.7 | ) |
| (2.8 | ) |
| (2.1 | ) | |||||
Net earnings attributable to Mylan N.V. ordinary shareholders | $ | 480.0 | |
| $ | 847.6 | |
| $ | 929.4 | |
| $ | 623.7 | |
| $ | 640.9 | |
Earnings per ordinary share attributable to Mylan N.V. ordinary shareholders: |
|
|
|
|
|
|
|
|
| ||||||||||
Basic | $ | 0.94 | |
| $ | 1.80 | |
| $ | 2.49 | |
| $ | 1.63 | |
| $ | 1.54 | |
Diluted | $ | 0.92 | |
| $ | 1.70 | |
| $ | 2.34 | |
| $ | 1.58 | |
| $ | 1.52 | |
Weighted average ordinary shares outstanding: |
|
|
|
|
|
|
|
|
| ||||||||||
Basic | 513.0 | |
| 472.2 | |
| 373.7 | |
| 383.3 | |
| 415.2 | | |||||
Diluted | 520.5 | |
| 497.4 | |
| 398.0 | |
| 394.5 | |
| 420.2 | | |||||
Selected Balance Sheet data: |
|
|
|
|
|
|
|
|
| ||||||||||
Total assets (2) (3) | $ | 34,726.2 | |
| $ | 22,267.7 | |
| $ | 15,820.5 | |
| $ | 15,086.6 | |
| $ | 11,847.8 | |
Working capital (2) (3) (4) | 2,481.8 | |
| 2,350.5 | |
| 1,137.2 | |
| 1,258.6 | |
| 1,485.4 | | |||||
Short-term borrowings | 46.4 | |
| 1.3 | |
| 330.7 | |
| 439.8 | |
| 299.0 | | |||||
Long-term debt, including current portion of long-term debt (2) | 15,426.2 | |
| 7,294.3 | |
| 8,104.1 | |
| 7,543.8 | |
| 5,395.6 | | |||||
Total equity | 11,117.6 | |
| 9,765.8 | |
| 3,276.0 | |
| 2,959.9 | |
| 3,355.8 | | |||||
55
Table of Contents
____________
(1) | Cost of sales includes the following amounts primarily related to the amortization of purchased intangibles from acquisitions : $1.32 billion , $854.2 million , $375.9 million , $351.1 million and $349.5 million for the years ended December 31, 2016 , 2015 , 2014 , 2013 and 2012 , respectively. In addition, cost of sales included the following amounts related to impairment charges to intangible assets: $68.3 million , $31.3 million , $27.7 million , $18.0 million and $41.6 million for the years ended December 31, 2016 , 2015 , 2014 , 2013 and 2012 , respectively. |
(2) | Pursuant to the Company's early adoption of ASU 2015-03, Interest - Imputation of Interest , as of December 31, 2015, deferred financing fees related to term debt have been retrospectively reclassified from other assets to long-term debt or the current portion of long-term debt, depending on the debt instrument, on the Consolidated Balance Sheets for all periods presented. The Company retrospectively reclassified approximately $34.4 million , $42.7 million and $36.3 million for the years ended December 31, 2014 , 2013 and 2012 , respectively. |
(3) | Pursuant to the Company's early adoption of ASU 2015-17, Balance Sheet Classification of Deferred Taxes , deferred tax assets and liabilities that had been previously classified as current have been retrospectively reclassified to noncurrent on the Consolidated Balance Sheets for all periods presented. The reclassification resulted in a decrease in current assets of approximately $345.7 million , $250.1 million and $229.3 million for the years ended December 31, 2014 , 2013 and 2012 , respectively. The reclassification resulted in a decrease in current liabilities of approximately $0.2 million , $1.5 million and $1.3 million for the years ended December 31, 2014 , 2013 and 2012 , respectively. |
(4) | Working capital is calculated as current assets minus current liabilities. |
56
Table of Contents
ITEM 7. | Management's Discussion and Analysis of Financial Condition And Results of Operations |
The following discussion and analysis addresses material changes in the financial condition and results of operations of Mylan N.V. and subsidiaries for the periods presented. Unless context requires otherwise, the "Company," "Mylan," "our," or "we" refer to Mylan N.V. and its subsidiaries. This discussion and analysis should be read in conjunction with the Consolidated Financial Statements , the related Notes to Consolidated Financial Statements and our other Securities and Exchange Commission (the "SEC") filings and public disclosures.
This Form 10-K contains "forward-looking statements." These statements are made pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995. Such forward-looking statements may include, without limitation, statements about the acquisition of Meda AB (publ.) ("Meda") by Mylan (the "Meda Transaction"), Mylan's acquisition (the "EPD Transaction") of Mylan Inc. and Abbott Laboratories' non-U.S. developed markets specialty and branded generics business (the "EPD Business"), the potential benefits and synergies of the EPD Transaction and the Meda Transaction, future opportunities for Mylan and products, and any other statements regarding Mylan's future operations, anticipated business levels, future earnings, planned activities, anticipated growth, market opportunities, strategies, competition, and other expectations and targets for future periods. These may often be identified by the use of words such as "will," "may," "could," "should," "would," "project," "believe," "anticipate," "expect," "plan," "estimate," "forecast," "potential," "intend," "continue," "target" and variations of these words or comparable words. Because forward-looking statements inherently involve risks and uncertainties, actual future results may differ materially from those expressed or implied by such forward-looking statements. Factors that could cause or contribute to such differences include, but are not limited to: the ability to meet expectations regarding the accounting and tax treatments of the EPD Transaction and the Meda Transaction; changes in relevant tax and other laws, including but not limited to changes in the U.S. tax code and healthcare and pharmaceutical laws and regulations in the U.S. and abroad; actions and decisions of healthcare and pharmaceutical regulators; the integration of the EPD Business and Meda being more difficult, time-consuming, or costly than expected; operating costs, customer loss, and business disruption (including, without limitation, difficulties in maintaining relationships with employees, customers, clients, or suppliers) being greater than expected following the EPD Transaction and the Meda Transaction; the retention of certain key employees of the EPD Business and Meda being difficult; the possibility that Mylan may be unable to achieve expected synergies and operating efficiencies in connection with the EPD Transaction, the Meda Transaction, and the December 2016 announced restructuring program in certain locations, within the expected time-frames or at all and to successfully integrate the EPD Business and Meda; with respect to the Medicaid Drug Rebate Program Settlement (as defined below), the inability or unwillingness on the part of any of the parties to agree to a final settlement, any legal or regulatory challenges to the settlement, and any failure by third parties to comply with their contractual obligations; expected or targeted future financial and operating performance and results; the capacity to bring new products to market, including but not limited to where Mylan uses its business judgment and decides to manufacture, market, and/or sell products, directly or through third parties, notwithstanding the fact that allegations of patent infringement(s) have not been finally resolved by the courts (i.e., an "at-risk launch"); any regulatory, legal, or other impediments to Mylan's ability to bring new products to market; success of clinical trials and Mylan's ability to execute on new product opportunities; any changes in or difficulties with our inventory of, and our ability to manufacture and distribute, the EpiPen® Auto-Injector and EpiPen Jr® Auto-Injector (collectively, "EpiPen® Auto-Injector") to meet anticipated demand; the potential impact of any change in patient access to the EpiPen® Auto-Injector and the introduction of a generic version of the EpiPen® Auto-Injector; the scope, timing, and outcome of any ongoing legal proceedings, including government investigations, and the impact of any such proceedings on financial condition, results of operations, and/or cash flows; the ability to protect intellectual property and preserve intellectual property rights; the effect of any changes in customer and supplier relationships and customer purchasing patterns; the ability to attract and retain key personnel; changes in third-party relationships; the impact of competition; changes in the economic and financial conditions of the businesses of Mylan; the inherent challenges, risks, and costs in identifying, acquiring, and integrating complementary or strategic acquisitions of other companies, products, or assets and in achieving anticipated synergies; uncertainties and matters beyond the control of management; and inherent uncertainties involved in the estimates and judgments used in the preparation of financial statements, and the providing of estimates of financial measures, in accordance with accounting principles generally accepted in the United States of America ("U.S. GAAP") and related standards or on an adjusted basis. For more detailed information on the risks and uncertainties associated with Mylan's business activities, see the risks described in this Annual Report on Form 10-K for the year ended December 31, 2016 and our other filings with the SEC. You can access Mylan's filings with the SEC through the SEC website at www.sec.gov, and Mylan strongly encourages you to do so. Mylan undertakes no obligation to update any statements herein for revisions or changes after the filing date of this Form 10-K.
Executive Overview
Mylan is a leading global pharmaceutical company, which develops, licenses, manufactures, markets and distributes generic, brand name and over-the-counter ("OTC") products in a variety of dosage forms and therapeutic categories. Mylan is committed to setting new standards in healthcare by creating better health for a better world, and our mission is to provide the
57
Table of Contents
world's 7 billion people access to high quality medicine. To do so, we innovate to satisfy unmet needs; make reliability and service excellence a habit; do what's right, not what's easy; and impact the future through passionate global leadership.
Mylan offers one of the industry's broadest product portfolios, including approximately 7,500 marketed products around the world, to customers in more than 165 countries and territories. We operate a global, high quality vertically-integrated manufacturing platform around the world and one of the world's largest active pharmaceutical ingredient ("API") operations. We also operate a strong research and development ("R&D") network that has consistently delivered a robust product pipeline.
As a result of our acquisition of Meda on August 5, 2016 and the integration of our portfolio across our branded, generics and OTC platforms in all of our regions, effective October 1, 2016, we expanded our reportable segments. We are reporting our results in three segments on a geographic basis as follows: North America, Europe and Rest of World. Our North America segment is made up of our operations in the U.S. and Canada and includes the results of our previously reported Specialty segment. Our Europe segment is made up of our operations in 35 countries within the region. Our Rest of World segment is primarily made up of our operations in India, Australia, Japan and New Zealand. Also included in our Rest of World segment are our operations in emerging markets, which includes countries in Africa (including South Africa), as well as Brazil and other countries throughout Asia and the Middle East. Our API business is conducted through Mylan Laboratories Limited (" Mylan India "), which is included within our Rest of World segment. This change in segment reporting is reflected in our Consolidated Financial Statements for the year ended December 31, 2016 in Item 8 included in this Form 10-K. Comparative segment financial information has been recast for prior periods to conform to this revised segment reporting. We also report in Corporate/Other certain R&D expenses, general and administrative expenses, litigation settlements and other contingencies, net, amortization of intangible assets and certain purchase accounting items, impairment charges, if any, and other items not directly attributable to the segments.
Since 2015, the Company has made a number of significant acquisitions, and as part of the holistic, global integration of these acquisitions, the Company is focused on how to best optimize and maximize all of its assets across the organization and across all geographies. On December 5, 2016, the Company announced restructuring programs in certain locations representing initial steps in a series of actions that are anticipated to further streamline our operations globally. The Company continues to develop the details of the cost reduction initiatives, including workforce actions and other potential restructuring activities beyond the programs already announced. A pre-tax charge of $149.7 million was recorded during the year ended December 31, 2016 , the majority of which represents employee severance and other employee related costs. The continued restructuring actions are expected to be implemented through fiscal year 2018. For the restructuring activities that have been approved to date, the Company estimates that it will incur aggregate pre-tax charges ranging between $175.0 million and $225.0 million , inclusive of the 2016 restructuring charge of $149.7 million , the majority of which are employee related costs. In addition, as a result of the restructuring activities that have been approved to date, management believes the potential annual savings will be between approximately $175.0 million and $225.0 million once fully implemented, with the majority of these savings improving operating cash flow. At this time, the expenses related to the additional restructuring activities cannot be reasonably estimated.
Significant Recent Acquisitions and Other Transactions
Meda AB
On February 10, 2016 , the Company issued an offer announcement under the Nasdaq Stockholm's Takeover Rules and the Swedish Takeover Act (collectively, the "Swedish Takeover Rules") setting forth a public offer to the shareholders of Meda AB (publ.) (" Meda ") to acquire all of the outstanding shares of Meda (the " Offer "), with an enterprise value, including the net debt of Meda , of approximately Swedish kronor ("SEK" or "kr") 83.6 billion (based on a SEK/USD exchange rate of 8.4158 ) or $9.9 billion at announcement. On August 2, 2016 , the Company announced that the Offer was accepted by Meda shareholders holding an aggregate of approximately 343 million shares, representing approximately 94% of the total number of outstanding Meda shares, as of July 29, 2016 , and the Company declared the Offer unconditional. On August 5, 2016 , settlement occurred with respect to the Meda shares duly tendered by July 29, 2016 and, as a result, Meda became a controlled subsidiary of the Company.
Subsequent to the August 5, 2016 closing, a compulsory acquisition proceeding was initiated in accordance with the Swedish Companies Act (Sw. aktiebolagslagen (2005:551), to acquire the remaining Meda shares. On November 1, 2016, the Company made an offer to the remaining Meda shareholders to tender all their Meda shares for cash consideration of 161.31kr per Meda share (the "November Offer") to provide such remaining shareholders with an opportunity to sell their shares in Meda to the Company in advance of the automatic acquisition of their shares for cash in connection with the compulsory acquisition proceeding. At the end of November 2016, Mylan completed the acquisition of approximately 19 million Meda
58
Table of Contents
shares duly tendered for aggregate cash consideration of approximately $330.3 million . As of March 1, 2017, Mylan obtained full legal ownership to the remaining Meda shares pursuant to the compulsory acquisition proceeding, and now owns 100% of the total number of outstanding Meda shares. The Meda shareholders whose shares are subject to the compulsory acquisition proceeding, representing approximately 1% of the total number of outstanding Meda shares, will automatically receive cash consideration plus statutory interest for their Meda shares as determined in the compulsory acquisition proceeding.
Renaissance Topicals Business
On June 15, 2016 , the Company completed the acquisition of the non-sterile, topicals-focused business (the " Topicals Business ") of Renaissance Acquisition Holdings, LLC (" Renaissance ") for approximately $1.0 billion in cash at closing, including amounts deposited into escrow for potential contingent payments, subject to customary adjustments. The Topicals Business provides the Company with a complementary portfolio of approximately 25 products, an active pipeline of approximately 25 products, and an established U.S. sales and marketing infrastructure targeting dermatologists. The Topicals Business also provides an integrated manufacturing and development platform. The U.S. GAAP purchase price was $972.7 million , which includes estimated contingent consideration of approximately $16 million related to the potential $50 million payment contingent on the achievement of certain 2016 financial targets. The $50 million contingent payment remains in escrow.
Momenta Collaboration
On January 8, 2016, the Company entered into an agreement with Momenta Pharmaceuticals, Inc. (" Momenta ") to develop, manufacture and commercialize up to six of Momenta 's current biosimilar candidates, including Momenta 's biosimilar candidate, ORENCIA® (abatacept). Mylan paid an up-front cash payment of $45 million to Momenta . Under the terms of the agreement, Momenta is eligible to receive additional contingent milestone payments of up to $200 million . The Company and Momenta are jointly responsible for product development and equally share in the costs and profits of the products. Under the agreement, Mylan is leading the worldwide commercialization efforts. On November 2, 2016, the Company and Momenta announced that dosing had begun in a Phase 1 study to compare the pharmacokinetics, safety and immunogenicity of M834, a proposed biosimilar of ORENCIA® (abatacept), to U.S. and European Union sourced ORENCIA® in normal healthy volunteers. Under the agreement, Mylan paid $60 million related to certain milestones in 2016.
Refer to Note 3 Acquisitions and Other Transactions in Item 8 in this Form 10-K for additional information regarding significant recent events, including other acquisitions and transactions.
Senior Credit Facilities and Issuance of Senior Notes
On November 22, 2016, the Company entered into a revolving credit agreement (the "2016 Senior Revolving Credit Agreement") among the Company, as borrower, Mylan Inc., as a guarantor ("the Guarantor"), certain lenders and issuing banks and Bank of America, N.A. as the administrative agent (in such capacity, the "Revolving Administrative Agent"). The 2016 Senior Revolving Credit Agreement contains a revolving credit facility (the "2016 Senior Revolving Facility") under which the Company may obtain extensions of credit in an aggregate principal amount not to exceed $2.0 billion , subject to the satisfaction of customary conditions, in U.S. Dollars or alternative currencies including Euro, Sterling, Yen and any other currency that is approved by the Revolving Administrative Agent and each lender under the 2016 Senior Revolving Facility. The 2016 Senior Revolving Facility includes a $200 million subfacility for the issuance of letters of credit and a $175 million sublimit for swingline borrowings. In conjunction with the effectiveness of the 2016 Senior Revolving Credit Agreement in the fourth quarter of 2016, the Revolving Credit Agreement, dated as of December 19, 2014 (the "2014 Revolving Credit Agreement"), among the Company, as guarantor, Mylan Inc., as borrower, the lenders and issuing banks from time to time party thereto, and Bank of America, N.A., as administrative agent, was terminated.
On November 22, 2016, the Company entered into a term loan credit agreement (the "2016 Senior Term Credit Agreement") among the Company, as borrower, the Guarantor, as a guarantor, certain lenders and Goldman Sachs Bank USA, as administrative agent (in such capacity, the "Term Administrative Agent") pursuant to which the Company borrowed $2.0 billion in term loans denominated in U.S. Dollars (the "2016 Term Loans"). The effectiveness of the 2016 Senior Term Credit Agreement was concurrent with, and contingent upon, the termination of (i) the term credit agreement entered into on December 19, 2014 (as amended, restated, supplemented or otherwise modified from time to time, the "2014 Term Credit Agreement"), among the Company, as guarantor, Mylan Inc., as borrower, certain lenders and Bank of America, N.A., as the administrative agent , and (ii) the term credit agreement entered into on July 15, 2015 (as amended, restated, supplemented or otherwise modified from time to time, the "2015 Term Credit Agreement"), among the Company, as guarantor, Mylan Inc., as borrower, certain lenders and PNC Bank, National Association, as the administrative agent.
59
Table of Contents
On November 22, 2016, the Company completed its offering of €500 million aggregate principal amount of the Company's Floating Rate Senior Notes due 2018 (the "Floating Rate Euro Notes"), €750 million aggregate principal amount of the Company's 1.250% Senior Notes due 2020 (the "2020 Euro Notes"), €1.0 billion aggregate principal amount of the Company's 2.250% Senior Notes due 2024 (the "2024 Euro Notes") and €750 million aggregate principal amount of the Company's 3.125% Senior Notes due 2028 (the "2028 Euro Notes," together with the 2020 Euro Notes and the 2024 Euro Notes, the "Fixed Rate Euro Notes"), issued to persons outside the U.S. pursuant to Regulation S under the Securities Act and pursuant to the indenture dated November 22, 2016 (the "Euro Notes Indenture"), among the Company, the Guarantor and Citibank, N.A., London Branch, as trustee, paying agent, transfer agent, registrar and calculation agent. The Floating Rate Euro Notes and the Fixed Rate Euro Notes, together, are referred to as the "Euro Notes."
During 2016, in anticipation of the completion of the Offer, Mylan N.V. issued $1.00 billion aggregate principal amount of 2.500% Senior Notes due 2019, $2.25 billion aggregate principal amount of 3.150% Senior Notes due 2021, $2.25 billion aggregate principal amount of 3.950% Senior Notes due 2026 and $1.00 billion aggregate principal amount of 5.250% Senior Notes due 2046 (collectively, the "June 2016 Senior Notes") in a private offering exempt from the registration requirements of the Securities Act of 1933, as amended (the "Securities Act"), to qualified institutional buyers in accordance with Rule 144A and to persons outside of the U.S. pursuant to Regulation S under the Securities Act. The June 2016 Senior Notes were issued pursuant to an indenture, dated as of June 9, 2016, among the Company, the Guarantor, and The Bank of New York Mellon, as trustee. The June 2016 Senior Notes were guaranteed by Mylan Inc. upon issuance. In addition, the Company entered into a registration rights agreement, requiring the Company and Mylan Inc. to use commercially reasonable efforts to file a registration statement with respect to an offer to exchange each series of the June 2016 Senior Notes for new notes with the same aggregate principal amount and terms identical in all material respects. In December 2016, Mylan N.V. and Mylan Inc. filed a registration statement with the SEC with respect to an offer to exchange these notes for registered notes with the same aggregate principal amount and terms substantially identical in all material respects, which was declared effective on January 3, 2017. The exchange offer expired on January 31, 2017 and settled on February 3, 2017.
Refer to Note 8 Debt in Item 8 in this Form 10-K for additional information regarding these items.
Financial Summary
The table below is a summary of the Company's financial results for the year ended December 31, 2016 compared to the prior year period:
| Year Ended December 31, |
|
|
|
| |||||||||
(In millions, except per share amounts) | 2016 |
| 2015 |
| Change |
| % Change | |||||||
Total revenues | $ | 11,076.9 | |
| $ | 9,429.3 | |
| $ | 1,647.6 | |
| 17 | % |
Gross profit | 4,697.0 | |
| 4,216.1 | |
| 480.9 | |
| 11 | % | |||
Earnings from operations | 701.6 | |
| 1,460.9 | |
| (759.3 | ) |
| (52 | )% | |||
Net earnings attributable to Mylan N.V. ordinary shareholders | 480.0 | |
| 847.6 | |
| (367.6 | ) |
| (43 | )% | |||
Diluted earnings per ordinary share attributable to Mylan N.V. ordinary shareholders | $ | 0.92 | |
| $ | 1.70 | |
| $ | (0.78 | ) |
| (46 | )% |
The decrease in earnings from operations for the year ended December 31, 2016 was primarily due to an increase in litigation settlements and other contingencies, net which included a charge of $465 million related to a settlement with the U.S. Department of Justice and other government agencies related to the classification of the EpiPen® Auto-Injector for purposes of the Medicaid Drug Rebate Program (the "Medicaid Drug Rebate Program Settlement"), a $165 million charge related to the Modafinil antitrust litigation settlement and a $90 million charge related to the Company's settlement with Strides Arcolab Limited ("Strides Arcolab") regarding substantially all outstanding regulatory, warranty and indemnity claims (the "Strides Settlement") related to the acquisition of Agila Specialties ("Agila"), partially offset by approximately $56 million of net fair value gains recorded to certain acquisition related contingent consideration. Earnings from operations was also impacted by higher amortization expense and operating expenses related to acquisitions completed during 2016 including approximately $107 million related to the impact of the amortization of the fair value adjustment of the acquired Meda inventory.
In addition, diluted earnings per ordinary share attributable to Mylan N.V. ordinary shareholders for the year ended December 31, 2016 compared to the prior year period was also impacted by increased interest expense, losses related to the Company's SEK denominated foreign currency contracts, the write off of financing fees related to the termination of the 2016 Bridge Credit Agreement (defined below) and a higher average share count due to the impact of the ordinary shares issued in
60
Table of Contents
the EPD Transaction and the Meda acquisition. These items were partially offset by the impact of an increase in total revenues and the tax benefit that the Company realized during the year ended December 31, 2016 .
A detailed discussion of the Company's financial results can be found below in the section titled "Results of Operations." As part of this discussion, we also report sales performance using the non-GAAP financial measure of "constant currency" third party net sales and total revenues. This measure provides information on the change in net sales assuming that foreign currency exchange rates had not changed between the prior and current period. The comparisons presented at constant currency rates reflect comparative local currency sales at the prior year's foreign exchange rates. We routinely evaluate our third party net sales performance at constant currency so that sales results can be viewed without the impact of foreign currency exchange rates, thereby facilitating a period-to-period comparison of our operational activities, and believe that this presentation also provides useful information to investors for the same reason. The following table compares third party net sales on an actual and constant currency basis for each reportable segment for the years ended December 31, 2016 , 2015 and 2014 .
More information about other non-GAAP measures used by the Company as part of this discussion, including adjusted third party net sales from Europe, adjusted third party net sales, adjusted total revenues, adjusted cost of sales, adjusted gross margins, adjusted earnings and adjusted EPS are discussed further in this Item 7 under Results of Operations and Results of Operations - Use of Non-GAAP Financial Measures .
Results of Operations
2016 Compared to 2015
| Year Ended December 31, | ||||||||||||||||||||
(In millions) | 2016 |
| 2015 |
| % Change |
| 2016 Currency Impact (1) |
| 2016 Constant Currency Revenues |
| Constant Currency % Change (2) | ||||||||||
Third party net sales |
|
|
|
|
|
|
|
|
|
|
| ||||||||||
North America (3) | $ | 5,629.5 | |
| $ | 5,100.4 | |
| 10 | % |
| $ | 6.9 | |
| $ | 5,636.4 | |
| 11 | % |
Europe (3)(4) | 2,953.8 | |
| 2,205.6 | |
| 34 | % |
| 30.1 | |
| 2,983.9 | |
| 35 | % | ||||
Rest of World (3) | 2,383.8 | |
| 2,056.6 | |
| 16 | % |
| (21.3 | ) |
| 2,362.5 | |
| 15 | % | ||||
Total third party net sales (3)(4) | 10,967.1 | |
| 9,362.6 | |
| 17 | % |
| 15.7 | |
| 10,982.8 | |
| 17 | % | ||||
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||
Other third party revenues | 109.8 | |
| 66.7 | |
| 65 | % |
| 0.8 | |
| 110.6 | |
| 66 | % | ||||
Consolidated total revenues (4) | $ | 11,076.9 | |
| $ | 9,429.3 | |
| 18 | % |
| $ | 16.5 | |
| $ | 11,093.4 | |
| 18 | % |
(1) | Currency impact is shown as unfavorable (favorable). |
(2) | The constant currency percentage change is derived by translating third party net sales or revenues for the current period at prior year comparative period exchange rates, and in doing so shows the percentage change from 2016 constant currency third party net sales or revenues to the corresponding amount in the prior year. |
(3) | Effective October 1, 2016, the Company expanded its reportable segments as follows: North America, Europe and Rest of World. As a result, the amounts previously reported under the Specialty segment have been recast to North America and amounts related to Brazil are included in Rest of World for all periods presented. |
(4) | For the year ended December 31, 2015, adjusted third party net sales in Europe totaled $2.22 billion , adjusted third party net sales totaled $9.38 billion , and adjusted total revenues were $9.45 billion . Adjusted third party net sales in Europe, adjusted third party net sales and adjusted total revenues are non-GAAP financial measures. |
Total Revenues
For the year ended December 31, 2016 , Mylan reported total revenues of $11.08 billion compared to $9.43 billion in the prior year. Total revenues include both net sales and other revenues from third parties. Third party net sales for the current year were $10.97 billion compared to $9.36 billion for the prior year, representing an increase of $1.60 billion , or 17% . Other third party revenues for the current year were $109.8 million compared to $66.7 million in the prior year, an increase of $43.1 million . The increase in other third party revenues was principally the result of an increase in royalty income, including due to current year acquisitions.
61
Table of Contents
The increase in total revenues was the result of third party net sales growth in all segments. Contributing to this increase were net sales from the acquisitions of Meda and the Topicals Business , net sales from new product introductions, and to a lesser extent, the two additional months of net sales from the EPD Business (the "incremental EPD Business sales") when compared to the prior year, all of which when combined totaled approximately $1.70 billion . Net sales on existing products decreased approximately $96 million as a result of a decrease in pricing of approximately $196 million , offset by an increase in volume of approximately $100 million . Mylan 's current year total revenues were unfavorably impacted by the effect of foreign currency translation, primarily reflecting strengthening in the U.S. Dollar as compared to the currencies of Mylan 's subsidiaries in Canada, the European Union, India, the United Kingdom and Brazil, partially offset by the strengthening of the Japanese Yen. The unfavorable impact of foreign currency translation on current year total revenues was approximately $17 million . As such, constant currency total revenues increased approximately $1.7 billion , or 18% .
In arriving at net sales, gross sales are reduced by provisions for estimates, including discounts, rebates, promotions, price adjustments, returns and chargebacks. See the Application of Critical Accounting Policies section in this Item 7 for a discussion of our methodology with respect to such provisions. For 2016 , the most significant amounts charged against gross sales were $4.33 billion related to chargebacks and $3.93 billion related to incentives offered to our customers, such as volume related incentives and promotions. For 2015 , the most significant amounts charged against gross sales were for chargebacks in the amount of $4.21 billion and incentives offered to our customers in the amount of $3.77 billion .
From time to time, a limited number of our products may represent a significant portion of our net sales, gross profit and net earnings. Generally, this is due to the timing of new product launches and the amount, if any, of additional competition in the market. Our top ten products in terms of net sales, in the aggregate, represented approximately 27% and 28% of the Company's third party net sales in 2016 and 2015 , respectively.
Products generally contribute most significantly to revenues and gross margins at the time of their launch, even more so in periods of market exclusivity, or in periods of limited generic competition. As such, the timing of new product introductions can have a significant impact on the Company's financial results. The entrance into the market of additional competition generally has a negative impact on the volume and pricing of the affected products. Additionally, pricing is often affected by factors outside of the Company's control.
Third party net sales are derived from our three geographic reporting segments: North America, Europe and Rest of World. The graph below shows third party net sales by segment for the years ended December 31, 2016 and 2015 and the increase period over period.
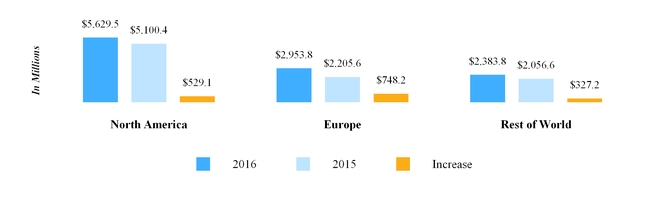
North America Segment
Third party net sales from North America increased $529.1 million , or 10% during the year ended December 31, 2016 when compared to the prior year. The increase was principally due to net sales from the acquisitions of Meda , the Topicals Business and the incremental EPD Business sales, and to a lesser extent, net sales from new product introductions, together totaling approximately $634.4 million . These increases were partially offset by lower volume and pricing on existing products of approximately $98.7 million . As anticipated, the U.S. generics products experienced price erosion in the mid-single digits. The unfavorable impact of foreign currency translation on current year third party net sales was approximately $7 million , or less than 1% within North America. As such, constant currency third party net sales increased by approximately $536 million , or 11% when compared to the prior year.
62
Table of Contents
Sales of the EpiPen® Auto-Injector are primarily included in the North America segment, and on a worldwide basis totaled approximately $1 billion for the years ended December 31, 2016 and 2015. On August 29, 2016, the Company announced its intent to launch the first authorized generic to EpiPen® Auto-Injector which was launched on December 16, 2016. The authorized generic has the same drug formulation and device functionality as the branded product. The Company also continues to market and distribute branded EpiPen® Auto-Injector .
Europe Segment
Third party net sales from Europe increased $748.2 million , or 34% during the year ended December 31, 2016 when compared to the prior year. This increase was primarily the result of net sales from the acquisition of Meda and the incremental EPD Business sales, and to a lesser extent, net sales from new product introductions, together totaling approximately $735.8 million . In addition, higher volumes on existing products were partially offset by lower pricing throughout Europe. Lower pricing is a result of government-imposed pricing reductions and competitive market conditions throughout the segment. The unfavorable impact of foreign currency translation on current year third party net sales was approximately $30 million , or 1% within Europe. As such, constant currency third party net sales increased by approximately $778 million , or 35% when compared to the prior year.
The acquisition of Meda significantly increased our operations and revenues throughout Europe, but particularly in France, Italy, Germany and Sweden. Third party net sales from Mylan 's business in France increased compared to the prior year as a result of net sales from the acquisition of Meda , the incremental EPD Business sales, higher volumes on existing products and new product introductions, partially offset by lower pricing. Our market share in France increased in 2016 as compared to 2015 , and we remain the generics market leader. In Italy, net sales increased compared to the prior year as a result of net sales from the acquisition of Meda , the incremental EPD Business sales and new product introductions, partially offset by lower pricing and lower volume. Sales in France and Italy continue to be negatively impacted by government-imposed pricing reductions and an increasingly competitive market.
Certain markets in Europe in which we do business have undergone government-imposed price reductions, and further government-imposed price reductions are expected in the future. Such measures, along with the tender systems discussed below, are likely to have a negative impact on sales and gross profit in these markets. However, government initiatives in certain markets that appear to favor generic products could help to mitigate this unfavorable effect by increasing rates of generic substitution and penetration.
A number of markets in which we operate in Europe have implemented, or may implement, tender systems for generic pharmaceuticals in an effort to lower prices. Generally speaking, tender systems can have an unfavorable impact on sales and profitability. Under such tender systems, manufacturers submit bids that establish prices for generic pharmaceutical products. Upon winning the tender, the winning company will be awarded an exclusive or semi-exclusive contract to supply the market for a period of time. The tender system often results in companies underbidding one another by proposing low pricing in order to win the tender. The loss of a tender by a third party to whom we supply API can also have a negative impact on our sales and profitability. Sales continue to be negatively affected by the impact of tender systems.
Rest of World Segment
In Rest of World, third party net sales increased $327.2 million , or 16% during the year ended December 31, 2016 when compared to the prior year. This increase was primarily driven by the acquisition of Meda , the incremental EPD Business sales, and to a lesser extent, new product introductions, together totaling approximately $328.7 million . In addition, higher sales volumes in Japan, India, Australia and emerging markets positively contributed to the sales growth in this segment. These increases were partially offset by lower pricing throughout the segment, including the antiretroviral ("ARV") franchise. However, sales within our ARV franchise increased progressively throughout 2016. The favorable impact of foreign currency translation on third party net sales was approximately $21 million , or 1% . As such, constant currency third party net sales increased by approximately $306 million , or 15% .
In addition to third party net sales, the Rest of World segment supplies both finished dosage form ("FDF") generic products and API, primarily from Mylan India, to Mylan subsidiaries in conjunction with the Company's vertical integration strategy. Intercompany product sales related to this strategy were $350.7 million in 2016 , compared to $319.9 million in the prior year. These intercompany sales eliminate within, and therefore are not included in the consolidated third party net sales.
In Japan, third party net sales increased as a result of the incremental EPD Business sales, higher volumes on existing products and net sales from new product introductions. In Australia, third party net sales increased as a result of net sales from new product introductions, the incremental EPD Business sales, and to a lesser extent, the acquisition of Meda and higher
63
Table of Contents
volumes on existing products. As in Europe, both Australia and Japan have undergone government-imposed price reductions which have had, and could continue to have, a negative impact on sales and gross profit in these markets.
As a result of the acquisition of Meda, we have significantly expanded and strengthened our presence in emerging markets including China, Southeast Asia and the Middle East. These markets provide opportunities for future growth and expansion and are complemented by Mylan's historical presence in India, Brazil and certain countries in Africa (including South Africa).
Cost of Sales and Gross Profit
Cost of sales for the year ended December 31, 2016 was $6.38 billion , compared to $5.21 billion in the prior year, corresponding to the increase in sales. Cost of sales was primarily impacted by purchase accounting related amortization of acquired intangible assets, acquisition related costs and restructuring and other special items, which are described further in the section titled Use of Non-GAAP Financial Measures . In addition to the increase in net sales, the increase in cost of sales was also impacted by acquisition related amortization expense of Meda, the Topicals Business and Jai Pharma Limited as well as an additional two months of amortization expense related to the EPD Business as compared to the prior year.
Gross profit for the current year was $4.70 billion and gross margins were 42% . For 2015 , gross profit was $4.22 billion and gross margins were 45% . Gross margins were negatively impacted in the current year by approximately 315 basis points due to increased amortization of intangible assets, inventory step-up and intangible asset impairment charges, including in-process research and development, (collectively, "purchase accounting amortization and other related items.") This negative impact was partially offset by approximately 110 basis points as a result of the positive impact of net sales from new products. Adjusted gross margins were approximately 56% in both 2016 and 2015 . Adjusted gross margins increased approximately 50 basis points and were positively impacted in the current year as a result of net sales from new products and the net impact of acquisitions.
A reconciliation between cost of sales, as reported under U.S. GAAP, and adjusted cost of sales and adjusted gross margin for the periods shown follows:
| Year Ended December 31, | ||||||
(In millions) | 2016 |
| 2015 | ||||
U.S. GAAP cost of sales | $ | 6,379.9 | |
| $ | 5,213.2 | |
Deduct: |
|
|
| ||||
Purchase accounting amortization and other related items | (1,389.3 | ) |
| (885.5 | ) | ||
Restructuring related items | (28.9 | ) |
| (0.2 | ) | ||
Acquisition related and other special items | (97.3 | ) |
| (134.6 | ) | ||
Adjusted cost of sales | $ | 4,864.4 | |
| $ | 4,192.9 | |
|
|
|
| ||||
Adjusted gross profit (a) | $ | 6,212.5 | |
| $ | 5,253.5 | |
|
|
|
| ||||
Adjusted gross margin (a) | 56 | % |
| 56 | % | ||
(a) | Adjusted gross profit is calculated as total revenues (adjusted total revenues for 2015) less adjusted cost of sales. Adjusted gross margin is calculated as adjusted gross profit divided by total revenues (adjusted total revenues for 2015). The reconciliation for 2015 adjusted total revenues can be found under "Use of Non-GAAP Financial Measurements." |
Operating Expenses
Research & Development Expense
R&D expense in 2016 was $826.8 million , compared to $671.9 million in the prior year, an increase of $154.9 million . R&D expense increased primarily due the Company's collaboration agreement with Momenta. As part of the collaboration agreement, the Company made a $45 million upfront payment and incurred approximately $29.2 million of additional R&D expense during 2016. The inclusion of Meda increased R&D expense by approximately $26 million . The remainder of the R&D expense increase was due to the continued development of our respiratory, insulin and biologics programs.
64
Table of Contents
Selling, General & Administrative Expense
Selling, general and administrative expense ("SG&A") for the current year was $2.50 billion , compared to $2.18 billion for the prior year, an increase of $315.4 million . The primary factors contributing to this increase were the additional expense related to the acquisition of Meda and the additional two months of expense from the EPD Business which together increased SG&A by approximately $295.1 million in 2016. In addition, we incurred approximately $113.1 million of restructuring charges which were offset by lower acquisition related costs, including consulting and legal costs, which totaled $106.1 million in 2016 , as compared to $209.4 million in the prior year.
Litigation Settlements and Other Contingencies, Net
During 2016 , the Company recorded a net charge of $672.5 million for litigation settlements and other contingencies, net, compared to a net gain of $97.4 million in the prior year. The following table includes the losses/(gains) recognized in litigation settlements and other contingencies, net during the year ended December 31, 2016:
(In millions) | Loss/(gain) | ||
Medicaid Drug Rebate Program Settlement | $ | 465.0 | |
Modafinil antitrust litigation settlement | 165.0 | | |
Strides Settlement | 90.0 | | |
Respiratory Delivery Platform contingent consideration adjustment | (68.5 | ) | |
Jai Pharma Limited contingent consideration adjustment | 12.6 | | |
Other litigation settlements | 8.4 | | |
Total litigation settlements and other contingencies, net | $ | 672.5 | |
The gain in the prior year was primarily related to the settlement of the Paroxetine CR matter with GlaxoSmithKline for approximately $113 million and the settlement of certain antitrust matters. This gain was partially offset by the settlement of patent infringement matters.
Interest Expense
Interest expense for 2016 totaled $454.8 million , compared to $339.4 million for 2015 . The increase in the current year is primarily due to approximately $132.5 million of interest related to the issuance of the June 2016 Senior Notes and approximately $34.0 million of interest related to borrowings acquired from Meda. Partially offsetting these increases was lower amortization of discounts as a result of the repayment of the Company's Cash Convertible Notes in September 2015 and the maturity and repayment of certain debt instruments in 2016.
Other Expense, Net
Other expense, net was $125.1 million in 2016 , compared to $206.1 million in the prior year. Other expense, net includes losses from equity affiliates, foreign exchange gains and losses and interest and dividend income. Other expense, net was comprised of the following for the years ended December 31, 2016 and 2015 , respectively:
| Year Ended December 31, | ||||||
(In millions) | 2016 |
| 2015 | ||||
Losses from equity affiliates, primarily clean energy investments | $ | 112.8 | |
| $ | 105.0 | |
Write off of deferred financing fees | 34.8 | |
| 54.3 | | ||
Interest income | (12.3 | ) |
| (3.0 | ) | ||
Foreign exchange gains, net | (0.5 | ) |
| (58.0 | ) | ||
Losses from termination of interest rate swaps | - | |
| 71.2 | | ||
Redemption premium on July 2020 Senior Notes | - | |
| 39.4 | | ||
Write off of unamortized premium on July 2020 Senior Notes | - | |
| (9.7 | ) | ||
Other (gains) losses, net | (9.7 | ) |
| 6.9 | | ||
| $ | 125.1 | |
| $ | 206.1 | |
65
Table of Contents
In the current year, foreign exchange gains, net of approximately $0.5 million included approximately $128.6 million of losses related to the Company's SEK non-designated foreign currency contracts that were entered into to economically hedge the foreign currency exposure associated with the expected payment of the Swedish krona-denominated cash portion of the purchase price of the Offer. This loss was offset by foreign exchange gains of approximately $30.5 million related to the mark-to-market impact for the settlement of the November Offer and the remaining obligation on non-tendered Meda shares, foreign exchange gains related to the mark-to-market on the Euro Notes of approximately $32.0 million and additional net gains as a result of the Company's foreign currency exchange risk management program.
Income Tax (Benefit) Provision
For the year ended December 31, 2016 , the Company recognized an income tax benefit of $358.3 million , compared to an income tax provision of $67.7 million in 2015 . During the year ended December 31, 2016 , the Company legally merged its wholly owned subsidiary, Jai Pharma Limited, into Mylan Laboratories Limited, resulting in the recognition of a deferred tax asset of $150 million for the tax deductible goodwill in excess of the book goodwill with a corresponding benefit to income tax provision for the year ended December 31, 2016 . In addition, the effective tax rate for 2016 was also impacted by lower income in the U.S., primarily as a result of litigation charges.
2015 Compared to 2014
| Year Ended December 31, | ||||||||||||||||||||
(In millions) | 2015 |
| 2014 |
| % Change |
| 2015 Currency Impact (1) |
| 2015 Constant Currency Revenues |
| Constant Currency % Change (2) | ||||||||||
Third party net sales |
|
|
|
|
|
|
|
|
|
|
| ||||||||||
North America (3) | $ | 5,100.4 | |
| $ | 4,548.4 | |
| 12 | % |
| $ | 17.0 | |
| $ | 5,117.4 | |
| 13 | % |
Europe (3)(4) | 2,205.6 | |
| 1,476.8 | |
| 49 | % |
| 233.8 | |
| 2,439.4 | |
| 65 | % | ||||
Rest of World (3) | 2,056.6 | |
| 1,621.3 | |
| 27 | % |
| 178.0 | |
| 2,234.6 | |
| 38 | % | ||||
Total third party net sales (3)(4) | 9,362.6 | |
| 7,646.5 | |
| 22 | % |
| 428.8 | |
| 9,791.4 | |
| 28 | % | ||||
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||
Other third party revenues | 66.7 | |
| 73.1 | |
| (9 | )% |
| 1.2 | |
| 67.9 | |
| (7 | )% | ||||
Consolidated total revenues (4) | $ | 9,429.3 | |
| $ | 7,719.6 | |
| 22 | % |
| $ | 430.0 | |
| $ | 9,859.3 | |
| 28 | % |
(1) | Currency impact is shown as unfavorable (favorable). |
(2) | The constant currency percentage change is derived by translating third party net sales or revenues for the current period at prior year comparative period exchange rates, and in doing so shows the percentage change from 2015 constant currency third party net sales or revenues to the corresponding amount in the prior year. |
(3) | Effective October 1, 2016, the Company expanded its reportable segments as follows: North America, Europe and Rest of World. As a result, the amounts previously reported under the Specialty segment have been recast to North America and amounts related to Brazil are included in Rest of World for all periods presented. |
(4) | For the year ended December 31, 2015, adjusted third party net sales in Europe totaled $2.22 billion , adjusted third party net sales totaled $9.38 billion , and adjusted total revenues were $9.45 billion . Adjusted third party net sales in Europe, adjusted third party net sales and adjusted total revenues are non-GAAP financial measures. |
Total Revenues
For the year ended December 31, 2015 , Mylan reported total revenues of $9.43 billion compared to $7.72 billion in 2014 . Total revenues include both net sales and other revenues from third parties. Third party net sales for 2015 were $9.36 billion compared to $7.65 billion for 2014 , representing an increase of $1.72 billion , or 22% . Other third party revenues for 2015 were $66.7 million compared to $73.1 million in 2014 , a decrease of $6.4 million .
The increase in total revenues included third party net sales growth in all segments. Contributing to this increase was net sales from the acquired EPD Business, which totaled approximately $1.47 billion and net sales from new product introductions that totaled approximately $438.1 million in 2015. Mylan 's 2015 revenues were unfavorably impacted by the effect of foreign currency translation, primarily reflecting changes in the U.S. Dollar as compared to the currencies of Mylan 's subsidiaries in the European Union, India, Japan and Australia. The unfavorable impact of foreign currency translation on 2015
66
Table of Contents
total revenues was approximately $430 million , or 6% . As such, constant currency total revenues increased approximately $2.1 billion , or 28% as a result of growth in all segments.
In arriving at net sales, gross sales are reduced by provisions for estimates, including discounts, rebates, promotions, price adjustments, returns and chargebacks. See the Application of Critical Accounting Policies section in this Item 7 for a discussion of our methodology with respect to such provisions. For 2015 , the most significant amounts charged against gross sales were $4.21 billion related to chargebacks and $3.77 billion related to incentives offered to our customers, such as volume related incentives and promotions. For 2014 , the most significant amounts charged against gross revenues were for chargebacks in the amount of $3.72 billion and incentives offered to our customers in the amount of $2.65 billion .
From time to time, a limited number of our products may represent a significant portion of our net sales, gross profit and net earnings. Generally, this is due to the timing of new product launches and the amount, if any, of additional competition in the market. Our top ten products in terms of net sales, in the aggregate, represented approximately 28% and 33% of the Company's third party net sales in 2015 and 2014 , respectively.
Products generally contribute most significantly to revenues and gross margins at the time of their launch, even more so in periods of market exclusivity, or in periods of limited generic competition. As such, the timing of new product introductions can have a significant impact on the Company's financial results. The entrance into the market of additional competition generally has a negative impact on the volume and pricing of the affected products. Additionally, pricing is often affected by factors outside of the Company's control.
Third party net sales are derived from our three geographic reporting segments: North America, Europe and Rest of World. The graph below details third party net sales by segment for the years ended December 31, 2015 and 2014 and the increase period over period.
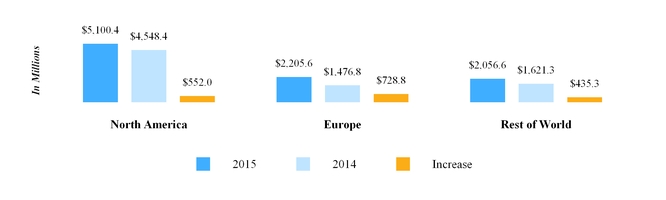
North America Segment
Third party net sales from North America increased $552.0 million , or 12% during the year ended December 31, 2015 when compared to 2014 . The increase in 2015 third party net sales was principally due to net sales from new product introductions, and to a lesser extent, net sales from the acquired EPD Business, together totaling approximately $469 million in 2015 . In addition, there was higher volumes on existing products, which was partially offset by lower pricing. In 2015, the EpiPen® Auto-Injector contributed $1 billion in annual sales for the second year in a row. The effect of foreign currency translation was insignificant within North America.
Europe Segment
Third party net sales from Europe increased $728.8 million , or 49% during the year ended December 31, 2015 when compared to 2014 . This increase was the result of net sales from the acquired EPD Business, and to a lesser extent, net sales from new product introductions, together totaling approximately $977 million in 2015 . In addition, higher volumes on existing products, primarily in France and Italy, partially offset lower pricing throughout Europe as a result of government-imposed pricing reductions and competitive market conditions. The unfavorable impact of foreign currency translation on 2015 third party net sales was approximately $234 million , or 16% within Europe. As such, constant currency third party net sales increased by approximately $963 million , or 65% when compared to 2014 .
67
Table of Contents
Third party net sales from Mylan 's businesses in France and Italy increased in 2015 as compared to 2014 as a result of net sales from the acquired EPD Business, higher volumes on existing products and new products, partially offset by lower pricing. Sales in France continued to be negatively impacted by government-imposed pricing reductions and an increasingly competitive market. Our market share in France, excluding the impact of the acquired EPD Business, remained relatively stable in 2015 as compared to 2014 , and we remained the generics market leader. In Italy, third party net sales increased in 2015 as a result of the impact of the acquired EPD Business. In addition, sales increased in 2015 as a result of higher volumes on existing products, which were partially offset by lower pricing.
In addition to France and Italy, certain other markets in which we do business, including Spain, have undergone government-imposed price reductions, and further government-imposed price reductions are expected in the future. Such measures, along with the tender systems discussed below, are likely to have a negative impact on revenues and gross profit in these markets. However, government initiatives in certain markets that appear to favor generic products could help to mitigate this unfavorable effect by increasing rates of generic substitution and penetration.
A number of markets in which we operate in Europe have implemented or may implement tender systems for generic pharmaceuticals in an effort to lower prices. Generally speaking, tender systems can have an unfavorable impact on revenue and profitability. Under such tender systems, manufacturers submit bids that establish prices for generic pharmaceutical products. Upon winning the tender, the winning company will receive a preferential reimbursement for a period of time. The tender system often results in companies underbidding one another by proposing low pricing in order to win the tender. Additionally, the loss of a tender by a third party to whom we supply API can also have a negative impact on our sales and profitability. Sales continue to be negatively affected by the impact of tender systems.
Rest of World Segment
In Rest of World, third party net sales increased $435.3 million , or 27% during the year ended December 31, 2015 when compared to the prior year. The increase was primarily driven by net sales from the acquired EPD Business, and to a lesser extent, net sales from new product introductions, mainly in Australia and Japan, together totaling approximately $439 million in 2015 . In addition, this increase was driven by higher third party net sales from our operations in India as a result of strong growth in the ARV franchise, partially offset by lower pricing throughout this segment. The unfavorable impact of foreign currency translation on third party net sales was approximately $178 million , or 11% . As such, constant currency third party net sales increased by approximately $613 million , or 38% .
In addition to third party net sales, the Rest of World segment supplied both FDF generic products and API, primarily from Mylan India, to Mylan subsidiaries in conjunction with the Company's vertical integration strategy. Rest of World segment intercompany product sales related to this strategy were $319.9 million in 2015 , compared to $320.3 million in 2014 . These intercompany sales eliminate within, and therefore are not included in consolidated third party net sales.
In Japan, third party net sales increased as a result of net sales from the acquired EPD Business, and to a lesser extent, new products, partially offset by a decline in volume on existing products. Pricing was essentially flat in 2015 when compared to 2014 . In Australia, third party net sales increased in 2015 versus 2014 as a result of net sales from the acquired EPD Business, new products and higher volumes, partially offset by decreases in pricing as a result of significant government-imposed pricing reform. As in Europe, both Australia and Japan have undergone government-imposed price reductions which have had a negative impact on sales and gross profit in these markets.
Cost of Sales and Gross Profit
Cost of sales for 2015 was $5.21 billion , compared to $4.19 billion in 2014 . Cost of sales in 2015 was impacted by purchase accounting related amortization of acquired intangible assets of approximately $885.5 million , acquisition related costs and restructuring and other special items of approximately $134.6 million as described further in the section titled Use of Non-GAAP Financial Measures . Cost of sales for 2014 included similar purchase accounting and restructuring and other special items in the amount of $403.6 million and $109.7 million , respectively. The increase in 2015 of purchase accounting related items was principally the result of the acquired EPD Business. Excluding these amounts, adjusted cost of sales increased in 2015 to $4.19 billion from $3.67 billion in 2014, corresponding to the increase in sales.
Gross profit for 2015 was $4.22 billion and gross margins were 45% . For 2014 , gross profit was $3.53 billion and gross margins were 46% . Gross margins were negatively impacted in 2015 versus 2014 by increased purchase accounting amortization and other related items as a result of acquiring the EPD Business, partially offset by the introduction of new products. Excluding the purchase accounting, restructuring, related amortization and other special items discussed in the paragraph above, adjusted gross margins were approximately 56% and 52% in 2015 and 2014 , respectively. Adjusted gross
68
Table of Contents
margins were positively impacted in 2015 by net sales from the acquired EPD Business by approximately 200 basis points, new product introductions and increased margins on existing products by approximately 100 basis points.
A reconciliation between cost of sales, as reported under U.S. GAAP, and adjusted cost of sales and adjusted gross margin for the periods shown follows:
| Year Ended December 31, | ||||||
(In millions) | 2015 |
| 2014 | ||||
U.S. GAAP cost of sales | $ | 5,213.2 | |
| $ | 4,191.6 | |
Deduct: |
|
|
| ||||
Purchase accounting amortization and other related items | (885.5 | ) |
| (403.6 | ) | ||
Restructuring related items | (0.2 | ) |
| (4.0 | ) | ||
Acquisition related and other special items | (134.6 | ) |
| (109.7 | ) | ||
Adjusted cost of sales | $ | 4,192.9 | |
| $ | 3,674.3 | |
|
|
|
| ||||
Adjusted gross profit (a) | $ | 5,253.5 | |
| $ | 4,045.3 | |
|
|
|
| ||||
Adjusted gross margin (a) | 56 | % |
| 52 | % | ||
(a) | Adjusted gross profit is calculated as total revenues (adjusted total revenues for 2015) less adjusted cost of sales. Adjusted gross margin is calculated as adjusted gross profit divided by total revenues (adjusted total revenues for 2015). The reconciliation for 2015 adjusted total revenues can be found under "Use of Non-GAAP Financial Measurements." |
Operating Expenses
Research & Development Expense
R&D expense in 2015 was $671.9 million , compared to $581.8 million in 2014 , an increase of $90.1 million . R&D expense increased in 2015 primarily due to the impact of the acquired EPD Business, which increased R&D expense by approximately $50 million in 2015 . In addition, R&D expense increased due to the continued development of our respiratory, insulin and biologics programs as well as the timing of internal and external product development projects. These increases were partially offset by a decline in up front licensing and milestone payments, which totaled approximately $15 million in 2015 , relating to the Theravance Biopharma agreement, compared to approximately $18 million in 2014 .
Selling, General & Administrative Expense
SG&A for 2015 was $2.18 billion , compared to $1.63 billion for 2014 , an increase of $555.0 million . Factors contributing to the increase in SG&A include the impact of the acquired EPD Business, which increased SG&A by approximately $379.4 million in 2015 and acquisition related costs of approximately $227.4 million in 2015 compared to $65.9 million in 2014 .
Litigation Settlements and Other Contingencies, Net
During 2015 , the Company recorded a net gain of $97.4 million for litigation settlements and other contingencies, net, compared to a net gain of $32.1 million during 2014 . The gain in 2015 was primarily related to the settlement of the Paroxetine CR matter with GlaxoSmithKline for approximately $113 million and the settlement of certain antitrust matters. This gain was partially offset by the settlement of patent infringement matters. During 2014 , the Company recognized a gain of $80.0 million as a result of an agreement with Strides Arcolab to settle a component of the contingent consideration related to the Agila acquisition. The gain recognized related to the recovery of lost revenues in 2014 arising from supply disruptions that resulted from on-going quality-enhancement activities initiated at certain Agila facilities prior to the Company's acquisition of Agila in 2013. Partially offsetting this gain were charges primarily related to the settlement of a European Commission matter of $21.7 million , the settlement of an intellectual property matter, and to a lesser extent, litigation settlements related to product liability claims.
69
Table of Contents
Interest Expense
Interest expense for 2015 totaled $339.4 million , compared to $333.2 million for 2014 . In 2015 , the Company recorded approximately $56.4 million of commitment and other fees related to the Bridge Credit Agreement, dated April 24, 2015 (as amended on April 29, 2015 and on August 6, 2015, the "Bridge Credit Agreement"), among the Company, as borrower, Mylan Inc., as guarantor, the lenders party thereto from time to time and Goldman Sachs Bank USA, as the administrative agent in connection with the Company's offer to acquire all of the issued and outstanding ordinary shares of Perrigo Company plc. The increase resulting from these fees was partially offset by a lower effective interest rate in 2015 versus 2014 due to the refinancing transactions undertaken late in 2014 and in 2015. Included in interest expense is non-cash interest, primarily made up of the amortization of the discounts and premiums on our convertible debt instruments and senior notes totaling $29.2 million for 2015 and $30.2 million for 2014 . Also included in interest expense is accretion of our contingent consideration liability related to certain acquisitions, which was $38.4 million in 2015 compared to $35.3 million in 2014 .
Other Expense, Net
Other expense, net , was $206.1 million in 2015 , compared to $44.9 million in 2014 . Other expense, net included losses from equity affiliates, foreign exchange gains and losses and interest and dividend income. In 2015 , the Company incurred losses of approximately $71.2 million related to the termination of certain interest rate swaps and charges of approximately $43.2 million related to the write-off of the Bridge Credit Agreement's deferred financing fees. Other expense, net also included charges of approximately $40.8 million related to the redemption of the Company's 7.875% Senior Notes due 2020 (the " July 2020 Senior Notes "), comprised of the $39.4 million redemption premium and the $11.1 million write-off of deferred financing fees offset by the write-off of the remaining $9.7 million unamortized premium related to the July 2020 Senior Notes . In addition, other expense (income), net includes losses from equity affiliates of approximately $105 million , principally related to the Company's clean energy investments, offset by foreign exchange gains, net of approximately $58.0 million . In 2014 , the Company incurred losses from equity affiliates of approximately $91 million , principally related to the Company's clean energy investments, charges of approximately $33 million related to the redemption of the 6.000% Senior Notes due 2018 and the termination of certain interest rate swaps, partially offset by foreign exchange gains of approximately $78 million .
Income Tax Expense
The Company recorded income tax expense of $67.7 million in 2015 , compared to $41.4 million in 2014 , a increase of $26.3 million . The effective tax rate was 7.4% and 4.2% for the years ended December 31, 2015 and 2014, respectively. During 2014, the Company received approvals from the relevant Indian regulatory authorities to legally merge its wholly owned subsidiaries, Agila Specialties Private Limited and Onco Therapies Limited, into Mylan Laboratories Limited . The merger resulted in the recognition of a deferred tax asset of approximately $156 million for the tax deductible goodwill in excess of the book goodwill with a corresponding benefit to income tax expense. The effective tax rate for the year ended December 31, 2015 was impacted by the changing mix of income earned in jurisdictions with differing tax rates, an increase in tax credits as a result of additional investments in facilities whose production is eligible for tax credits under Section 45 of the Code, increases in valuation allowances for net operating losses in foreign jurisdictions, lower net foreign tax credit benefits and lower uncertain tax positions in 2015 .
Use of Non-GAAP Financial Measures
Whenever the Company uses non-GAAP financial measures, we provide a reconciliation of the non-GAAP financial measures to their most directly comparable U.S. GAAP financial measure. Investors and other readers are encouraged to review the related U.S. GAAP financial measures and the reconciliation of non-GAAP measures to their most directly comparable U.S. GAAP measure and should consider non-GAAP measures only as a supplement to, not as a substitute for or as a superior measure to, measures of financial performance prepared in accordance with U.S. GAAP. Additionally, since these are not measures determined in accordance with U.S. GAAP, non-GAAP financial measures have no standardized meaning across companies, or as prescribed by U.S. GAAP and, therefore, may not be comparable to the calculation of similar measures or measures with the same title used by other companies.
Management uses these measures internally for forecasting, budgeting, measuring its operating performance, and incentive-based awards. In addition, primarily due to acquisitions, we believe that an evaluation of our ongoing operations (and comparisons of our current operations with historical and future operations) would be difficult if the disclosure of our financial results was limited to financial measures prepared only in accordance with U.S. GAAP. We believe that non-GAAP financial measures are useful supplemental information for our investors and when considered together with our U.S. GAAP financial
70
Table of Contents
measures and the reconciliation to the most directly comparable U.S. GAAP financial measure, provide a more complete understanding of the factors and trends affecting our operations. The financial performance of the Company is measured by senior management, in part, using adjusted metrics as described below, along with other performance metrics. Management's annual incentive compensation is derived, in part, based on the adjusted EPS metric.
Adjusted Third Party Net Sales from Europe, Adjusted Third Party Net Sales and Adjusted Total Revenues
| Year Ended December 31, | ||||||||||
(In millions) | 2016 |
| 2015 |
| 2014 | ||||||
U.S. GAAP third party net sales from Europe | $ | 2,953.8 | |
| $ | 2,205.6 | |
| $ | 1,476.8 | |
Add: |
|
|
|
|
| ||||||
Acquisition related customer incentive | - | |
| 17.1 | |
| - | | |||
Adjusted third party net sales from Europe | $ | 2,953.8 | |
| $ | 2,222.7 | |
| $ | 1,476.8 | |
U.S. GAAP third party net sales | $ | 10,967.1 | |
| $ | 9,362.6 | |
| $ | 7,646.5 | |
Add: |
|
|
|
|
| ||||||
Acquisition related customer incentive | - | |
| 17.1 | |
| - | | |||
Adjusted third party net sales | $ | 10,967.1 | |
| $ | 9,379.7 | |
| $ | 7,646.5 | |
U.S. GAAP total revenues | $ | 11,076.9 | |
| $ | 9,429.3 | |
| $ | 7,719.6 | |
Add: |
|
|
|
|
| ||||||
Acquisition related customer incentive | - | |
| 17.1 | |
| - | | |||
Adjusted total revenues | $ | 11,076.9 | |
| $ | 9,446.4 | |
| $ | 7,719.6 | |
Adjusted Cost of Sales and Adjusted Gross Margin
We use the non-GAAP financial measure "adjusted cost of sales" and the corresponding non-GAAP financial measure "adjusted gross margin." The principal items excluded from adjusted cost of sales include restructuring, acquisition related and other special items and purchase accounting amortization and other related items, which are described in greater detail above.
Adjusted Earnings and Adjusted EPS
Adjusted net earnings attributable to Mylan N.V. ("adjusted earnings") is a non-GAAP financial measure and provides an alternative view of performance used by management. Management believes that, primarily due to acquisition activity, an evaluation of the Company's ongoing operations (and comparisons of its current operations with historical and future operations) would be difficult if the disclosure of its financial results were limited to financial measures prepared only in accordance with U.S. GAAP. Management believes that adjusted earnings and adjusted earnings per diluted share ("adjusted EPS") are two of the most important internal financial metrics related to the ongoing operating performance of the Company, and are therefore useful to investors and that their understanding of our performance is enhanced by these adjusted measures. Actual internal and forecasted operating results and annual budgets used by management include adjusted earnings and adjusted EPS.
The significant items excluded from adjusted cost of sales, adjusted earnings and adjusted EPS include:
Purchase Accounting Amortization and Other Related Items
The ongoing impact of certain amounts recorded in connection with acquisitions is excluded from adjusted cost of sales, adjusted earnings and adjusted EPS. These amounts include the amortization of intangible assets, inventory step-up and intangible asset impairment charges, including in-process research and development.
71
Table of Contents
Upfront and Milestone-Related R&D Expenses
These expenses and payments are excluded from adjusted earnings and adjusted EPS because they generally occur at irregular intervals and are not indicative of the Company's ongoing operations. Also included in this adjustment are certain expenses related to the Company's collaboration agreement with Momenta including certain milestone related costs. Such costs include payments related to Mylan's future decisions, on a product by product basis, to continue with the development of such product in the collaboration after certain R&D work is performed. Related amounts are excluded from adjusted earnings as Mylan considers such payments as additional upfront buy-in payments for the products.
Accretion of Contingent Consideration Liability and Other Fair Value Adjustments
The impact of changes to the fair value of contingent consideration and accretion expense are excluded from adjusted earnings and adjusted EPS because they are not indicative of the Company's ongoing operations due to the variability of the amounts and the lack of predictability as to occurrence and/or timing and management believes it is helpful to understanding the underlying, ongoing operational performance of the business.
Restructuring, Acquisition Related and Other Special Items
Costs related to restructuring, acquisition and integration activities and other actions are excluded from adjusted cost of sales, adjusted earnings and adjusted EPS, as applicable. These amounts include items such as:
• | Costs related to formal restructuring programs and actions, including costs associated with facilities to be closed or divested, employee separation costs, impairment charges, accelerated depreciation, incremental manufacturing variances, equipment relocation costs and other restructuring related costs; |
• | Certain acquisition related remediation and integration and planning costs, as well as other costs associated with acquisitions such as advisory and legal fees and certain financing related costs, and other business transformation and/or optimization initiatives, which are not part of a formal restructuring program, including employee separation and post-employment costs; |
• | The pre-tax loss of the Company's clean energy investments, whose activities qualify for income tax credits under Section 45 of the U.S. Internal Revenue Code of 1986, as amended (the "Code"); only included in adjusted earnings and adjusted EPS is the net tax effect of the entity's activities; and |
• | Certain costs to further develop and optimize our global enterprise resource planning systems, operations and supply chain. |
The Company has undertaken restructurings and other optimization initiatives of differing types, scope and amount during the covered periods and, therefore, these charges should not be considered non-recurring; however, management excludes these amounts from adjusted earnings and adjusted EPS because it believes it is helpful to understanding the underlying, ongoing operational performance of the business.
Litigation Settlements, Net
Charges and gains related to legal matters, such as those discussed in the Notes to Consolidated Financial Statements - Note 18 Litigation are generally excluded from adjusted earnings and adjusted EPS. Normal, ongoing defense costs of the Company made in the normal course of our business are not excluded.
Reconciliation of Adjusted Earnings and Adjusted EPS
A reconciliation between net earnings attributable to Mylan N.V. ordinary shareholders and diluted earnings per share attributable to Mylan N.V. ordinary shareholders, as reported under U.S. GAAP, and adjusted earnings and adjusted EPS for the periods shown follows:
72
Table of Contents
| Year Ended December 31, | ||||||||||||||||||||||
(In millions, except per share amounts) | 2016 |
| 2015 |
| 2014 | ||||||||||||||||||
U.S. GAAP net earnings attributable to Mylan N.V. and U.S. GAAP diluted EPS | $ | 480.0 | |
| $ | 0.92 | |
| $ | 847.6 | |
| $ | 1.70 | |
| $ | 929.4 | |
| $ | 2.34 | |
Purchase accounting amortization and other related items (primarily included in cost of sales) (a) | 1,412.3 | |
|
|
| 900.9 | |
|
|
| 419.0 | |
|
| |||||||||
Litigation settlements, net (b) | 638.5 | |
|
|
| (97.4 | ) |
|
|
| 47.9 | |
|
| |||||||||
Interest expense (primarily related to clean energy investment financing) | 24.4 | |
|
|
| 45.6 | |
|
|
| 46.0 | |
|
| |||||||||
Accretion of contingent consideration liability and other fair value adjustments (c) | 75.4 | |
|
|
| 38.4 | |
|
|
| 35.3 | |
|
| |||||||||
Clean energy investment pre-tax loss (d) | 92.3 | |
|
|
| 93.2 | |
|
|
| 78.9 | |
|
| |||||||||
Financing related costs (included in other expense, net) | - | |
|
|
| 112.0 | |
|
|
| 33.3 | |
|
| |||||||||
Acquisition related costs (primarily included in SG&A, other expense, net and interest expense) (e) | 335.3 | |
|
|
| 419.8 | |
|
|
| 139.5 | |
|
| |||||||||
Acquisition related customer incentive (included in third party net sales) | - | |
|
|
| 17.1 | |
|
|
| - | |
|
| |||||||||
Restructuring related costs (f) | 149.7 | |
|
|
| 18.7 | |
|
|
| 10.4 | |
|
| |||||||||
Other special items included in: |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Cost of sales | 44.6 | |
|
|
| 36.3 | |
|
|
| 41.1 | |
|
| |||||||||
Research and development expense (g) | 121.3 | |
|
|
| 20.3 | |
|
|
| 17.7 | |
|
| |||||||||
Selling, general and administrative expense | 35.5 | |
|
|
| 47.8 | |
|
|
| 60.7 | |
|
| |||||||||
Other expense, net (h) | (18.5 | ) |
|
|
| 7.2 | |
|
|
| (10.9 | ) |
|
| |||||||||
Tax effect of the above items and other income tax related items | (843.5 | ) |
|
|
| (370.1 | ) |
|
|
| (432.0 | ) |
|
| |||||||||
Adjusted net earnings attributable to Mylan N.V. and adjusted diluted EPS | $ | 2,547.3 | |
| $ | 4.89 | |
| $ | 2,137.4 | |
| $ | 4.30 | |
| $ | 1,416.3 | |
| $ | 3.56 | |
Weighted average diluted ordinary shares outstanding | 520.5 | |
|
|
| 497.4 | |
|
|
| 398.0 | |
|
| |||||||||
Significant items for the year ended December 31, 2016 include the following:
(a) | Includes amortization of the purchase accounting inventory fair value adjustments for Meda and the Topicals Business totaling approximately $121.3 million , and intangible asset impairment charges totaling approximately $68.3 million . |
(b) | Includes $465 million related to the Medicaid Drug Rebate Program Settlement and $165 million settlement related to the Modafinil antitrust litigation. Refer to Note 18 Litigation included in Item 8 in this Form 10-K for further information regarding these settlements. |
(c) | Includes approximately $90 million related to the Strides Settlement and $55.9 million of fair value gains, net recognized on contingent consideration. The remaining amount relates to interest expense for the accretion of contingent consideration liability. |
(d) | Adjustment represents exclusion of the pre-tax loss related to Mylan's clean energy investments and related financing, the activities of which qualify for income tax credits under Section 45 of the Code. The amount is included in other expense, net in the Consolidated Statements of Operations. |
(e) | Acquisition related costs primarily relate to acquisition and integration, including ongoing activities. Included in SG&A is approximately $106.1 million , which primarily related to consulting, professional and legal costs. Included in interest expense, net is approximately $30.3 million of interest expense, net of interest income, which related to the issuance of June 2016 Senior Notes for the period prior to the completion date of the Offer. Such costs included in other expense, net is approximately $128.6 million of losses related to the Company's SEK non-designated foreign currency contracts. Also included in other expense, net is $34.8 million related to 2016 Bridge Credit Agreement. In addition, other acquisition related costs are offset by certain mark-to-market gains, including $30.5 million related to the settlement of the November Offer and the remaining compulsory acquisition proceeding liability. |
(f) | Refer to Note 16 Restructuring included in Item 8 in this Form 10-K. Of the total amount, approximately $28.9 million is included in cost of sales, $7.7 million is included in R&D and $113.1 million is included in SG&A. |
(g) | R&D expense includes a $45 million upfront payment to Momenta and $15 million of milestone payments to Theravance Biopharma. In addition, included in this amount is approximately $29.2 million of R&D expense incurred |
73
Table of Contents
related to the Company's collaboration with Momenta and approximately $32 million of upfront and milestone payments related to several smaller collaboration agreements.
(h) | Includes a $32 million mark-to-market foreign currency gains on the Euro Notes, partially offset by the other foreign currency losses. |
Liquidity and Capital Resources
Our primary source of liquidity is cash provided by operations, which was $2.05 billion for the year ended December 31, 2016 . We believe that cash provided by operating activities and available liquidity will continue to allow us to meet our needs for working capital, capital expenditures and interest and principal payments on debt obligations. Nevertheless, our ability to satisfy our working capital requirements and debt service obligations, or fund planned capital expenditures, will substantially depend upon our future operating performance (which will be affected by prevailing economic conditions), and financial, business and other factors, some of which are beyond our control.
Operating Activities
Net cash provided by operating activities increased by $38.7 million to $2.05 billion for the year ended December 31, 2016 , as compared to $2.01 billion for the year ended December 31, 2015 . Cash provided by operating activities is derived by net earnings adjusted for non-cash operating items, gains and losses attributed to investing and financing activities and changes in operating assets and liabilities resulting from timing differences between the receipts and payments of cash. As a result, changes in cash from operating activities primarily reflect the timing of cash collections from customers, payments to vendors and employees and tax payments in the ordinary course of business.

The net increase in cash provided by operating activities was principally due to the following:
• | net earnings for the year ended December 31, 2016 decreased $367.7 million when compared to the prior year, principally as a result of an increase in non-cash expenses of $884.7 million from the prior year. Significant changes in the current year include the following: |
◦ | increased depreciation and amortization of $491 million as a result of current year acquisitions; |
◦ | higher litigation settlements and other contingencies, net including $465 million related to the Medicaid Drug Rebate Program Settlement and $96.5 million related to the $165 million Modafinil antitrust litigation settlement, of which $68.5 million was paid during 2016; |
◦ | the loss of $128.6 million recognized on acquisition-related foreign currency derivatives associated with the Meda acquisition, the cash impact of which is included in investing activities; |
◦ | other non-cash charges including restructuring charges, the write off of financing fees and accretion and fair value adjustments of contingent consideration; and |
◦ | these amounts were partially offset by an increase in the benefit of deferred tax assets. |
74
Table of Contents
Significant changes in operating assets and liabilities that increased operating cash flow included the following:
• | a net increase in the amount of cash provided by changes in income taxes of $201.7 million as a result of the level and timing of estimated tax payments made during the current year; and |
• | a net decrease of $41.1 million in the amount of cash used through changes in inventory balances. The decrease in cash utilized for inventory in 2016 (as compared to 2015) primarily relates to the timing of product launches and forecasted demand. |
These items were offset by the following:
• | a net increase in the amount of cash used in accounts receivable, including estimated sales allowances, of $197.6 million reflecting increased sales, the timing of cash collections and disbursements related to sales allowances; |
• | a net decrease in the amount of cash provided by changes in trade accounts payable of $44.1 million as a result of the timing of cash disbursements; and |
• | an increase in the amount of cash used in other operating assets and liabilities, net of $479.4 million , principally due to payments of employee benefits and the timing of payments for consulting and transaction costs. |
Investing Activities
Cash used in investing activities was $7.62 billion for the year ended December 31, 2016 , as compared to cash used in investing activities of $1.57 billion for the year ended December 31, 2015 , an increase of $6.05 billion .

In 2016, significant items in investing activities included the following:
• | cash paid for acquisitions, net totaling approximately $6.48 billion related to the Company's acquisitions of Meda and the Topicals Business; |
• | capital expenditures, primarily for equipment and facilities, which totaled $390.4 million . While there can be no assurance that current expectations will be realized, we expect to continue to invest in our future growth and expect capital expenditures for 2017 to be between $400 million and $500 million ; |
• | payments of product rights and other investing activities, net, which totaled $360.2 million . In 2016, the Company paid $57.9 million to acquire a marketed pharmaceutical product and $165 million to acquire certain European intellectual property rights and marketing authorizations which were accrued for at December 31, 2015; |
• | a deferred purchase price payment of $308 million relating to Meda's acquisition of Rottapharm S.p.A paid in the third quarter of 2016 which was assumed as part of the acquisition of Meda; and |
• | a $128.6 million settlement of the Company's non-designated foreign exchange forward and option contracts used to economically hedge the foreign currency exposure associated with the payment of the Swedish krona-denominated cash portion of the purchase price of Meda. |
75
Table of Contents
In 2015, significant items in investing activities included the following:
• | cash paid for acquisitions, net totaling approximately $693.1 million related to the Company's acquisition of Jai Pharma Limited; |
• | payments of product rights and other investing activities, net, which totaled $506.5 million , primarily related to the acquisition of certain commercialization rights in the U.S. and other countries; and |
• | capital expenditures, primarily for equipment and facilities, which totaled $362.9 million . |
Financing Activities
Cash provided by financing activities was $5.34 billion for the year ended December 31, 2016 , as compared to cash provided by financing activities of $604.8 million for the year ended December 31, 2015 , a net increase of $4.74 billion .

In 2016, significant items in financing activities included the following:
• | proceeds from long-term debt which totaled approximately $11.75 billion and included the following: |
◦ | the Company borrowed $2.0 billion in term loans denominated in U.S. Dollars (the "2016 Term Loans"); |
◦ | the Company received proceeds of approximately $3.27 billion related to its offering of €500 million aggregate principal amount of the Floating Rate Euro Notes, €750 million aggregate principal amount of the 2020 Euro Notes, €1.0 billion aggregate principal amount of the 2024 Euro Notes and €750 million aggregate principal amount of the 2028 Euro Notes; and |
◦ | the Company received proceeds of approximately $6.48 billion related to the issuance of $1.00 billion aggregate principal amount of 2.500% Senior Notes due 2019, $2.25 billion aggregate principal amount of 3.150% Senior Notes due 2021, $2.25 billion aggregate principal amount of 3.950% Senior Notes due 2026 and $1.00 billion aggregate principal amount of 5.250% Senior Notes due 2046 (collectively, the "June 2016 Senior Notes"). |
• | payments of long-term debt, which totaled $6.30 billion and included the following: |
◦ | the Company repaid the $1.6 billion aggregate principal amount outstanding under the 2015 Term Credit Agreement and the $800 million aggregate principal amount outstanding under the 2014 Term Credit Agreement in conjunction with the effectiveness of the 2016 Senior Term Credit Agreement; |
◦ | the Company voluntarily prepaid $400 million of the aggregate principal amount of the 2016 Term Loans; |
◦ | the Company paid the principal amount of $500.0 million on the 1.350% Senior Notes due 2016, which matured on November 29, 2016, and the aggregate principal amount of $500 million on the 1.800% Senior Notes due 2016 which matured on June 24, 2016; and |
◦ | the Company repaid approximately $1.8 billion of borrowings under Meda's 25kr billion facility and approximately $567 million of Meda's bank loans. |
• | payment of financing fees totaled approximately $112.6 million . |
76
Table of Contents
In 2015, significant items in financing activities included the following:
• | proceeds from the issuance of long-term debt which totaled approximately $3.54 billion and included the following: |
◦ | the Company received proceeds of approximately $1.0 billion related to the issuance of the December 2015 Senior Notes (as defined in Note 8 Debt ); and |
◦ | the Company received proceeds of approximately $1.6 billion related to borrowings under the 2015 Term Credit Agreement and had borrowings under the 2014 Revolving Credit Agreement totaling approximately $940 million. |
• | payments of long-term debt, which totaled $4.48 billion and included the following: |
◦ | the Company paid $2.54 billion in connection with the maturity of the Cash Convertible Notes on September 15, 2015; |
◦ | the Company paid $1.08 billion in connection with the redemption of the July 2020 Senior Notes; and |
◦ | the Company repaid its borrowings of $940 million under the 2014 Revolving Credit Agreement. |
• | proceeds from the cash convertible note hedge which totaled $1.97 billion . The cash convertible note hedge settled in the third quarter of 2015 in conjunction with the maturity and full redemption of the Cash Convertible Notes; |
• | repayments of short-term borrowings totaling approximately $329.2 million primarily related to repayments under the accounts receivable securitization facility (the "Receivables Facility") of approximately $325 million, net; and |
• | the Company repurchased $67.5 million of ordinary shares. The Company did not repurchase any ordinary shares in 2016, and at December 31, 2016 , the Share Repurchase Program has approximately $932.5 million remaining for ordinary share repurchases. |
Capital Resources
Our cash and cash equivalents at December 31, 2016 total approximately $1.0 billion , with approximately $912.2 million of cash and cash equivalents held at our non-U.S. operations. The majority of these funds represented earnings considered to be permanently reinvested to support the growth strategies of our non-U.S. subsidiaries. The Company anticipates having sufficient U.S. liquidity, including existing borrowing capacity under the 2016 Senior Revolving Facility and the Receivables Facility combined with cash to be generated from operations, to fund foreseeable U.S. cash needs without requiring the repatriation of non-U.S. cash. If these funds are ultimately needed for the Company's operations in the U.S., the Company may be required to accrue and pay U.S. taxes to repatriate these funds. If funds are needed from the Company's subsidiaries that do not have an ultimate U.S. parent, the Company will generally not be required to accrue and pay taxes to repatriate these funds because its foreign parent would not be subject to tax on receipt of these distributions.
The Company has access to $2.0 billion under the 2016 Senior Revolving Facility which also includes a $200 million subfacility for the issuance of letters of credit and a $175 million sublimit for swingline borrowings. At December 31, 2016, the Company had no amounts outstanding on the 2016 Senior Revolving Facility. At December 31, 2016 , we had $11.2 million outstanding under existing letters of credit. Additionally, as of December 31, 2016 , we had $193.6 million available under the $200.0 million subfacility on our 2016 Senior Revolving Facility for the issuance of letters of credit.
In addition to the 2016 Senior Revolving Facility, Mylan Pharmaceuticals Inc. (" MPI "), a wholly owned subsidiary of the Company, has a $400 million Receivables Facility, which will expire in January 2018. Although from time-to-time, the available amount of the Receivables Facility may be less than $400 million based on accounts receivable concentration limits and other eligibility requirements. Under the terms of the Receivables Facility, MPI sells certain accounts receivable to Mylan Securitization LLC, a wholly owned special purpose entity which in turn sells a percentage ownership interest in the receivables to financial institutions and commercial paper conduits sponsored by financial institutions. As of December 31, 2016 , the Company had no amounts outstanding under the Receivables Facility.
At December 31, 2016 our long-term debt totaled $15.20 billion , as compared to $6.30 billion at December 31, 2015. The increase in long-term debt was due to several debt issuances in 2016 to facilitate the acquisition of Meda and the refinancing of certain debt, as well as the long-term debt assumed in the acquisition of Meda. The total long-term debt balance at December 31, 2016 was comprised primarily of $1.60 billion of term loans, $146.4 million of Medium Term Notes acquired from Meda, $13.02 billion of fixed rate senior notes and $526.0 million of floating rate senior notes. In addition, at December 31, 2016, we had $223.3 million of long-term debt classified as current and payable within the next twelve months, as compared to $998.7 million at December 31, 2015. The decrease in current portion of long-term debt was primarily due to the
77
Table of Contents
maturities and repayments of the 1.350% Senior Notes due 2016 and 1.800% Senior Notes due 2016. In addition to the current portion of long-term debt, the Company has significant debt maturities in the second and fourth quarters of 2018, as the 2.600% Senior Notes due 2018 mature in June 2018, the Floating Rate Euro Notes mature in November 2018 and the $3.000% Senior Notes due 2018 mature in December 2018. The Company intends to utilize available liquidity to fund these repayments.
For additional information regarding our debt agreements, refer to Note 8 Debt in Item 8 in this Form 10-K.
Long-term Debt Maturity
Mandatory minimum repayments remaining on the outstanding long-term debt at December 31, 2016 , excluding the discounts and premiums, are as follows for each of the periods ending December 31:
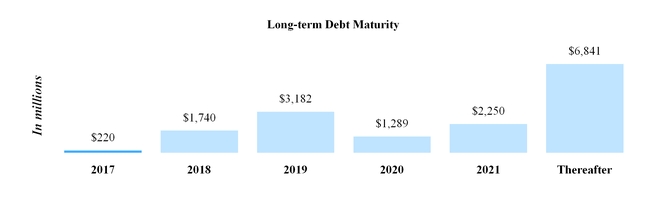
The Company's 2016 Term Loans and 2016 Senior Revolving Facility contain customary affirmative covenants for facilities of this type, including among others, covenants pertaining to the delivery of financial statements, notices of default and certain material events, maintenance of corporate existence and rights, property, and insurance and compliance with laws, as well as customary negative covenants for facilities of this type, including limitations on the incurrence of subsidiary indebtedness, liens, mergers and certain other fundamental changes, investments and loans, acquisitions, transactions with affiliates, payments of dividends and other restricted payments and changes in our lines of business. The 2016 Term Loans and 2016 Senior Revolving Facility contain maximum consolidated leverage ratio financial covenants. We have been compliant with these financial covenants during the year ended December 31, 2016 , and we expect to remain in compliance for the next twelve months.
Other Commitments
We are involved in various legal proceedings that are considered normal to our business. While it is not possible to predict the outcome of such proceedings, an adverse outcome in any of these proceedings could materially affect our financial condition, results of operations and operating cash flow and could cause the market value of our ordinary shares to decline. We have approximately $525 million accrued for such legal contingencies. For certain contingencies assumed in conjunction with the acquisition of the former Merck Generics business, Merck KGaA, the seller, has indemnified Mylan. Strides Arcolab has also agreed to indemnify Mylan for certain contingencies related to our acquisition of Agila . The inability or denial of Merck KGaA, Strides Arcolab , or another indemnitor or insurer to pay on an indemnified claim could have a material adverse effect on our business, financial condition, results of operations, cash flows and/or, our ordinary share price.
We are continuously evaluating the potential acquisition of products, as well as companies, as a strategic part of our future growth. Consequently, we may utilize current cash reserves or incur additional indebtedness to finance any such acquisitions, which could impact future liquidity. In addition, on an ongoing basis, we review our operations including the evaluation of potential divestitures of products and businesses as part of our future strategy. Any divestitures could impact future liquidity.
78
Table of Contents
Contractual Obligations
The following table summarizes our contractual obligations at December 31, 2016 and the effect that such obligations are expected to have on our liquidity and cash flows in future periods:
(In millions) | Total |
| Less than |
| One- Three |
| Three- Five |
| Thereafter | ||||||||||
Long-term debt | $ | 15,522.0 | |
| $ | 220.0 | |
| $ | 4,922.0 | |
| $ | 3,539.0 | |
| $ | 6,841.0 | |
Scheduled interest payments (1) | 4,622.0 | |
| 477.8 | |
| 879.4 | |
| 663.9 | |
| 2,600.9 | | |||||
Operating leases (2) | 295.8 | |
| 295.8 | |
| 114.4 | |
| 57.7 | |
| 48.1 | | |||||
Other Commitments (3) | 2,113.0 | |
| 948.1 | |
| 580.0 | |
| 577.3 | |
| 7.6 | | |||||
| $ | 22,552.8 | |
| $ | 1,721.5 | |
| $ | 6,495.8 | |
| $ | 4,837.9 | |
| $ | 9,497.6 | |
____________
(1) | Scheduled interest payments represent the estimated interest payments related to our outstanding borrowings under term loans, senior notes, the Meda borrowings and other long-term debt. Variable debt interest payments are estimated using current interest rates. |
(2) | We lease certain property under various operating lease arrangements that expire generally over the next five to seven years. These leases generally provide us with the option to renew the lease at the end of the lease term. |
(3) | Other commitments include funding commitments related to the Company's clean energy investments , agreements to purchase third-party manufactured products, open purchase orders and capital leases at December 31, 2016 . |
Due to the uncertainty with respect to the timing of future payments, if any, the following contingent payments have not been included in the table above.
We are contractually obligated to make potential future development, regulatory and commercial milestone, royalty and/or profit sharing payments in conjunction with acquisitions we have entered into with third parties. The most significant of these relates to the potential future consideration related to the respiratory delivery platform. These payments are contingent upon the occurrence of certain future events and, given the nature of these events, it is unclear when, if ever, we may be required to pay such amounts. The amount of the contingent consideration liability was $564.6 million at December 31, 2016 . In addition, the Company expects to incur approximately $30 million to $35 million of non-cash accretion expense related to the increase in the net present value of the contingent consideration liability in 2017.
The fair value measurement of contingent consideration is determined using unobservable inputs based on the Company's own assumptions. Significant unobservable inputs in the valuation include the probability and timing of future development and commercial milestones and future profit sharing payments. For the respiratory delivery platform, Jai Pharma Limited, the Topicals Business and certain other acquisitions, significant unobservable inputs in the valuation include the probability and timing of future development and commercial milestones and future profit sharing payments. When valuing the contingent consideration related to the respiratory delivery platform and Jai Pharma Limited, the value of the obligations are derived from a probability assessment based on expectations of when certain milestones or profit sharing payments occur which are discounted using a market rate of return. At December 31, 2016 and 2015 , discount rates ranging from 0.9% to 9.8% were utilized in such valuations. Significant changes in unobservable inputs could result in material changes to the contingent consideration liability. The Company recorded a $68.5 million reduction in contingent consideration related to the respiratory delivery system and a $12.6 million increase in contingent consideration related to Jai Pharma Limited in 2016.
With respect to the timing of future cash flows associated with our unrecognized tax benefits at December 31, 2016 , we are unable to make reasonably reliable estimates of the period of cash settlement with the respective taxing authority. As such, $190.9 million of unrecognized tax benefits have been excluded from the contractual obligations table above.
We have entered into employment and other agreements with certain executives and other employees that provide for compensation and certain other benefits. These agreements provide for severance payments under certain circumstances. Certain commercial agreements require us to provide performance bonds and/or indemnification; while it is difficult to forecast the amount of payments, if any, to be made over the next few years, we do not believe the amount would be material to our results of operations, cash flows or financial condition.
79
Table of Contents
Collaboration and Licensing Agreements
We periodically enter into collaboration and licensing agreements with other pharmaceutical companies for the development, manufacture, marketing and/or sale of pharmaceutical products. Our significant collaboration agreements are focused on the development, manufacturing, supply and commercialization of multiple, high value generic biologic compounds, insulin analog products and respiratory products. Under these agreements, we have future potential milestone payments and co-development expenses payable to third parties as part of our licensing, development and co-development programs. Payments under these agreements generally become due and are payable upon the satisfaction or achievement of certain developmental, regulatory or commercial milestones or as development expenses are incurred on defined projects. Milestone payment obligations are uncertain, including the prediction of timing and the occurrence of events triggering a future obligation and are not reflected as liabilities in the Consolidated Balance Sheets, except for milestone and royalty obligations reflected as acquisition related contingent consideration. Our potential maximum development milestones not accrued for at December 31, 2016 totaled approximately $596 million . We estimate that the amounts that may be paid in the next twelve months to be approximately $172 million . These agreements may also include potential sales based milestones and call for us to pay a percentage of amounts earned from the sale of the product as a royalty or a profit share. The amounts disclosed do not include sales based milestones or royalty obligations on future sales of product as the timing and amount of future sales levels and costs to produce products subject to these obligations is not reasonably estimable. These sales based milestones or royalty obligations may be significant depending upon the level of commercial sales for each product. A summary of our most significant collaboration and licensing agreements include the following:
On January 8, 2016, the Company entered into an agreement with Momenta to develop, manufacture and commercialize up to six of Momenta 's current biosimilar candidates, including Momenta 's biosimilar candidate, ORENCIA® (abatacept). Mylan paid an up-front cash payment of $45 million to Momenta . Under the terms of the agreement, the Company and Momenta are jointly responsible for product development and equally share in the costs and profits of the products with Mylan leading the worldwide commercialization efforts. Under the terms of the agreement, Momenta is eligible to receive additional contingent milestone payments of up to $200 million .
On November 2, 2016, the Company and Momenta announced that dosing had begun in a Phase 1 study to compare the pharmacokinetics, safety and immunogenicity of M834, a proposed biosimilar of ORENCIA® (abatacept), to U.S. and European Union sourced ORENCIA® in normal healthy volunteers. Under the agreement, Mylan paid $60 million related to certain milestones in 2016.
In accordance with ASC 730, Research and Development and based upon the cost sharing provisions of the agreement, the Company is accounting for the contingent milestone payments related to the Momenta agreement as non-refundable advance payments for services to be used in future R&D activities, which are required to be capitalized until the related services have been performed. More specifically, as costs are incurred within the scope of the collaboration, the Company will record its share of the costs as R&D expense. In addition to the upfront cash payment, during the year ended December 31, 2016 the Company incurred approximately $29.2 million of R&D expense related to this collaboration. To the extent the contingent milestone payments made by the Company exceed the liability incurred, a prepaid asset will be reflected on the Company's Consolidated Balance Sheet. To the extent the contingent milestone payments made by the Company are less than the expense incurred, the difference between the payment and the expense will be recorded as a liability on the Company's Consolidated Balance Sheet. At December 31, 2016, approximately $30.8 million was recorded as a prepaid asset on the Consolidated Balance Sheet.
On January 30, 2015 , the Company entered into a development and commercialization collaboration with Theravance Biopharma, Inc. (" Theravance Biopharma ") for the development and, subject to FDA approval, commercialization of Revefenacin ("TD-4208"), a novel once-daily nebulized long-acting muscarinic antagonist for chronic obstructive pulmonary disease ("COPD") and other respiratory diseases. Under the terms of the agreement, Mylan and Theravance Biopharma are co-developing nebulized TD-4208 for COPD and other respiratory diseases. Theravance Biopharma is leading the U.S. registrational development program and Mylan is responsible for the reimbursement of Theravance Biopharma 's development costs for that program up until the approval of the first new drug application, after which costs will be shared. In addition, Mylan is responsible for commercial manufacturing. In the U.S., Mylan is leading commercialization and Theravance Biopharma retains the right to co-promote the product under a profit-sharing arrangement. On September 14, 2015, Mylan announced the initiation of the Phase 3 program that will support the registrational development program of TD-4208 in the U.S. In addition to funding the U.S. registrational development program, the Company made a $30 million investment in Theravance Biopharma 's common stock during the first quarter of 2015, which is being accounted for as an available-for-sale security. The Company also incurred $15 million in upfront development costs during the year ended December 31, 2015. Under the terms of the agreement, Theravance Biopharma is eligible to receive potential development and sales milestone
80
Table of Contents
payments totaling $220 million in the aggregate. As of December 31, 2016, Mylan has paid a total of $15 million in milestone payments to Theravance Biopharma .
We have entered into exclusive collaborations with Biocon Limited on the development, manufacturing, supply and commercialization of multiple, high value generic biologic compounds and three insulin analog products for the global marketplace. We plan to provide funding related to the collaborations over the next several years. As the timing of cash expenditures is dependent upon a number of factors, many of which are outside of our control, it is difficult to forecast the amount of payments to be made over the next few years, which could be significant.
Impact of Currency Fluctuations and Inflation
Because our results are reported in U.S. Dollars, changes in the rate of exchange between the U.S. Dollar and the local currencies in the markets in which we operate, mainly the Euro, Swedish Krona, Indian Rupee, Japanese Yen, Australian Dollar, Canadian Dollar, Pound Sterling and Brazilian Real affect our results as previously noted. We do not believe that inflation has had a material impact on our revenues or operations in any of the past three years.
Application of Critical Accounting Policies
Our significant accounting policies are described in Note 2 Summary of Significant Accounting Policies in Item 8 in this Form 10-K and are in accordance with U.S. GAAP.
Included within these policies are certain policies which contain critical accounting estimates and, therefore, have been deemed to be "critical accounting policies." Critical accounting estimates are those which require management to make assumptions about matters that were uncertain at the time the estimate was made and for which the use of different estimates, which reasonably could have been used, or changes in the accounting estimates that are reasonably likely to occur from period to period could have a material impact on our financial condition or results of operations. We have identified the following to be our critical accounting policies: the determination of net revenue provisions, acquisitions, intangible assets, goodwill and contingent consideration, income taxes and the impact of existing legal matters.
Net Revenue Provisions
Net revenues are recognized for product sales when title and risk of loss have transferred to the customer and when provisions for estimates, including discounts, sales allowances, price adjustments, returns, chargebacks and other promotional programs are reasonably determinable. Accruals for these provisions are presented in the Consolidated Financial Statements as reductions in determining net revenues and in accounts receivable and other current liabilities. Accounts receivable are presented net of allowances relating to these provisions, which were $2.05 billion and $1.84 billion at December 31, 2016 and 2015 , respectively. Other current liabilities include $809.0 million and $681.8 million at December 31, 2016 and 2015 , respectively, for certain sales allowances and other adjustments that are paid to indirect customers. The following is a rollforward of the most significant provisions for estimated sales allowances during 2016 :
(In millions) | Balance at |
| Current Provision Related to |
| Balances Acquired Through Acquisition (1) |
| Checks/ Credits Issued to Third Parties |
| Effects of Foreign Exchange |
| Balance at | ||||||||
December 31, 2015 |
|
|
|
|
| December 31, 2016 | |||||||||||||
Incentives offered to customers | $ | 1,142.0 | |
| 3,929.7 | |
| 25.1 | |
| (3,866.2 | ) |
| (1.6 | ) |
| $ | 1,229.0 | |
Chargebacks | $ | 584.2 | |
| 4,334.3 | |
| 20.4 | |
| (4,328.3 | ) |
| (0.1 | ) |
| $ | 610.5 | |
Returns | $ | 317.3 | |
| 293.6 | |
| 138.9 | |
| (276.6 | ) |
| (2.5 | ) |
| $ | 470.7 | |
(1) | Principally includes the acquisitions of Meda and the Topicals Business . |
We have not made and do not anticipate making any significant changes to the methodologies that we use to measure sales provisions; however, the balances within these reserves can fluctuate significantly through the consistent application of our methodologies. In the current year, accruals for incentives offered to customers increased as a result of acquisitions and an increase in related sales and overall higher rebate rates, mainly in response to the competitive environment in various markets. Historically, we have not recorded in any current period any material amounts related to adjustments made to prior period reserves.
81
Table of Contents
Provisions for incentives offered to customers, including estimated discounts, sales allowances, promotional and other credits require a lower degree of subjectivity and are less complex in nature, yet, when combined, represent a significant portion of the overall provisions. These provisions are estimated based on historical payment experience, historical relationships to revenues, estimated customer inventory levels and contract terms. Such provisions are determinable due to the limited number of assumptions and consistency of historical experience.
Others, such as chargebacks and returns, require management to make more subjective judgments and evaluate current market conditions. These provisions are discussed in further detail below.
Chargebacks - The provision for chargebacks is the most significant and complex estimate used in the recognition of revenue. Mylan markets products directly to wholesalers, distributors, retail pharmacy chains, mail order pharmacies and group purchasing organizations. We also market products indirectly to independent pharmacies, managed care organizations, hospitals, nursing homes and pharmacy benefit managers, collectively referred to as "indirect customers." Mylan enters into agreements with its indirect customers to establish contract pricing for certain products. The indirect customers then independently select a wholesaler from which to actually purchase the products at these contracted prices. Alternatively, certain wholesalers may enter into agreements with indirect customers that establish contract pricing for certain products, which the wholesalers provide. Under either arrangement, Mylan will provide credit to the wholesaler for any difference between the contracted price with the indirect party and the wholesaler's invoice price. Such credit is called a chargeback, while the difference between the contracted price and the wholesaler's invoice price is referred to as the chargeback rate. The provision for chargebacks is based on expected sell-through levels by our wholesaler customers to indirect customers, as well as estimated wholesaler inventory levels. For the latter, in most cases, inventory levels are obtained directly from certain of our largest wholesalers. Additionally, internal estimates are prepared based upon historical buying patterns and estimated end-user demand. Such information allows us to estimate the potential chargeback that we may ultimately owe to our customers given the quantity of inventory on hand. We continually monitor our provision for chargebacks and evaluate our reserve and estimates as additional information becomes available. A change of 5% in the estimated sell-through levels by our wholesaler customers and in the estimated wholesaler inventory levels would have an effect on our reserve balance of approximately $40 million .
Returns - Consistent with industry practice, we maintain a return policy that allows our customers to return product within a specified period prior to and subsequent to the expiration date. Although application of the policy varies from country to country in accordance with local practices, generally, product may be returned for a period beginning six months prior to its expiration date to up to one year after its expiration date. The majority of our product returns occur as a result of product dating, which falls within the range set by our policy, and are settled through the issuance of a credit to our customer. Although the introduction of additional generic competition does not give our customers the right to return product outside of our established policy, we do recognize that such competition could ultimately lead to increased returns. We analyze this on a case-by-case basis, when significant, and make adjustments to increase our reserve for product returns as necessary. Our estimate of the provision for returns is based upon our historical experience with actual returns, which is applied to the level of sales for the period that corresponds to the period during which our customers may return product. This period is known by us based on the shelf lives of our products at the time of shipment. Additionally, we consider factors such as levels of inventory in the distribution channel, product dating and expiration period, size and maturity of the market prior to a product launch, entrance into the market of additional generic competition, changes in formularies or launch of OTC products, and make adjustments to the provision for returns in the event that it appears that actual product returns may differ from our established reserves. We obtain data with respect to the level of inventory in the channel directly from certain of our largest customers. A change of 5% in the estimated product return rate used in our calculation of our return reserve would have an effect on our reserve balance of approximately $24 million .
Acquisitions, Intangible Assets, Goodwill and Contingent Consideration
We account for acquired businesses using the acquisition method of accounting, which requires that the assets acquired and liabilities assumed be recorded at the date of acquisition at their respective estimated fair values. The cost to acquire businesses has been allocated to the underlying net assets of the acquired businesses based on estimates of their respective fair values. Amounts allocated to acquired IPR&D are capitalized at the date of an acquisition and, at that time, such IPR&D assets have indefinite lives. As products in development are approved for sale, amounts will be allocated to product rights and licenses and will be amortized over their estimated useful lives. Definite-lived intangible assets are amortized over the expected life of the asset. Any excess of the purchase price over the estimated fair values of the net assets acquired is recorded as goodwill.
Purchases of developed products and licenses that are accounted for as an asset acquisition are capitalized as intangible assets and amortized over an estimated useful life. IPR&D assets acquired as part of an asset acquisition are expensed immediately if they have no alternative future uses.
82
Table of Contents
The judgments made in determining the estimated fair value assigned to each class of assets acquired and liabilities assumed, as well as asset lives, can materially impact our results of operations. Fair values and useful lives are determined based on, among other factors, the expected future period of benefit of the asset, the various characteristics of the asset and projected cash flows. Because this process involves management making estimates with respect to future sales volumes, pricing, new product launches, government reform actions, anticipated cost environment and overall market conditions, and because these estimates form the basis for the determination of whether or not an impairment charge should be recorded, these estimates are considered to be critical accounting estimates.
We record contingent consideration resulting from a business acquisition at its estimated fair value on the acquisition date. Each reporting period thereafter, we revalue these obligations and record increases or decreases in their fair value as an adjustment to litigation settlements and other contingencies, net within the Consolidated Statements of Operations. Changes in the fair value of the contingent consideration obligations can result from adjustments to the discount rates, payment periods and adjustments in the probability of achieving future development steps, regulatory approvals, market launches, sales targets and profitability. These fair value measurements represent Level 3 measurements as they are based on significant inputs not observable in the market.
Significant judgment is employed in determining the assumptions utilized as of the acquisition date and for each subsequent measurement period. Accordingly, changes in assumptions described above, could have a material impact on our consolidated results of operations.
Goodwill and intangible assets, including IPR&D, are reviewed for impairment annually and/or when events or other changes in circumstances indicate that the carrying amount of the assets may not be recoverable. Impairment of goodwill and indefinite-lived intangibles is determined to exist when the fair value is less than the carrying value of the net assets being tested. Impairment of definite-lived intangibles is determined to exist when undiscounted cash flows related to the assets are less than the carrying value of the assets being tested. Future events and decisions may lead to asset impairment and/or related costs.
Goodwill is allocated and evaluated for impairment at the reporting unit level, which is defined as an operating segment or one level below an operating segment. As of April 1, 2016 , Mylan had four reporting units, of which three were included in the former Generics segment with the remaining reporting unit consisting of our former Specialty segment. As of the date of our annual impairment test, April 1, 2016 , the allocation of Mylan's total goodwill was as follows: North America $2.46 billion , Europe $1.05 billion and Rest of World $1.66 billion , with $349.1 million allocated to our Specialty segment and reporting unit. As a result of our acquisition of Meda on August 5, 2016 and the integration of our portfolio across our branded, generics and OTC platforms in all of our regions, effective October 1, 2016, the Company expanded its reportable segments. As of October 1, 2016, the Company now has three reportable segments on a geographic basis, North America, Europe and Rest of World. Our reporting units for allocating and evaluating Goodwill impairment remains unchanged subsequent to the change in segments. At December 31, 2016, we have performed a qualitative assessment, excluding the goodwill related to the recent acquisition of Meda, and concluded that it was more likely than not that the fair value of all the reporting units continues to be greater than the carrying amounts.
For our North American and Specialty reporting units, we have utilized the Financial Accounting Standards Board ("FASB") amended guidance on goodwill impairment testing as part of our annual impairment test at April 1, 2016 . Under this guidance, entities testing goodwill for impairment have the option of performing a qualitative assessment before calculating the fair value of the reporting unit ("step 1"). We concluded that it was more likely than not that the fair value of North America and Specialty reporting units is greater than the carrying amount, therefore no step 1 quantitative analysis was performed. During 2016 , the Company performed a step 1 quantitative analysis for the Europe and Rest of World reporting units. Step 1 of the impairment analysis consists of a comparison of the estimated fair value of the individual reporting units with their carrying amount, including goodwill. In estimating each reporting unit's fair value, we performed extensive valuation analysis utilizing both income and market-based approaches, in our goodwill assessment process. We utilized an average of the two methods in estimating the fair value of the individual reporting units. The following describes the valuation methodologies used to derive the estimated fair value of the reporting units.
Income Approach : Under this approach, to determine fair value, we discounted the expected future cash flows of each reporting unit. We used a discount rate, which reflected the overall level of inherent risk and the rate of return an outside investor would have expected to earn. To estimate cash flows beyond the final year of our model, we used a terminal value approach. Under this approach, we used estimated earnings before interest, taxes, depreciation and amortization ("EBITDA") in the final year of our model, adjusted to estimate a normalized cash flow, applied a perpetuity growth assumption, and discounted by a perpetuity discount factor to determine the terminal value. We incorporated the present value of the resulting terminal value into our estimate of fair value.
83
Table of Contents
Market-Based Approach : The Company also utilizes a market-based approach to estimate fair value, principally utilizing the guideline company method which focuses on comparing our risk profile and growth prospects to a select group of publicly traded companies with reasonably similar guidelines.
The Company performed its annual impairment test as of April 1, 2016 . The estimated fair value of the two reporting units tested on a quantitative basis, Europe and Rest of World, were in excess of the respective carrying values of each reporting unit. For the Europe reporting unit, the estimated fair value of this business exceeded its carrying value by approximately 18% . As it relates to the income approach for the Europe reporting unit at April 1, 2016 , we forecasted cash flows for the next ten years. During the forecast period, the revenue compound annual growth rate ("CAGR") was approximately 4% . A terminal value year was calculated with a 1% revenue growth rate applied. The CAGR in EBITDA was approximately 3% . The discount rate utilized was 8.5% . Under the market-based approach, we utilized an estimated range of market multiples of 8.0 to 9.5 times EBITDA plus a control premium of 15% .
As it relates to the income approach for the Rest of World reporting unit at April 1, 2016 , we forecasted cash flows for the next ten years. During the forecast period, the revenue CAGR was approximately 8% . A terminal value year was calculated with a 3% revenue growth rate applied. The CAGR in EBITDA was approximately 12% . The discount rate utilized was 11.0% . Under the market-based approach, we utilized an estimated range of market multiples of 8.0 to 11.5 times EBITDA plus a control premium of 15% . The estimated fair value of the Rest of World reporting unit exceeded its carrying value by approximately 19% .
The determination of the fair value of the reporting units requires us to make significant estimates and assumptions that affect the reporting unit's expected future cash flows. These estimates and assumptions primarily include, but are not limited to, market multiples, control premiums, the discount rate, terminal growth rates, operating income before depreciation and amortization, and capital expenditures forecasts. Due to the inherent uncertainty involved in making these estimates, actual results could differ from those estimates. In addition, changes in underlying assumptions, especially as it relates to the key assumptions detailed, could have a significant impact on the fair value of the reporting units.
We have also assessed the recoverability of certain long-lived assets contained within the reporting units. Any impairment of these assets must be considered prior to our impairment review of goodwill. The assessment for impairment is based on our ability to recover the carrying value of the long-lived assets by analyzing the expected future undiscounted pre-tax cash flows specific to the asset grouping.
We assess the recoverability of the carrying value of long-lived assets at the lowest level for which identifiable undiscounted cash flows are largely independent of the cash flows of other assets and liabilities. For Rest of World and Europe reporting units, this assessment is generally performed at the country level within the reporting units. If these undiscounted cash flows are less than the carrying value of long-lived assets within the asset group, an impairment loss is measured based on the difference between the estimated fair value and carrying value. Significant management judgment is involved in estimating the recoverability of these assets and is dependent upon the accuracy of the assumptions used in making these estimates, as well as how the estimates compare to the eventual future operating performance of the specific asset grouping. The Company's Australia operation in Rest of World reporting unit and certain asset groupings in the Europe reporting unit, principally Portugal, Belgium and Germany, remain at risk for potential impairment charges if the projected operating results are not achieved. Any future long-lived assets impairment charges would likely materially impact the Company's reported financial condition and results of operations.
The Company performs its annual impairment review of IPR&D assets during the third and fourth quarters of each fiscal year. The impairment test for IPR&D consists of a comparison of the asset's fair value with its carrying value. Impairment is determined to exist when the fair value is less than the carrying value of the assets being tested. This review of IPR&D assets principally relates to assets acquired as part of the Jai Pharma Limited acquisition in November 2015, Agila acquisition in December 2013, the respiratory delivery platform acquisition in December 2011 and the Bioniche Pharma acquisition in September 2010. The Company calculates the fair value based upon detailed valuations employing the income approach utilizing Level 3 inputs, as defined in Note 7 Financial Instruments and Risk Management . The fair value of IPR&D is calculated as the present value of the estimated future net cash flows using a market rate of return. The assumptions inherent in the estimated future cash flows include, among other things, the impact of changes to the development programs, the projected development and regulatory time frames and the current competitive environment. For the years ended December 31, 2016 , 2015 and 2014, the Company recorded $49.9 million , $31.3 million , and $27.7 million , respectively, of impairment charges, which were recorded as a component of amortization expense. At December 31, 2016 and 2015 , the Company's IPR&D assets totaled $921.1 million and $737.7 million , respectively.
84
Table of Contents
Income Taxes
We compute our income taxes based on the statutory tax rates and tax planning opportunities available to Mylan in the various jurisdictions in which we generate income. Significant judgment is required in determining our income taxes and in evaluating our tax positions. We establish reserves in accordance with Mylan 's policy regarding accounting for uncertainty in income taxes. Our policy provides that the tax effects from an uncertain tax position be recognized in Mylan 's financial statements, only if the position is more likely than not of being sustained upon audit, based on the technical merits of the position. We adjust these reserves in light of changing facts and circumstances, such as the settlement of a tax audit. Our provision for income taxes includes the impact of reserve provisions and changes to reserves. Favorable resolution would be recognized as a reduction to our provision for income taxes in the period of resolution. Based on this evaluation, as of December 31, 2016 , our reserve for unrecognized tax benefits totaled $190.9 million .
Management assesses the available positive and negative evidence to estimate if sufficient future taxable income will be generated to utilize the existing deferred tax assets. A significant piece of objective negative evidence evaluated was the cumulative loss incurred in certain taxing jurisdictions over the three-year period ended December 31, 2016 . Such objective evidence limits the ability to consider other subjective evidence such as our projections for future growth.
Based on this evaluation, as of December 31, 2016 , a valuation allowance of $460.7 million has been recorded in order to measure only the portion of the deferred tax asset that more likely than not will be realized. The amount of the deferred tax asset considered realizable, however, could be adjusted if estimates of future taxable income during the carryforward period are reduced or if objective negative evidence in the form of cumulative losses is no longer present and additional weight may be given to subjective evidence such as projections for growth.
The resolution of tax reserves and changes in valuation allowances could be material to Mylan's results of operations or financial condition. A variance of 5% between estimated reserves and valuation allowances and actual resolution and realization of these tax items would have an effect on our reserve balance and valuation allowance of approximately $33 million .
Legal Matters
Mylan is involved in various legal proceedings, some of which involve claims for substantial amounts. An estimate is made to accrue for a loss contingency relating to any of these legal proceedings if it is probable that a liability was incurred as of the date of the financial statements and the amount of loss can be reasonably estimated. Because of the subjective nature inherent in assessing the outcome of litigation and because of the potential that an adverse outcome in a legal proceeding could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or share price, such estimates are considered to be critical accounting estimates.
A variance of 5% between estimated and recorded litigation reserves (excluding indemnified claims) and actual resolution of certain legal matters would have an effect on our litigation reserve balance of approximately $26 million . Refer to Note 18 Litigation in Item 8 in this Form 10-K for further discussion of litigation matters.
Recent Accounting Pronouncements
Refer to Note 2 Summary of Significant Accounting Policies in Item 8 in this Form 10-K for recently adopted accounting pronouncements and recently issued accounting pronouncements not yet adopted.
ITEM 7A. | Quantitative and Qualitative Disclosures About Market Risk |
Foreign Currency Exchange Risk
A significant portion of our revenues and earnings are exposed to changes in foreign currency exchange rates. We seek to manage this foreign exchange risk in part through operational means, including managing same currency revenues in relation to same currency costs and same currency assets in relation to same currency liabilities.
From time to time, foreign exchange risk is managed through the use of foreign currency forward-exchange contracts. These contracts are used to offset the potential earnings effects from mostly intercompany foreign currency assets and liabilities that arise from operations and from intercompany loans. Mylan 's primary areas of foreign exchange risk relative to the U.S. Dollar are the Euro, Swedish Krona, Indian Rupee, Japanese Yen, Australian Dollar, Canadian Dollar, Pound Sterling and Brazilian Real . Any unhedged foreign exchange exposures continue to be subject to market fluctuations.
85
Table of Contents
Our financial instrument holdings at year end were analyzed to determine their sensitivity to foreign exchange rate changes. The fair values of these instruments were determined as follows:
• | foreign currency forward-exchange contracts - net present values |
• | foreign currency denominated receivables, payables, debt and loans - changes in exchange rates |
In this sensitivity analysis, we assumed that the change in one currency's rate relative to the U.S. Dollar would not have an effect on other currencies' rates relative to the U.S. Dollar. All other factors were held constant.
If there were an adverse change in foreign currency exchange rates of 10% , the expected net effect on net income related to Mylan 's foreign currency denominated financial instruments would not be material .
Interest Rate and Long-Term Debt Risk
Mylan 's exposure to interest rate risk arises primarily from our U.S. Dollar and Euro borrowings and U.S. Dollar investments. We invest primarily on a variable-rate basis and we borrow on both a fixed and variable basis. In order to maintain a certain ratio of fixed to variable rate debt, from time to time, depending on market conditions, Mylan will use derivative financial instruments such as interest rate swaps to fix interest rates on variable-rate borrowings or to convert fixed-rate borrowings to variable interest rates.
As of December 31, 2016 , Mylan 's long-term fixed rate borrowings consist principally of $13.0 billion notional amount of Senior Notes and Euro Notes. Generally, the fair value of fixed interest rate debt will decrease as interest rates rise and increase as interest rates fall. As of December 31, 2016 , the fair value of our fixed rate Senior Notes and Euro Notes was approximately $13.2 billion . A 100 basis point change in interest rates on Mylan 's variable rate debt, net of interest rate swaps, would result in a change in interest expense of approximately $32.9 million per year.
86
Table of Contents
ITEM 8. | Financial Statements And Supplementary Data |
Index to Consolidated Financial Statements and
Supplementary Financial Information
| Page |
|
|
Management's Report on Internal Control over Financial Reporting | 88 |
|
|
Reports of Independent Registered Public Accounting Firm | 89 |
|
|
Consolidated Balance Sheets as of December 31, 2016 and 2015 | 91 |
|
|
Consolidated Statements of Operations for the Years Ended December 31, 2016, 2015 and 2014 | 92 |
|
|
Consolidated Statements of Comprehensive Earnings for the Years Ended December 31, 2016, 2015 and 2014 | 93 |
|
|
Consolidated Statements of Equity for the Years Ended December 31, 2016, 2015 and 2014 | 94 |
|
|
Consolidated Statements of Cash Flows for the Years Ended December 31, 2016, 2015 and 2014 | 95 |
|
|
Notes to Consolidated Financial Statements | 96 |
|
|
Supplementary Financial Information | 176 |
87
Table of Contents
Management's Report on Internal Control over Financial Reporting
Management of Mylan N.V. (the "Company") is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America. In order to evaluate the effectiveness of internal control over financial reporting, management has conducted an assessment, including testing, using the criteria in Internal Control - Integrated Framework ( 2013 ) , issued by the Committee of Sponsoring Organizations of the Treadway Commission ("COSO"). Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions or that the degree of compliance with the policies or procedures may deteriorate.
On August 5, 2016, the Company completed its acquisition of Meda AB (publ.) ("Meda"). Meda represented 8% of the Company's consolidated total revenues for the year ended December 31, 2016 , and assets (including intangible assets and goodwill) represented 34% of the Company's consolidated total assets, as of December 31, 2016 . Management did not include Meda when conducting its assessment of the effectiveness of the Company's internal control over financial reporting as of December 31, 2016 .
As a result of this assessment, management has concluded that the Company maintained effective internal control over financial reporting as of December 31, 2016 based on the criteria in Internal Control - Integrated Framework ( 2013 ) issued by COSO.
Our independent registered public accounting firm, Deloitte & Touche LLP, has audited the effectiveness of the Company's internal control over financial reporting. Deloitte & Touche LLP's opinion on the Company's internal control over financial reporting appears on page 90 of this Form 10-K.
88
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Shareholders of Mylan N.V.:
We have audited the accompanying consolidated balance sheets of Mylan N.V. and subsidiaries (the "Company") as of December 31, 2016 and 2015 , and the related consolidated statements of operations, comprehensive earnings, equity, and cash flows for each of the three years in the period ended December 31, 2016 . Our audits also included the consolidated financial statement schedule listed in the Index at Item 15. These consolidated financial statements and consolidated financial statement schedule are the responsibility of the Company's management. Our responsibility is to express an opinion on the consolidated financial statements and consolidated financial statement schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the consolidated financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, such consolidated financial statements present fairly, in all material respects, the financial position of Mylan N.V. and subsidiaries as of December 31, 2016 and 2015 , and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2016 , in conformity with accounting principles generally accepted in the United States of America. Also, in our opinion, such consolidated financial statement schedule, when considered in relation to the basic consolidated financial statements taken as a whole, presents fairly, in all material respects, the information set forth therein.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the Company's internal control over financial reporting as of December 31, 2016 , based on the criteria established in Internal Control - Integrated Framework ( 2013 ) issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 1, 2017 expressed an unqualified opinion on the Company's internal control over financial reporting.
/s/ DELOITTE & TOUCHE LLP
Pittsburgh, Pennsylvania
March 1, 2017
89
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Shareholders of Mylan N.V.:
We have audited the internal control over financial reporting of Mylan N.V. and subsidiaries (the "Company") as of December 31, 2016 , based on criteria established in Internal Control - Integrated Framework ( 2013 ) issued by the Committee of Sponsoring Organizations of the Treadway Commission. As described in Management's Report on Internal Control over Financial Reporting, management excluded from its assessment the internal control over financial reporting at Meda AB (publ.) ("Meda"), which was acquired on August 5, 2016. Meda represented 8% of the Company's consolidated total revenues for the year ended December 31, 2016, and assets (including intangible assets and goodwill) represented 34% of the Company's consolidated total assets as of December 31, 2016. Accordingly, our audit did not include the internal control over financial reporting at Meda. The Company's management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management's Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company's internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company's internal control over financial reporting is a process designed by, or under the supervision of, the company's principal executive and principal financial officers, or persons performing similar functions, and effected by the company's board of directors, management, and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of the inherent limitations of internal control over financial reporting, including the possibility of collusion or improper management override of controls, material misstatements due to error or fraud may not be prevented or detected on a timely basis. Also, projections of any evaluation of the effectiveness of the internal control over financial reporting to future periods are subject to the risk that the controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2016 , based on the criteria established in Internal Control - Integrated Framework ( 2013 ) issued by the Committee of Sponsoring Organizations of the Treadway Commission.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated financial statements and consolidated financial statement schedule as of and for the year ended December 31, 2016 of the Company and our report dated March 1, 2017 expressed an unqualified opinion on those consolidated financial statements and consolidated financial statement schedule.
/s/ DELOITTE & TOUCHE LLP
Pittsburgh, Pennsylvania
March 1, 2017
90
Table of Contents
MYLAN N.V. AND SUBSIDIARIES
Consolidated Balance Sheets
(In millions, except share and per share amounts)
| December 31, |
| December 31, | ||||
ASSETS |
|
|
| ||||
Assets |
|
|
| ||||
Current assets: |
|
|
| ||||
Cash and cash equivalents | $ | 998.8 | |
| $ | 1,236.0 | |
Accounts receivable, net | 3,310.9 | |
| 2,689.1 | | ||
Inventories | 2,456.4 | |
| 1,951.0 | | ||
Prepaid expenses and other current assets | 756.4 | |
| 596.6 | | ||
Total current assets | 7,522.5 | |
| 6,472.7 | | ||
Property, plant and equipment, net | 2,322.2 | |
| 1,983.9 | | ||
Intangible assets, net | 14,447.8 | |
| 7,221.9 | | ||
Goodwill | 9,231.9 | |
| 5,380.1 | | ||
Deferred income tax benefit | 633.2 | |
| 457.6 | | ||
Other assets | 568.6 | |
| 751.5 | | ||
Total assets | $ | 34,726.2 | |
| $ | 22,267.7 | |
LIABILITIES AND EQUITY |
|
|
| ||||
Liabilities |
|
|
| ||||
Current liabilities: |
|
|
| ||||
Trade accounts payable | $ | 1,348.1 | |
| $ | 1,109.6 | |
Short-term borrowings | 46.4 | |
| 1.3 | | ||
Income taxes payable | 97.7 | |
| 92.4 | | ||
Current portion of long-term debt and other long-term obligations | 290.0 | |
| 1,077.0 | | ||
Other current liabilities | 3,258.5 | |
| 1,841.9 | | ||
Total current liabilities | 5,040.7 | |
| 4,122.2 | | ||
Long-term debt | 15,202.9 | |
| 6,295.6 | | ||
Deferred income tax liability | 2,006.4 | |
| 718.1 | | ||
Other long-term obligations | 1,358.6 | |
| 1,366.0 | | ||
Total liabilities | 23,608.6 | |
| 12,501.9 | | ||
Equity |
|
|
| ||||
Mylan N.V. shareholders' equity |
|
|
| ||||
Ordinary shares - nominal value €0.01 per share as of December 31, 2016 and December 31, 2015 |
|
|
| ||||
Shares authorized: 1,200,000,000 as of December 31, 2016 and December 31, 2015 |
|
|
| ||||
Shares issued: 536,639,291 and 491,928,095 as of December 31, 2016 and December 31, 2015 | 6.0 | |
| 5.5 | | ||
Additional paid-in capital | 8,499.3 | |
| 7,128.6 | | ||
Retained earnings | 4,942.1 | |
| 4,462.1 | | ||
Accumulated other comprehensive loss | (2,263.7 | ) |
| (1,764.3 | ) | ||
| 11,183.7 | |
| 9,831.9 | | ||
Noncontrolling interest | 1.4 | |
| 1.4 | | ||
Less: Treasury stock - at cost |
|
|
| ||||
Ordinary shares: 1,311,193 as of December 31, 2016 and December 31, 2015 | 67.5 | |
| 67.5 | | ||
Total equity | 11,117.6 | |
| 9,765.8 | | ||
Total liabilities and equity | $ | 34,726.2 | |
| $ | 22,267.7 | |
See Notes to Consolidated Financial Statements
91
Table of Contents
MYLAN N.V. AND SUBSIDIARIES
Consolidated Statements of Operations
(In millions, except per share amounts)
| Year Ended December 31, | ||||||||||
| 2016 |
| 2015 |
| 2014 | ||||||
Revenues: |
|
|
|
|
| ||||||
Net sales | $ | 10,967.1 | |
| $ | 9,362.6 | |
| $ | 7,646.5 | |
Other revenues | 109.8 | |
| 66.7 | |
| 73.1 | | |||
Total revenues | 11,076.9 | |
| 9,429.3 | |
| 7,719.6 | | |||
Cost of sales | 6,379.9 | |
| 5,213.2 | |
| 4,191.6 | | |||
Gross profit | 4,697.0 | |
| 4,216.1 | |
| 3,528.0 | | |||
Operating expenses: |
|
|
|
|
| ||||||
Research and development | 826.8 | |
| 671.9 | |
| 581.8 | | |||
Selling, general and administrative | 2,496.1 | |
| 2,180.7 | |
| 1,625.7 | | |||
Litigation settlements and other contingencies, net | 672.5 | |
| (97.4 | ) |
| (32.1 | ) | |||
Total operating expenses | 3,995.4 | |
| 2,755.2 | |
| 2,175.4 | | |||
Earnings from operations | 701.6 | |
| 1,460.9 | |
| 1,352.6 | | |||
Interest expense | 454.8 | |
| 339.4 | |
| 333.2 | | |||
Other expense, net | 125.1 | |
| 206.1 | |
| 44.9 | | |||
Earnings before income taxes and noncontrolling interest | 121.7 | |
| 915.4 | |
| 974.5 | | |||
Income tax (benefit) provision | (358.3 | ) |
| 67.7 | |
| 41.4 | | |||
Net earnings | 480.0 | |
| 847.7 | |
| 933.1 | | |||
Net earnings attributable to the noncontrolling interest | - | |
| (0.1 | ) |
| (3.7 | ) | |||
Net earnings attributable to Mylan N.V. ordinary shareholders | $ | 480.0 | |
| $ | 847.6 | |
| $ | 929.4 | |
Earnings per ordinary share attributable to Mylan N.V. ordinary shareholders: |
|
|
|
|
| ||||||
Basic | $ | 0.94 | |
| $ | 1.80 | |
| $ | 2.49 | |
Diluted | $ | 0.92 | |
| $ | 1.70 | |
| $ | 2.34 | |
Weighted average ordinary shares outstanding: |
|
|
|
|
| ||||||
Basic | 513.0 | |
| 472.2 | |
| 373.7 | | |||
Diluted | 520.5 | |
| 497.4 | |
| 398.0 | | |||
See Notes to Consolidated Financial Statements
92
Table of Contents
MYLAN N.V. AND SUBSIDIARIES
Consolidated Statements of Comprehensive Earnings
(In millions)
| Year Ended December 31, | ||||||||||
| 2016 |
| 2015 |
| 2014 | ||||||
Net earnings | $ | 480.0 | |
| $ | 847.7 | |
| $ | 933.1 | |
Other comprehensive (loss) earnings, before tax: |
|
|
|
|
| ||||||
Foreign currency translation adjustment | (507.4 | ) |
| (790.9 | ) |
| (622.9 | ) | |||
Change in unrecognized gain (loss) and prior service cost related to defined benefit plans | 21.4 | |
| 3.1 | |
| (11.8 | ) | |||
Net unrecognized gain (loss) on derivatives in cash flow hedging relationships | (31.2 | ) |
| 16.7 | |
| (182.6 | ) | |||
Net unrecognized loss on derivatives in net investment hedging relationships | (1.8 | ) |
| - | |
| - | | |||
Net unrealized gain (loss) on marketable securities | 24.6 | |
| (2.0 | ) |
| - | | |||
Other comprehensive loss, before tax | (494.4 | ) |
| (773.1 | ) |
| (817.3 | ) | |||
Income tax provision (benefit) | 5.0 | |
| 4.2 | |
| (70.4 | ) | |||
Other comprehensive loss, net of tax | (499.4 | ) |
| (777.3 | ) |
| (746.9 | ) | |||
Comprehensive earnings | (19.4 | ) |
| 70.4 | |
| 186.2 | | |||
Comprehensive earnings attributable to the noncontrolling interest | - | |
| (0.1 | ) |
| (3.7 | ) | |||
Comprehensive earnings attributable to Mylan N.V. ordinary shareholders | $ | (19.4 | ) |
| $ | 70.3 | |
| $ | 182.5 | |
See Notes to Consolidated Financial Statements
93
Table of Contents
MYLAN N.V. AND SUBSIDIARIES Consolidated Statements of Equity (In millions, except share amounts) | |||||||||||||||||||||||||||||||||
|
|
|
|
| Additional Paid-In Capital |
| Retained |
|
|
|
|
| Accumulated Other Comprehensive Loss |
| Noncontrolling |
| Total | ||||||||||||||||
| Ordinary Shares (1) |
|
|
| Treasury Stock |
|
|
| |||||||||||||||||||||||||
| Shares |
| Cost |
|
|
| Shares |
| Cost |
|
|
| |||||||||||||||||||||
Balance at December 31, 2013 | 543,978,030 | |
| $ | 272.0 | |
| $ | 4,103.6 | |
| $ | 2,685.1 | |
| (172,373,899 | ) |
| $ | (3,878.8 | ) |
| $ | (240.1 | ) |
| $ | 18.1 | |
| $ | 2,959.9 | |
Net earnings | - | |
| - | |
| - | |
| 929.4 | |
| - | |
| - | |
| - | |
| 3.7 | |
| 933.1 | | |||||||
Other comprehensive loss, net of tax | - | |
| - | |
| - | |
| - | |
| - | |
| - | |
| (746.9 | ) |
| - | |
| (746.9 | ) | |||||||
Stock options exercised, net of shares tendered for payment | 2,680,477 | |
| 1.3 | |
| 52.5 | |
| - | |
| - | |
| - | |
| - | |
| - | |
| 53.8 | | |||||||
Share-based compensation expense | - | |
| - | |
| 66.0 | |
| - | |
| - | |
| - | |
| - | |
| - | |
| 66.0 | | |||||||
Issuance of restricted stock, net of shares withheld | - | |
| - | |
| (40.2 | ) |
| - | |
| 938,699 | |
| 21.1 | |
| - | |
| - | |
| (19.1 | ) | |||||||
Tax benefit of stock option plans | - | |
| - | |
| 30.9 | |
| - | |
| - | |
| - | |
| - | |
| - | |
| 30.9 | | |||||||
Other | - | |
| - | |
| - | |
| - | |
| - | |
| - | |
| - | |
| (1.7 | ) |
| (1.7 | ) | |||||||
Balance at December 31, 2014 | 546,658,507 | |
| $ | 273.3 | |
| $ | 4,212.8 | |
| $ | 3,614.5 | |
| (171,435,200 | ) |
| $ | (3,857.7 | ) |
| $ | (987.0 | ) |
| $ | 20.1 | |
| $ | 3,276.0 | |
Net earnings | - | |
| $ | - | |
| $ | - | |
| $ | 847.6 | |
| - | |
| $ | - | |
| $ | - | |
| $ | 0.1 | |
| $ | 847.7 | |
Other comprehensive loss, net of tax | - | |
| - | |
| - | |
| - | |
| - | |
| - | |
| (777.3 | ) |
| - | |
| (777.3 | ) | |||||||
Ordinary shares repurchase | - | |
| - | |
| - | |
| - | |
| 1,311,193 | |
| (67.5 | ) |
| - | |
| - | |
| (67.5 | ) | |||||||
Stock options exercised, net of shares tendered for payment | 6,086,450 | |
| 1.3 | |
| 96.7 | |
| - | |
| - | |
| - | |
| - | |
| - | |
| 98.0 | | |||||||
Share-based compensation expense | - | |
| - | |
| 92.8 | |
| - | |
| - | |
| - | |
| - | |
| - | |
| 92.8 | | |||||||
Issuance of restricted stock, net of shares withheld | - | |
| - | |
| (56.2 | ) |
| - | |
| 618,338 | |
| 14.5 | |
| - | |
| - | |
| (41.7 | ) | |||||||
Purchase of subsidiary shares from noncontrolling interest | - | |
| - | |
| - | |
| - | |
| - | |
| - | |
| - | |
| (18.7 | ) |
| (18.7 | ) | |||||||
Tax benefit of stock option plans | - | |
| - | |
| 52.5 | |
| - | |
| - | |
| - | |
| - | |
| - | |
| 52.5 | | |||||||
Exchange of Mylan Inc. common stock into Mylan N.V. ordinary shares | (378,388,431 | ) |
| (185.0 | ) |
| 185.0 | |
| - | |
| - | |
| - | |
| - | |
| - | |
| - | | |||||||
Issuance of ordinary shares to Mylan N.V. | 378,388,431 | |
| - | |
| - | |
| - | |
| - | |
| - | |
| - | |
| - | |
| - | | |||||||
Issuance of ordinary shares to purchase the EPD Business | 110,000,000 | |
| 1.3 | |
| 6,304.5 | |
| - | |
| - | |
| - | |
| - | |
| - | |
| 6,305.8 | | |||||||
Retirement of Mylan Inc. treasury stock, net | (170,816,862 | ) |
| (85.4 | ) |
| (3,757.7 | ) |
| - | |
| 170,816,862 | |
| 3,843.1 | |
| - | |
| - | |
| - | | |||||||
Other | - | |
| - | |
| (1.8 | ) |
| - | |
| - | |
| 0.1 | |
| - | |
| (0.1 | ) |
| (1.8 | ) | |||||||
Balance at December 31, 2015 | 491,928,095 | |
| $ | 5.5 | |
| $ | 7,128.6 | |
| $ | 4,462.1 | |
| 1,311,193 | |
| $ | (67.5 | ) |
| $ | (1,764.3 | ) |
| $ | 1.4 | |
| $ | 9,765.8 | |
Net earnings | - | |
| $ | - | |
| $ | - | |
| $ | 480.0 | |
| - | |
| $ | - | |
| $ | - | |
| $ | - | |
| $ | 480.0 | |
Other comprehensive loss, net of tax | - | |
| - | |
| - | |
| - | |
| - | |
| - | |
| (499.4 | ) |
| - | |
| (499.4 | ) | |||||||
Stock options exercised, net of shares tendered for payment | 1,283,580 | |
| - | |
| 13.6 | |
| - | |
| - | |
| - | |
| - | |
| - | |
| 13.6 | | |||||||
Share-based compensation expense | - | |
| - | |
| 88.9 | |
| - | |
| - | |
| - | |
| - | |
| - | |
| 88.9 | | |||||||
Issuance of restricted stock, net of shares withheld | | |
| - | |
| (14.2 | ) |
| - | |
| - | |
| - | |
| - | |
| - | |
| (14.2 | ) | |||||||
Shares issued for warrant settlement | 16,979,984 | |
| 0.2 | |
| (0.2 | ) |
| - | |
| - | |
| - | |
| - | |
| - | |
| - | | |||||||
Tax benefit of stock option plans | - | |
| - | |
| 1.2 | |
| - | |
| - | |
| - | |
| - | |
| - | |
| 1.2 | | |||||||
Issuance of ordinary shares to purchase Meda | 26,447,632 | |
| 0.3 | |
| 1,281.4 | |
| - | |
| - | |
| - | |
| - | |
| - | |
| 1,281.7 | | |||||||
Balance at December 31, 2016 | 536,639,291 | |
| $ | 6.0 | |
| $ | 8,499.3 | |
| $ | 4,942.1 | |
| 1,311,193 | |
| $ | (67.5 | ) |
| $ | (2,263.7 | ) |
| $ | 1.4 | |
| $ | 11,117.6 | |
____________
(1) | Common stock prior to February 27, 2015 . |
See Notes to Consolidated Financial Statements
94
Table of Contents
MYLAN N.V. AND SUBSIDIARIES
Consolidated Statements of Cash Flows
| Year Ended December 31, | ||||||||||
| 2016 |
| 2015 |
| 2014 | ||||||
Cash flows from operating activities: |
|
|
|
|
| ||||||
Net earnings | $ | 480.0 | |
| $ | 847.7 | |
| $ | 933.1 | |
Adjustments to reconcile net earnings to net cash provided by operating activities: |
|
|
|
|
| ||||||
Depreciation and amortization | 1,523.0 | |
| 1,032.1 | |
| 566.6 | | |||
Deferred income tax benefit | (609.5 | ) |
| (115.9 | ) |
| (315.2 | ) | |||
Litigation settlements and other contingencies, net | 597.7 | |
| 15.1 | |
| 7.4 | | |||
Losses on acquisition-related foreign currency derivatives | 128.6 | |
| - | |
| - | | |||
Loss from equity method investments | 112.8 | |
| 105.1 | |
| 91.4 | | |||
Share-based compensation expense | 88.9 | |
| 92.8 | |
| 66.0 | | |||
Write off of financing fees | 35.8 | |
| 99.6 | |
| - | | |||
Other non-cash items | 499.4 | |
| 263.2 | |
| 139.1 | | |||
Changes in operating assets and liabilities: |
|
|
|
|
| ||||||
Accounts receivable | (131.8 | ) |
| 65.8 | |
| (231.2 | ) | |||
Inventories | (279.3 | ) |
| (320.4 | ) |
| (147.5 | ) | |||
Trade accounts payable | 87.7 | |
| 131.8 | |
| (0.3 | ) | |||
Income taxes | 37.5 | |
| (164.2 | ) |
| 78.5 | | |||
Other operating assets and liabilities, net | (523.6 | ) |
| (44.2 | ) |
| (173.1 | ) | |||
Net cash provided by operating activities | 2,047.2 | |
| 2,008.5 | |
| 1,014.8 | | |||
Cash flows from investing activities: |
|
|
|
|
| ||||||
Cash paid for acquisitions, net | (6,481.9 | ) |
| (693.1 | ) |
| (50.0 | ) | |||
Capital expenditures | (390.4 | ) |
| (362.9 | ) |
| (325.3 | ) | |||
Payments for product rights and other, net | (360.2 | ) |
| (506.5 | ) |
| (420.2 | ) | |||
Cash paid for Meda's unconditional deferred payment | (308.0 | ) |
| - | |
| - | | |||
Settlement of acquisition-related foreign currency derivatives | (128.6 | ) |
| - | |
| - | | |||
Purchase of marketable securities | (30.2 | ) |
| (62.1 | ) |
| (19.9 | ) | |||
Change in restricted cash | 57.1 | |
| 21.8 | |
| (5.1 | ) | |||
Proceeds from sale of marketable securities | 21.5 | |
| 33.1 | |
| 20.2 | | |||
Net cash used in investing activities | (7,620.7 | ) |
| (1,569.7 | ) |
| (800.3 | ) | |||
Cash flows from financing activities: |
|
|
|
|
| ||||||
Proceeds from issuance of long-term debt | 11,752.2 | |
| 3,539.2 | |
| 2,235.0 | | |||
Payments of long-term debt | (6,296.3 | ) |
| (4,484.1 | ) |
| (2,295.8 | ) | |||
Payments of financing fees | (112.6 | ) |
| (130.4 | ) |
| (5.8 | ) | |||
Proceeds from convertible note hedge | - | |
| 1,970.8 | |
| - | | |||
Change in short-term borrowings, net | 40.8 | |
| (329.2 | ) |
| (107.8 | ) | |||
Purchase of ordinary shares | - | |
| (67.5 | ) |
| - | | |||
Proceeds from exercise of stock options | 13.8 | |
| 97.7 | |
| 53.8 | | |||
Taxes paid related to net share settlement of equity awards | (17.5 | ) |
| (31.8 | ) |
| (27.7 | ) | |||
Contingent consideration payments | (35.5 | ) |
| - | |
| (150.0 | ) | |||
Acquisition of noncontrolling interest | (1.1 | ) |
| (11.7 | ) |
| - | | |||
Other items, net | 0.8 | |
| 51.8 | |
| 30.9 | | |||
Net cash provided by (used in) financing activities | 5,344.6 | |
| 604.8 | |
| (267.4 | ) | |||
Effect on cash of changes in exchange rates | (8.3 | ) |
| (33.1 | ) |
| (12.9 | ) | |||
Net (decrease) increase in cash and cash equivalents | (237.2 | ) |
| 1,010.5 | |
| (65.8 | ) | |||
Cash and cash equivalents - beginning of period | 1,236.0 | |
| 225.5 | |
| 291.3 | | |||
Cash and cash equivalents - end of period | $ | 998.8 | |
| $ | 1,236.0 | |
| $ | 225.5 | |
Supplemental disclosures of cash flow information - |
|
|
|
|
| ||||||
Non-cash transactions: |
|
|
|
|
| ||||||
Contingent consideration | $ | 16.0 | |
| $ | 18.0 | |
| $ | - | |
Ordinary shares issued for acquisition | $ | 1,281.7 | |
| $ | 6,305.8 | |
| $ | - | |
Cash paid during the period for: |
|
|
|
|
| ||||||
Income taxes | $ | 285.6 | |
| $ | 302.9 | |
| $ | 210.5 | |
Interest | $ | 357.2 | |
| $ | 254.7 | |
| $ | 273.8 | |
See Notes to Consolidated Financial Statements
95
Table of Contents
Mylan N.V. and Subsidiaries
Notes to Consolidated Financial Statements
1. | Nature of Operations |
Mylan N.V. and its subsidiaries (collectively, the "Company," " Mylan ," "our" or "we") are engaged in the global development, licensing, manufacture, marketing and distribution of generic, brand and branded generic pharmaceutical products for resale by others and active pharmaceutical ingredients ("API") through three reportable segments on a geographic basis, North America, Europe and Rest of World. Our North America segment is primarily made up of our operations in the United States ("U.S.") and Canada and includes the operations of our previously reported Specialty segment. Our Europe segment is made up our of operations in 35 countries within the region. Our Rest of World segment is primarily made up of our operations in India, Australia, Japan and New Zealand. Also included in our Rest of World segment are our operations in emerging markets, which includes countries in Africa (including South Africa) as well as Brazil and other countries throughout Asia and the Middle East. Our API business is conducted through Mylan Laboratories Limited (" Mylan India "), which is included within our Rest of World segment. Effective October 1, 2016, the Company expanded its reporting segments as identified above; refer to Note 13 Segment Information for further information regarding the Company's change in reporting segments.
2. | Summary of Significant Accounting Policies |
Principles of Consolidation. The Consolidated Financial Statements include the accounts of Mylan and those of its wholly owned and majority-owned subsidiaries. All intercompany accounts and transactions have been eliminated in consolidation. Investments in equity method affiliates are recorded at cost and adjusted for the Company's share of the affiliates' cumulative results of operations, capital contributions and distributions. Noncontrolling interests in the Company's subsidiaries are generally recorded net of tax as net earnings attributable to noncontrolling interests. Certain prior period amounts have been reclassified to conform to the presentation for the current year. The reclassifications had no impact on the previously reported net earnings attributable to Mylan N.V. ordinary shareholders.
Use of Estimates in the Preparation of Financial Statements. The preparation of financial statements, in conformity with accounting principles generally accepted in the United States of America ("U.S. GAAP"), requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Because of the uncertainty inherent in such estimates, actual results could differ from those estimates.
Foreign Currencies. The Consolidated Financial Statements are presented in U.S. Dollars, the reporting currency of Mylan . Statements of Operations and Cash Flows of all of the Company's subsidiaries that have functional currencies other than U.S. Dollars are translated at a weighted average exchange rate for the period for inclusion in the Consolidated Statements of Operations and Cash Flows, whereas assets and liabilities are translated at the end of the period exchange rates for inclusion in the Consolidated Balance Sheets. Translation differences are recorded directly in shareholders' equity as foreign currency translation adjustments. Gains or losses on transactions denominated in a currency other than the subsidiaries' functional currency, which arise as a result of changes in foreign currency exchange rates, are recorded in the Consolidated Statements of Operations .
Cash and Cash Equivalents. Cash and cash equivalents are comprised of highly liquid investments with an original maturity of three months or less at the date of purchase.
Marketable Securities. Marketable equity and debt securities classified as available-for-sale are recorded at fair value, with net unrealized gains and losses, net of income taxes, reflected in accumulated other comprehensive loss as a component of shareholders' equity. Net realized gains and losses on sales of available-for-sale securities are computed on a specific security basis and are included in other expense, net , in the Consolidated Statements of Operations . Marketable equity and debt securities classified as trading securities are valued at the quoted market price from broker or dealer quotations or transparent pricing sources at the reporting date, and realized and unrealized gains and losses are included in other expense, net , in the Consolidated Statements of Operations .
Concentrations of Credit Risk. Financial instruments that potentially subject the Company to credit risk consist principally of interest-bearing investments, derivatives and accounts receivable.
96
Table of Contents
Mylan invests its excess cash in high-quality, liquid money market instruments, principally overnight deposits and highly rated money market funds. The Company maintains deposit balances at certain financial institutions in excess of federally insured amounts. Periodically, the Company reviews the creditworthiness of its counterparties to derivative transactions, and it does not expect to incur a loss from failure of any counterparties to perform under agreements it has with such counterparties.
Inventories. Inventories are stated at the lower of cost or market, with cost principally determined by the first-in, first-out method. Provisions for potentially obsolete or slow-moving inventory, including pre-launch inventory, are made based on our analysis of inventory levels, historical obsolescence and future sales forecasts. Included as a component of cost of sales is expense related to the net realizable value of inventories.
Property, Plant and Equipment. Property, plant and equipment are stated at cost less accumulated depreciation. Depreciation is computed and recorded on a straight-line basis over the assets' estimated service lives ( three to 18 years for machinery and equipment and other fixed assets and 15 to 39 years for buildings and improvements). Capitalized software is included in property, plant and equipment and is amortized over estimated useful lives ranging from three to seven years .
Intangible Assets and Goodwill. Intangible assets are stated at cost less accumulated amortization. Amortization is generally recorded on a straight-line basis over estimated useful lives ranging from three to 20 years . The Company periodically reviews the original estimated useful lives of intangible assets and makes adjustments when events indicate that a shorter life is appropriate.
The Company accounts for acquired businesses using the acquisition method of accounting, which requires that the assets acquired and liabilities assumed be recorded at the date of acquisition at their respective fair values. The cost to acquire a business is allocated to the underlying net assets of the acquired business in proportion to their respective fair values. Amounts allocated to acquired in-process research and development ("IPR&D") are capitalized at the date of an acquisition and are not amortized. As products in development are approved for sale, amounts are allocated to product rights and licenses and amortized over their estimated useful lives. Definite-lived intangible assets are amortized over the expected life of the asset. Any excess of the purchase price over the estimated fair values of the net assets acquired is recorded as goodwill.
Purchases of developed products and licenses that are accounted for as an asset acquisition are capitalized as intangible assets and amortized over an estimated useful life. IPR&D assets acquired as part of an asset acquisition are expensed immediately if they have no alternative future uses.
The Company reviews goodwill for impairment at least annually or more frequently if events or changes in circumstances indicate that the carrying value of goodwill may not be recoverable based on management's assessment of the fair value of the Company's reporting units as compared to their related carrying value. Under the authoritative guidance issued by the Financial Accounting Standards Board ("FASB"), we have the option to first assess the qualitative factors to determine whether it is more likely than not that the fair value of the reporting unit is less than its carrying amount as a basis for determining whether it is necessary to perform a quantitative goodwill impairment test. If we determine that it is more likely than not that the fair value of a reporting unit is less than its carrying amount, then the goodwill impairment test is performed. The goodwill impairment test requires the Company to estimate the fair value of the reporting unit and to compare the fair value of the reporting unit with its carrying amount. If the carrying amount exceeds its fair value then there is no impairment recognized. If the carrying value recorded exceeds the fair value calculated, an impairment charge is recorded for the difference. The judgments made in determining the projected cash flows used to estimate the fair value can materially impact the Company's financial condition and results of operations.
Contingent Consideration. Mylan records contingent consideration resulting from business acquisitions at fair value on the acquisition date. Each reporting period thereafter, the Company revalues these obligations and records increases or decreases in their fair value as a charge (credit) to litigation settlements and other contingencies, net within the Consolidated Statements of Operations. Changes in the fair value of the contingent consideration obligations can result from adjustments to the discount rates, payment periods and adjustments in the probability of achieving future development steps, regulatory approvals, market launches, sales targets and profitability. These fair value measurements represent Level 3 measurements, as they are based on significant inputs not observable in the market. Refer to Note 7 Financial Instruments and Risk Management for further information regarding changes recorded to contingent consideration.
Significant judgment is employed in determining the assumptions utilized as of the acquisition date and for each subsequent measurement period. Accordingly, changes in the assumptions described above could have a material impact on the Company's consolidated financial condition and results of operations.
97
Table of Contents
Impairment of Long-Lived Assets. The carrying values of long-lived assets, which include property, plant and equipment and intangible assets with finite lives, are evaluated periodically in relation to the expected future undiscounted cash flows of the underlying assets and monitored for other potential triggering events. Adjustments are made in the event that estimated undiscounted net cash flows are less than the carrying value.
Indefinite-lived intangibles, principally IPR&D, are tested at least annually for impairment or upon the occurrence of a triggering event. The impairment test for IPR&D consists of a comparison of the asset's fair value with its carrying value. Impairment is determined to exist when the fair value is less than the carrying value of the assets being tested.
Revenue Recognition. Mylan recognizes net revenue for product sales when title and risk of loss pass to its customers and when provisions for estimates, including discounts, sales allowances, rebates, Medicaid and other government rebates, price adjustments, returns, chargebacks and other promotional programs, are reasonably determinable. Accruals for these provisions are presented in the Consolidated Financial Statements as reductions in determining net revenues and as a contra asset in accounts receivable, net (if settled via credit) and other current liabilities (if paid in cash). No significant revisions were made to the methodology used in determining these provisions during the years ended December 31, 2016 and 2015 . The following briefly describes the nature of our significant provisions and how such provisions are estimated.
• | Chargebacks: the Company has agreements with certain indirect customers, such as independent pharmacies, managed care organizations, hospitals, nursing homes, governmental agencies and pharmacy benefit managers, which establish contract prices for certain products. The indirect customers then independently select a wholesaler from which to actually purchase the products at these contracted prices. Alternatively, certain wholesalers may enter into agreements with indirect customers that establish contract pricing for certain products, which the wholesalers provide. Under either arrangement, Mylan will provide credit to the wholesaler for any difference between the contracted price with the indirect party and the wholesaler's invoice price. Such credits are called chargebacks. The provision for chargebacks is based on expected sell-through levels by our wholesaler customers to indirect customers, as well as estimated wholesaler inventory levels. |
• | Provision for returns: consistent with industry practice, Mylan maintains a return policy that allows customers to return a product generally within a specified period prior (six months) and subsequent to the expiration date (twelve months). The Company's estimate of the provision for returns is generally based upon historical experience with actual returns. |
• | Incentives offered to customers: these are offered to key customers to promote customer loyalty and encourage greater product sales. These programs generally provide that upon the attainment of pre-established volumes or the attainment of revenue milestones for a specified period, the customer receives credit against purchases. |
The following briefly describes the nature of our other sales reserves and allowances and how such provisions are estimated:
• | Discounts: these are reductions to invoiced amounts offered to customers for payment within a specified period and are estimated upon sale utilizing historical customer payment experience. |
• | Price adjustments : these are credits issued to reflect decreases in the selling prices of products and based upon the amount of product which the customer has remaining in its inventory at the time of the price reduction. In addition, there are decreases in selling prices that are discretionary decisions made by the Company to reflect market conditions. Amounts recorded for estimated price adjustments are based upon specified terms with customers, estimated launch dates of competing products and estimated declines in market price. |
• | Governmental rebate program s: government reimbursement programs include Medicare, Medicaid, and State Pharmacy Assistance Programs established according to statute, regulations and policy. Manufacturers of pharmaceutical products that are covered by the Medicaid program are required to rebate to each state a percentage of their average manufacturer's price for the products dispensed. Medicare beneficiaries are eligible to obtain discounted prescription drug coverage from private sector providers. In addition, certain states have also implemented supplemental rebate programs that obligate manufacturers to pay rebates in excess of those required under federal law. Our estimate of these rebates is based on the historical trends of rebates paid as well as on changes in wholesaler inventory levels and increases or decreases in the level of sales. |
• | Other promotional programs: these are incentive programs periodically offered to our customers. The Company is able to estimate provisions for volume-based sales allowances and other promotional programs based on the specific terms in each agreement at the time of sale. |
98
Table of Contents
Royalty or profit share revenue from licensees, which are based on third-party sales of licensed products and technology, is recorded in accordance with the contract terms, when third-party sales can be reliably measured and collection of the funds is reasonably assured. Royalty revenue is included in other revenue in the Consolidated Statements of Operations.
The Company recognizes contract manufacturing and other service revenue when the service is performed or when the Company's partners take ownership and title has passed, collectability is reasonably assured, the sales price is fixed or determinable, and there is persuasive evidence of an arrangement.
Research and Development. Research and development ("R&D") expenses are charged to operations as incurred.
Income Taxes. Income taxes have been provided for using an asset and liability approach in which deferred income taxes reflect the tax consequences on future years of events that the Company has already recognized in the financial statements or tax returns. Changes in enacted tax rates or laws may result in adjustments to the recorded tax assets or liabilities in the period that the new tax law is enacted.
Earnings per Ordinary Share. Basic earnings per ordinary share is computed by dividing net earnings attributable to Mylan N.V. ordinary shareholders by the weighted average number of ordinary shares outstanding during the period. Diluted earnings per ordinary share is computed by dividing net earnings attributable to Mylan N.V. ordinary shareholders by the weighted average number of ordinary shares outstanding during the period increased by the number of additional shares that would have been outstanding related to potentially dilutive securities or instruments, if the impact is dilutive.
On August 5, 2016 , in conjunction with its acquisition of Meda AB (publ.) (" Meda "), the Company issued approximately 26.4 million Mylan N.V. ordinary shares to Meda shareholders. The impact of the issuance of these ordinary shares is included in the calculation of basic earnings per share. The weighted average impact for the year ended December 31, 2016 , was approximately 10.8 million ordinary shares.
On September 15, 2008, concurrent with the sale of $575 million aggregate principal amount of Cash Convertible Notes due 2015 (the "Cash Convertible Notes"), Mylan Inc. entered into convertible note hedge and warrant transactions with certain counterparties. In connection with the consummation of the EPD Transaction (as defined below in Note 3 Acquisitions and Other Transactions ), the terms of the convertible note hedge were adjusted so that the cash settlement value would be based on Mylan N.V. ordinary shares. The terms of the warrant transactions were also adjusted so that, from and after the consummation of the EPD Transaction, the Company could settle the obligations under the warrant transactions by delivering Mylan N.V. ordinary shares. Pursuant to the warrant transactions, and a subsequent amendment in 2011, there were approximately 43.2 million warrants outstanding, with approximately 41.0 million of those warrants having an exercise price of $30.00 . The remaining warrants had an exercise price of $20.00 . The warrants met the definition of derivatives under the guidance in the FASB Accounting Standards Codification ("ASC") 815 Derivatives and Hedging ("ASC 815"); however, because these instruments had been determined to be indexed to the Company's own ordinary shares and met the criteria for equity classification under ASC 815-40 Contracts in Entity's Own Equity ("ASC 815-40"), the warrants were recorded in shareholders' equity in the Consolidated Balance Sheets.
On April 15, 2016, in connection with the expiration and settlement of the warrants, the Company issued approximately 17.0 million Mylan N.V. ordinary shares. The impact of the issuance of these ordinary shares is included in the calculation of basic earnings per share from the date of issuance. For the year ended December 31, 2016, 12.1 million ordinary shares is the weighted average impact included in the calculation of basic earnings per ordinary share. The dilutive impact of the warrants, prior to settlement, is included in the calculation of diluted earnings per ordinary share based upon the average market value of the Company's ordinary shares during the period as compared to the exercise price. For the years ended December 31, 2016 , 2015 and 2014 , warrants included in the calculation of diluted earnings per ordinary share were 4.9 million , 20.7 million and 17.7 million , respectively.
The Board of Directors periodically authorizes the Company to repurchase ordinary shares in the open market or through other methods. The Company may repurchase up to $1 billion of the Company's ordinary shares under its current repurchase program that was announced on November 16, 2015 (the "Share Repurchase Program"), but is not obligated to acquire any particular amount of ordinary shares. During 2016 , the Company did not repurchase any common shares under the Share Repurchase Program. In 2015 , the Company repurchased approximately 1.3 million common shares at a cost of approximately $67.5 million and approximately 28.5 million common shares at a cost of approximately $1.0 billion in 2014 . These amounts reflect transactions executed through December 31 st of each year.
99
Table of Contents
Basic and diluted earnings per ordinary share attributable to Mylan N.V. are calculated as follows:
| Year Ended December 31, | ||||||||||
(In millions, except per share amounts) | 2016 |
| 2015 (1) |
| 2014 | ||||||
Basic earnings attributable to Mylan N.V. ordinary shareholders (numerator): |
|
|
|
|
| ||||||
Net earnings attributable to Mylan N.V. ordinary shareholders | $ | 480.0 | |
| $ | 847.6 | |
| $ | 929.4 | |
Shares (denominator): |
|
|
|
|
| ||||||
Weighted average ordinary shares outstanding | 513.0 | |
| 472.2 | |
| 373.7 | | |||
Basic earnings per ordinary share attributable to Mylan N.V. ordinary shareholders | $ | 0.94 | |
| $ | 1.80 | |
| $ | 2.49 | |
|
|
|
|
|
| ||||||
Diluted earnings attributable to Mylan N.V. ordinary shareholders (numerator): |
|
|
|
|
| ||||||
Net earnings attributable to Mylan N.V. ordinary shareholders | $ | 480.0 | |
| $ | 847.6 | |
| $ | 929.4 | |
Shares (denominator): |
|
|
|
|
| ||||||
Weighted average ordinary shares outstanding | 513.0 | |
| 472.2 | |
| 373.7 | | |||
Share-based awards and warrants | 7.5 | |
| 25.2 | |
| 24.3 | | |||
Total dilutive shares outstanding | 520.5 | |
| 497.4 | |
| 398.0 | | |||
Diluted earnings per ordinary share attributable to Mylan N.V. ordinary shareholders | $ | 0.92 | |
| $ | 1.70 | |
| $ | 2.34 | |
____________
(1) | As Mylan N.V. is the successor to Mylan Inc., the information set forth above refers to Mylan Inc. for periods prior to February 27, 2015, and to Mylan N.V. on and after February 27, 2015. |
Additional stock awards and restricted ordinary shares were outstanding during the years ended December 31, 2016 , 2015 and 2014 but were not included in the computation of diluted earnings per ordinary share for each respective period because the effect would be anti-dilutive. Excluded shares at December 31, 2016 also include certain share-based compensation awards and restricted ordinary shares whose performance conditions had not been fully met. Such excluded shares and anti-dilutive awards represented 7.8 million , 5.9 million and 6.1 million shares for the years ended December 31, 2016 , 2015 and 2014 , respectively.
Share-Based Compensation. The fair value of share-based compensation is recognized as expense in the Consolidated Statements of Operations over the vesting period.
Derivatives. From time to time the Company may enter into derivative financial instruments (mainly foreign currency exchange forward contracts, interest rate swaps and purchased equity call options) designed to: 1) hedge the cash flows resulting from existing assets and liabilities and transactions expected to be entered into over the next 24 months in currencies other than the functional currency, 2) hedge the variability in interest expense on floating rate debt, 3) hedge the fair value of fixed-rate notes, 4) hedge against changes in interest rates that could impact future debt issuances, 5) hedge cash or share payments required on conversion of issued convertible notes, or 6) economically hedge the foreign currency exposure associated with the purchase price of non-U.S. acquisitions. Derivatives are recognized as assets or liabilities in the Consolidated Balance Sheets at their fair value. When the derivative instrument qualifies as a cash flow hedge, changes in the fair value are included in earnings or deferred through other comprehensive earnings depending on the nature and effectiveness of the offset. If a derivative instrument qualifies as a fair value hedge, the changes in the fair value, as well as the offsetting changes in the fair value of the hedged items, are generally included in interest expense. When such instruments do not qualify for hedge accounting the changes in fair value are recorded in the Consolidated Statements of Operations within other expense, net .
Financial Instruments. The Company's financial instruments consist primarily of short-term and long-term debt, interest rate swaps, forward contracts and option contracts. The Company's financial instruments also include cash and cash equivalents as well as accounts and other receivables and accounts payable, the fair values of which approximate their carrying values. As a policy, the Company does not engage in speculative or leveraged transactions.
The Company uses derivative financial instruments for the purpose of hedging foreign currency and interest rate exposures, which exist as part of ongoing business operations or to hedge cash or share payments required on conversion of issued convertible notes. The Company carries derivative instruments on the Consolidated Balance Sheets at fair value, determined by reference to market data such as forward rates for currencies, implied volatilities, and interest rate swap yield
100
Table of Contents
curves. The accounting for changes in the fair value of a derivative instrument depends on whether it has been designated and qualifies as part of a hedging relationship and, if so, the reason for holding it.
Recent Accounting Pronouncements. In January 2017, the FASB issued Accounting Standards Update 2017-04, Intangibles - Goodwill and Other (Topic 350): Simplifying the Test for Goodwill Impairment ("ASU 2017-04"), which simplifies the subsequent measurement of goodwill by eliminating Step 2 from the goodwill impairment test which previously required measurement of any goodwill impairment loss by comparing the implied fair value of a reporting unit's goodwill with the carrying amount of that goodwill. Under ASU 2017-04, an entity should perform its annual, or interim, goodwill impairment test by comparing the fair value of a reporting unit with its carrying value and recognize an impairment charge for the amount by which the carrying amount exceeds the reporting unit's fair value; without exceeding the total amount of goodwill allocated to that reporting unit. This guidance is effective for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2019, with early adoption permitted. The Company has elected to early adopt this guidance as of January 1, 2017 and will apply it on a prospective basis. The Company does not believe that the adoption will have a material impact on its consolidated financial statements.
In January 2017, the FASB issued Accounting Standards Update 2017-01, Business Combinations (Topic 805) Clarifying the Definition of a Business ("ASU 2017-01"), which narrows the definition of a business and requires an entity to evaluate if substantially all of the fair value of the gross assets acquired is concentrated in a single identifiable asset or a group of similar identifiable assets, which would not constitute the acquisition of a business. The guidance also requires a business to include at least one substantive process and narrows the definition of outputs. This guidance is effective for fiscal years beginning after December 15, 2017, and interim periods within those fiscal years, with early adoption permitted. The Company has elected to early adopt this guidance as of January 1, 2017 and will apply it on a prospective basis. The Company does not believe that the adoption will have a material impact on its consolidated financial statements.
In November 2016, the FASB issued Accounting Standards Update 2016-18, Statement of Cash Flows (Topic 230) Restricted Cash ("ASU 2016-18"), which requires that the reconciliation of the beginning of period and end of period amounts shown in the statement of cash flows include restricted cash and restricted cash equivalents. If restricted cash is presented separately from cash and cash equivalents on the balance sheet, companies will be required to reconcile the amounts presented on the statement of cash flows to the amounts on the balance sheet. This guidance is effective for fiscal years beginning after December 15, 2017, and interim periods within those fiscal years. The Company is currently assessing the impact of the adoption of this guidance on its consolidated statements of cash flows and disclosures.
In October 2016, the FASB issued Accounting Standards Update 2016-16, Income Taxes (Topic 740) ("ASU 2016-16"), which reduces the complexity in the accounting standards by allowing the recognition of current and deferred income taxes for an intra-entity asset transfer, other than inventory, when the transfer occurs. This guidance is effective for fiscal years beginning after December 15, 2017, and interim periods within those fiscal years, with early adoption permitted using a modified retrospective transition approach. The Company is currently assessing the impact of the adoption of this guidance on its consolidated financial statements and disclosures.
In August 2016, the FASB issued Accounting Standards Update 2016-15, Statement of Cash Flows (Topic 230) ("ASU 2016-15"), which clarifies how certain cash receipts and cash payments should be presented in the Statement of Cash Flows. This guidance is effective for fiscal years beginning after December 15, 2017, and interim periods within those fiscal years, with early adoption permitted using a retrospective transition approach. The Company is currently assessing the impact of the adoption of this guidance on its consolidated statements of cash flows.
In March 2016, the FASB issued Accounting Standards Update 2016-09, Compensation - Stock Compensation (Topic 718) ("ASU 2016-09"), which simplifies the accounting for share-based compensation payments. The new standard requires all excess tax benefits and tax deficiencies (including tax benefits of dividends on share-based payment awards) to be recognized as income tax expense or benefit on the income statement. The tax effects of exercised or vested awards should be treated as discrete items in the reporting period in which they occur. This guidance is effective for fiscal years, and interim periods within those years, beginning after December 15, 2016, with early adoption permitted. The Company will adopt this guidance at January 1, 2017 and does not believe the adoption of this guidance will have a material impact on its consolidated financial statements and disclosures.
In February 2016, the FASB issued Accounting Standards Update 2016-02, Leases (Topic 840) ("ASU 2016-02"), which provides principles for the recognition, measurement, presentation and disclosure of leases for both lessees and lessors. The new standard requires lessees to apply a dual approach, classifying leases as either finance or operating leases based on the principle of whether or not the lease is effectively a financed purchase by the lessee. This classification will determine whether lease expense is recognized based on an effective interest method or on a straight-line basis over the term of the lease. A lessee
101
Table of Contents
is also required to record a right-of-use asset and a lease liability for all leases with a term of greater than twelve months regardless of classification. Leases with a term of twelve months or less will be accounted for similar to existing guidance for operating leases. This guidance is effective for annual and interim periods beginning after December 15, 2018, with early adoption permitted. The Company is currently assessing the impact of the adoption of this guidance on its consolidated financial statements and disclosures.
In January 2016, the FASB issued Accounting Standards Update 2016-01, Recognition and Measurement of Financial Assets and Financial Liabilities ("ASU 2016-01"), which requires that most equity investments be measured at fair value, with subsequent changes in fair value recognized in net income (other than those accounted for under equity method of accounting). The amendments in this update also require an entity to present separately in other comprehensive earnings the portion of the total change in the fair value of a liability resulting from a change in the instrument-specific credit risk when the entity has elected to measure the liability at fair value in accordance with the fair value option for financial instruments. ASU 2016-01 also impacts financial liabilities under the fair value option and the presentation and disclosure requirements for financial instruments. This guidance is effective for fiscal years, and interim periods within those years, beginning after December 15, 2017. The Company is currently assessing the impact of the adoption of this guidance on its consolidated financial statements and disclosures.
In May 2014, the FASB issued Accounting Standards Update 2014-09, Revenue from Contracts with Customers ("ASU 2014-09" updated with "ASU 2015-14", "ASU 2016-08", "ASU 2016-10", "ASU 2016-12" and "ASU 2016-20"), which revises accounting guidance on revenue recognition that will supersede nearly all existing revenue recognition guidance under U.S. GAAP. The core principal of this guidance is that an entity should recognize revenue when it transfers promised goods or services to customers in an amount that reflects the consideration which the entity expects to receive in exchange for those goods or services. This guidance also requires additional disclosure about the nature, amount, timing and uncertainty of revenue and cash flows arising from customer contracts, including significant judgments and changes in judgments and assets recognized from costs incurred to obtain or fulfill a contract. This guidance is effective for fiscal years beginning after December 15, 2017, and for interim periods within those fiscal years, and can be applied using a full retrospective or modified retrospective approach. The Company is in the process of reviewing specific revenue arrangements and expects to complete the review in the third quarter of 2017. The Company is still evaluating the adoption method it will elect upon implementation.
3. | Acquisitions and Other Transactions |
Meda AB
On February 10, 2016 , the Company issued an offer announcement under the Nasdaq Stockholm's Takeover Rules and the Swedish Takeover Act (collectively, the "Swedish Takeover Rules") setting forth a public offer to the shareholders of Meda to acquire all of the outstanding shares of Meda (the " Offer "), with an enterprise value, including the net debt of Meda , of approximately Swedish kronor ("SEK" or "kr") 83.6 billion (based on a SEK/USD exchange rate of 8.4158 ) or $9.9 billion at announcement. On August 2, 2016 , the Company announced that the Offer was accepted by Meda shareholders holding an aggregate of approximately 343 million shares, representing approximately 94% of the total number of outstanding Meda shares, as of July 29, 2016 , and the Company declared the Offer unconditional. On August 5, 2016 , settlement occurred with respect to the Meda shares duly tendered by July 29, 2016 and, as a result, Meda became a controlled subsidiary of the Company. Pursuant to the terms of the Offer, each Meda shareholder that duly tendered Meda shares into the Offer received at settlement (1) in respect of 80% of the number of Meda shares tendered by such shareholder, 165kr in cash per Meda share, and (2) in respect of the remaining 20% of the number of Meda shares tendered by such shareholder, 0.386 of the Company's ordinary shares per Meda share (subject to treatment of fractional shares as described in the offer document published on June 16, 2016). The non-tendered shares are required to be acquired for cash through a compulsory acquisition proceeding, in accordance with the Swedish Companies Act (Sw. aktiebolagslagen (2005:551)). The compulsory acquisition proceeding price accrues interest as required by the Swedish Companies Act. Meda's shares were delisted from the Nasdaq Stockholm exchange on August 23, 2016.
On November 1, 2016, the Company made an offer to the remaining Meda shareholders to tender all their Meda shares for cash consideration of 161.31kr per Meda share (the "November Offer") to provide such remaining shareholders with an opportunity to sell their shares in Meda to the Company in advance of the automatic acquisition of their shares for cash in connection with the compulsory acquisition proceeding. At the end of November 2016, Mylan completed the acquisition of approximately 19 million Meda shares duly tendered for aggregate cash consideration of approximately $330.3 million . As of March 1, 2017, Mylan obtained full legal ownership to the remaining Meda shares pursuant to the compulsory acquisition proceeding, and now owns 100% of the total number of outstanding Meda shares. During the year ended December 31, 2016, the Company recognized a foreign currency gain of approximately $30.5 million included in other expense, net on the Consolidated Statements of Operations, related to the settlement of the November Offer and the remaining obligation. At
102
Table of Contents
December 31, 2016 , the Company's current liability associated with the compulsory acquisition proceeding was approximately $70.2 million . As of December 31, 2016, the Company has not hedged the foreign currency risk associated with the remaining liability for the compulsory acquisition proceeding. The Meda shareholders whose shares are subject to the compulsory acquisition proceeding, representing approximately 1% of the total number of outstanding Meda shares, will automatically receive cash consideration plus statutory interest for their Meda shares as determined in the compulsory acquisition proceeding.
On August 5, 2016, the total purchase price was approximately $6.92 billion , net of cash acquired, which includes cash consideration paid of approximately $5.3 billion , the issuance of approximately 26.4 million Mylan N.V. ordinary shares at a fair value of approximately $1.3 billion based on the closing price of the Company's ordinary shares on August 5, 2016, as reported by the NASDAQ Global Select Stock Market ("NASDAQ"), and an assumed liability of approximately $431.0 million related to the compulsory acquisition proceeding of the non-tendered Meda shares. In accordance with U.S. GAAP, the Company used the acquisition method of accounting to account for this transaction. Under the acquisition method of accounting, the assets acquired and liabilities assumed in the transaction have been recorded at their respective estimated fair values at the acquisition date. Acquisition related costs of approximately $182 million were incurred during the year ended December 31, 2016 , which were recorded as components of R&D expense, selling, general and administrative expense ("SG&A"), interest expense and other expense, net in the Consolidated Statements of Operations. These costs included approximately $128.6 million of losses on non-designated foreign currency forward and option contracts entered into in order to economically hedge the SEK purchase price of the Offer (explained further in Note 7 Financial Instruments and Risk Management ) and approximately $45.2 million of financing fees related to the terminated 2016 Bridge Credit Agreement (explained further in Note 8 Debt ).
During the year ended December 31, 2016, adjustments were made to the preliminary purchase price recorded at August 5, 2016, and are reflected as "Measurement Period Adjustments" in the table below. The preliminary allocation of the $6.92 billion purchase price to the assets acquired and liabilities assumed for Meda is as follows:
(In millions) | Preliminary Purchase Price Allocation as of August 5, 2016 (a) |
| Measurement Period Adjustments (b) |
| Preliminary Purchase Price Allocation as of December 31, 2016 (as adjusted) | ||||||
Current assets (excluding inventories and net of cash acquired) | $ | 470.2 | |
| $ | 12.3 | |
| $ | 482.5 | |
Inventories | 465.7 | |
| (2.6 | ) |
| 463.1 | | |||
Property, plant and equipment | 177.5 | |
| - | |
| 177.5 | | |||
Identified intangible assets | 8,060.7 | |
| - | |
| 8,060.7 | | |||
Goodwill | 3,677.6 | |
| (0.7 | ) |
| 3,676.9 | | |||
Other assets | 9.3 | |
| 0.2 | |
| 9.5 | | |||
Total assets acquired | 12,861.0 | |
| 9.2 | |
| 12,870.2 | | |||
Current liabilities | (1,088.4 | ) |
| (17.5 | ) |
| (1,105.9 | ) | |||
Long-term debt, including current portion | (2,864.6 | ) |
| - | |
| (2,864.6 | ) | |||
Deferred tax liabilities | (1,628.1 | ) |
| 14.2 | |
| (1,613.9 | ) | |||
Pension and other postretirement benefits | (322.3 | ) |
| - | |
| (322.3 | ) | |||
Other noncurrent liabilities | (36.5 | ) |
| (5.9 | ) |
| (42.4 | ) | |||
Net assets acquired | $ | 6,921.1 | |
| $ | - | |
| $ | 6,921.1 | |
____________
(a) | As previously reported in the Company's Quarterly Report on Form 10-Q for the nine months ended September 30, 2016. |
(b) | The measurement period adjustments were recorded in the fourth quarter of 2016 and are primarily related to certain working capital adjustments to reflect facts and circumstances that existed as of the acquisition date, and adjustments to deferred tax liabilities. |
The preliminary fair value estimates for the assets acquired and liabilities assumed were based upon preliminary calculations, valuations and assumptions that are subject to change as the Company obtains additional information during the
103
Table of Contents
measurement period (up to one year from the acquisition date). The primary areas subject to change relate to the finalization of the working capital components and income taxes.
The acquisition of Meda creates a more diversified and expansive portfolio of branded and generic medicines along with a strong and growing portfolio of over-the-counter ("OTC") products. Meda has a balanced global footprint with significant scale in key geographic markets, particularly the U.S. and Europe. The acquisition of Meda expanded our presence in emerging markets, which includes countries in Africa, as well as countries throughout Asia and the Middle East, and is complemented by Mylan's presence in India, Brazil and Africa (including South Africa). The Company recorded a step-up in the fair value of inventory of approximately $107 million at the acquisition date which was fully amortized as of December 31, 2016 . The amortization of the inventory step-up was included in cost of sales in the Consolidated Statements of Operations.
The identified intangible assets of $8.06 billion are comprised of product rights and licenses that have a weighted average useful life of 20 years . Significant assumptions utilized in the valuation of identified intangible assets were based on company specific information and projections which are not observable in the market and are thus considered Level 3 measurements as defined by U.S. GAAP. Refer to Note 7 "Financial Instruments and Risk Management" for more information on the U.S. GAAP fair value hierarchy. The goodwill of $3.68 billion arising from the acquisition consisted largely of the value of the employee workforce, and the expected value of products to be developed in the future. The final allocation of goodwill to Mylan's reportable segments has not been completed; however, the majority of goodwill is expected to be allocated to the Europe segment. None of the goodwill recognized in this transaction is currently expected to be deductible for income tax purposes.
The settlement of the Offer constituted an Acceleration Event (as defined in the Rottapharm Agreement referred to below) under the Sale and Purchase Agreement, dated as of July 30, 2014 (the "Rottapharm Agreement"), among Fidim S.r.l., Meda Pharma S.p.A and Meda, the occurrence of which accelerated an unconditional deferred purchase price payment of approximately $308 million ( €275 million ) relating to Meda's acquisition of Rottapharm S.p.A. which otherwise would have been payable in January 2017. The amount was paid during the year ended December 31, 2016.
The operating results of Meda have been included in the Company's Consolidated Statements of Operations since the acquisition date. The total revenues of Meda for the period from the acquisition date to December 31, 2016 were $833.9 million and the net loss, net of tax, was $208.7 million , which includes the effects of the purchase accounting adjustments and acquisition related costs.
Renaissance Topicals Business
On June 15, 2016 , the Company completed the acquisition of the non-sterile, topicals-focused business (the " Topicals Business ") of Renaissance Acquisition Holdings, LLC (" Renaissance ") for approximately $1.0 billion in cash at closing, including amounts deposited into escrow for potential contingent payments, subject to customary adjustments. The Topicals Business provides the Company with a complementary portfolio of approximately 25 products, an active pipeline of approximately 25 products, and an established U.S. sales and marketing infrastructure targeting dermatologists. The Topicals Business also provides an integrated manufacturing and development platform. In accordance with U.S. GAAP, the Company used the acquisition method of accounting to account for this transaction. Under the acquisition method of accounting, the assets acquired and liabilities assumed in the transaction were recorded at their respective estimated fair values at the acquisition date. The U.S. GAAP purchase price was $972.7 million , which includes estimated contingent consideration of approximately $16 million at the date of acquisition related to the potential $50 million payment contingent on the achievement of certain 2016 financial targets. The $50 million contingent payment remains in escrow and is classified as restricted cash included in prepaid expenses and other current assets on the Consolidated Balance Sheets at December 31, 2016.
During the year ended December 31, 2016, adjustments were made to the preliminary purchase price recorded at June 15, 2016 and are reflected as "Measurement Period Adjustments" in the table below. The preliminary allocation of the $972.7 million purchase price to the assets acquired and liabilities assumed for the Topicals Business is as follows:
104
Table of Contents
(In millions) | Preliminary Purchase Price Allocation as of June 15, 2016 (a) |
| Measurement Period Adjustments (b) |
| Preliminary Purchase Price Allocation as of December 31, 2016 (as adjusted) | ||||||
Current assets (excluding inventories) | $ | 68.8 | |
| $ | (11.1 | ) |
| $ | 57.7 | |
Inventories | 74.2 | |
| - | |
| 74.2 | | |||
Property, plant and equipment | 54.8 | |
| - | |
| 54.8 | | |||
Identified intangible assets | 467.0 | |
| - | |
| 467.0 | | |||
In-process research and development | 275.0 | |
| - | |
| 275.0 | | |||
Goodwill | 307.3 | |
| 11.3 | |
| 318.6 | | |||
Other assets | 0.9 | |
| (0.8 | ) |
| 0.1 | | |||
Total assets acquired | 1,248.0 | |
| (0.6 | ) |
| 1,247.4 | | |||
Current liabilities | (65.0 | ) |
| (9.2 | ) |
| (74.2 | ) | |||
Deferred tax liabilities | (203.6 | ) |
| 9.0 | |
| (194.6 | ) | |||
Other noncurrent liabilities | (6.7 | ) |
| 0.8 | |
| (5.9 | ) | |||
Net assets acquired | $ | 972.7 | |
| $ | - | |
| $ | 972.7 | |
____________
(a) | As previously reported in the Company's Quarterly Report on Form 10-Q for the six months ended June 30, 2016. |
(b) | The measurement period adjustments were recorded in the fourth quarter of 2016 and are primarily related to certain working capital adjustments to reflect facts and circumstances that existed as of the acquisition date, and adjustments to deferred tax liabilities and goodwill. |
The preliminary fair value estimates for the assets acquired and liabilities assumed were based upon preliminary calculations, valuations and assumptions that are subject to change as the Company obtains additional information during the measurement period (up to one year from the acquisition date). The primary areas subject to change relate to the finalization of the working capital components and income taxes.
The acquisition of the Topicals Business broadened the Company's dermatological portfolio. The amount allocated to IPR&D represents an estimate of the fair value of purchased in-process technology for research projects that, as of the closing date of the acquisition, had not reached technological feasibility and had no alternative future use. The fair value of IPR&D of $275.0 million was based on the excess earnings method, which utilizes forecasts of expected cash inflows (including estimates for ongoing costs) and other contributory charges. A discount rate of 12.5% was utilized to discount net cash inflows to present values. IPR&D is accounted for as an indefinite-lived intangible asset and will be subject to impairment testing until completion or abandonment of the projects. Upon successful completion and launch of each product, the Company will make a determination of the estimated useful life of the individual IPR&D asset and amounts will be allocated to product rights and licenses in intangible assets. The acquired IPR&D projects are in various stages of completion and the estimated costs to complete these projects total approximately $65 million , which is expected to be incurred through 2018. There are risks and uncertainties associated with the timely and successful completion of the projects included in IPR&D, and no assurances can be given that the underlying assumptions used to estimate the fair value of IPR&D will not change or the timely completion of each project to commercial success will occur.
The identified intangible assets of $467.0 million are comprised of $454.0 million of product rights and licenses that have a weighted average useful life 14 years and $13.0 million of contract manufacturing agreements that have a weighted average useful life of five years . Significant assumptions utilized in the valuation of identified intangible assets were based on company specific information and projections which are not observable in the market and are thus considered Level 3 measurements as defined by U.S. GAAP. Refer to Note 7 "Financial Instruments and Risk Management" for more information on the U.S. GAAP fair value hierarchy.
The goodwill of $318.6 million arising from the acquisition consisted largely of the value of the employee workforce and the expected value of products to be developed in the future. All of the goodwill was assigned to the North America segment. None of the goodwill recognized in this transaction is currently expected to be deductible for income tax purposes. Acquisition related costs of approximately $3.6 million were incurred during the year ended December 31, 2016 related to this transaction, which were recorded as a component of SG&A in the Consolidated Statements of Operations. The acquisition did
105
Table of Contents
not have a material impact on the Company's results of operations since the acquisition date or on a pro forma basis for the twelve months ended December 31, 2016 and 2015 .
Jai Pharma Limited
On November 20, 2015, the Company completed the acquisition of certain female healthcare businesses from Famy Care Limited (such businesses, " Jai Pharma Limited ") through its wholly owned subsidiary Mylan Laboratories Limited for a cash payment of $750 million plus additional contingent payments of up to $50 million for the filing for approval with, and receipt of approval from, the U.S. Food and Drug Administration ("FDA") of a product under development with Jai Pharma Limited .
In accordance with U.S. GAAP, the Company used the acquisition method of accounting to account for this transaction. Under the acquisition method of accounting, the assets acquired and liabilities assumed in the transaction were recorded at their respective estimated fair values at the acquisition date. The U.S. GAAP purchase price was $711.1 million , which excludes the $50 million paid into escrow at closing that is contingent upon at least one of two former principal shareholders of Jai Pharma Limited continuing to provide consulting services to the acquired business for the two -year post-closing period which is being treated as compensation expense over the service period. The U.S. GAAP purchase price also excludes $7 million of working capital and other adjustments and includes estimated contingent consideration at the date of acquisition of approximately $18 million related to the $50 million contingent payment.
(In millions) | Preliminary Purchase Price Allocation as of November 20, 2015 (a) |
| Measurement Period Adjustments (b) |
| Purchase Price Allocation as of December 31, 2016 (as adjusted) | ||||||
Current assets (excluding inventories) | $ | 25.7 | |
| $ | 2.9 | |
| $ | 28.6 | |
Inventories | 4.9 | |
| - | |
| 4.9 | | |||
Property, plant and equipment | 17.2 | |
| - | |
| 17.2 | | |||
Identified intangible assets | 437.0 | |
| - | |
| 437.0 | | |||
In-process research and development | 98.0 | |
| - | |
| 98.0 | | |||
Goodwill | 317.2 | |
| 6.7 | |
| 323.9 | | |||
Other assets | 0.7 | |
| - | |
| 0.7 | | |||
Total assets acquired | 900.7 | |
| 9.6 | |
| 910.3 | | |||
Current liabilities | (9.1 | ) |
| (5.4 | ) |
| (14.5 | ) | |||
Deferred tax liabilities | (180.5 | ) |
| (4.2 | ) |
| (184.7 | ) | |||
Net assets acquired | $ | 711.1 | |
| $ | - | |
| $ | 711.1 | |
____________
(a) | As previously reported in the Company's Annual Report on Form 10-K for the year ended December 31, 2015, as amended. |
(b) | The measurement period adjustments were recorded in the first and fourth quarters of 2016 and are related to the recognition of goodwill, deferred tax liabilities, current liabilities and adjustments to working capital components to reflect facts and circumstances that existed as of the acquisition date. |
The acquisition of Jai Pharma Limited significantly broadened the Company's women's healthcare portfolio and strengthened its technical and manufacturing capabilities. The amount allocated to IPR&D represents an estimate of the fair value of purchased in-process technology for research projects that, as of the closing date of the acquisition, had not reached technological feasibility and had no alternative future use. The fair value of IPR&D was based on the excess earnings method, which utilizes forecasts of expected cash inflows (including estimates for ongoing costs) and other contributory charges. Disco unt rates of 10% and 11% were utilized to discount net cash inflows to present values. IPR&D is accounted for as an indefinite-lived intangible asset and will be subject to impairment testing until completion or abandonment of the projects. Upon successful completion and launch of each product, the Company will make a determination of the estimated useful life of
106
Table of Contents
the individual IPR&D asset and amounts will be allocated to product rights and licenses in intangible assets. The acquired IPR&D projects are in various stages of completion and the estimated costs to complete these products will total approximately $5 million and are expected to be incurred through 2019. There are risks and uncertainties associated with the timely and successful completion of the projects included in IPR&D, and no assurances can be given that the underlying assumptions used to estimate the fair value of IPR&D will not change or the timely completion of each project to commercial success will occur.
The identified intangible assets of $437.0 million are comprised of product rights and licenses that have weighted average useful lives of nine years . Significant assumptions utilized in the valuation of identified intangible assets were based on company specific information and projections which are not observable in the market and are thus considered Level 3 measurements as defined by U.S. GAAP. Refer to Note 7 "Financial Instruments and Risk Management" for more information on the U.S. GAAP fair value hierarchy. The goodwill of $323.9 million arising from the acquisition consisted largely of the value of the employee workforce and the value of products to be developed in the future. A majority of the goodwill was assigned to Mylan's Rest of World segment. During the year ended December 31, 2016, the Company received approvals from the relevant Indian regulatory authorities to legally merge its wholly owned subsidiary, Jai Pharma Limited, into Mylan Laboratories Limited. The merger resulted in the recognition of a deferred tax asset of $150 million for the tax deductible goodwill in excess of the book goodwill with a corresponding benefit to income tax provision for the year ended December 31, 2016. Acquisition related costs of approximately $8.5 million were incurred during the year ended December 31, 2015, which were recorded as a component of SG&A expense in the Consolidated Statements of Operations . On a pro forma basis, the acquisition did not have a material impact on the Company's results of operations for the year ended December 31, 2015.
EPD Business
On July 13, 2014 , Mylan N.V. , Mylan Inc., and Moon of PA Inc. entered into a definitive agreement with Abbott Laboratories (" Abbott ") to acquire the EPD Business in an all-stock transaction (the "EPD Transaction"). On November 4, 2014 , Mylan N.V. , Mylan Inc., Moon of PA Inc. and Abbott entered into an amended and restated definitive agreement implementing the transaction. The EPD Transaction closed on February 27, 2015 (the " EPD Transaction Closing Date "), after receiving approval from Mylan Inc.'s shareholders on January 29, 2015 . At closing, Abbott transferred the EPD Business to Mylan N.V. , in exchange for 110 million ordinary shares of Mylan N.V. Immediately after the transfer of the EPD Business , Mylan Inc. merged with Moon of PA Inc., an indirect wholly owned subsidiary of Mylan N.V. , with Mylan Inc. becoming an indirect wholly owned subsidiary of Mylan N.V. In addition, Mylan Inc.'s outstanding common stock was exchanged on a one to one basis for Mylan N.V. ordinary shares. Following the EPD Transaction , Mylan N.V. 's corporate seat is located in Amsterdam, the Netherlands , its principal executive offices are located in Hatfield, Hertfordshire, England and Mylan N.V. group's global headquarters are located in Canonsburg, Pennsylvania .
The acquired EPD Business included more than 100 specialty and branded generic pharmaceutical products in five major therapeutic areas and included several patent protected, novel and/or hard-to-manufacture products. As a result of the acquisition, Mylan has significantly expanded and strengthened its product portfolio in Europe, Japan, Canada, Australia and New Zealand.
The purchase price for Mylan N.V. of the acquired EPD Business , which was on a debt-free basis, was $6.31 billion base d on the closing price of Mylan Inc.'s stock as of the EPD Transaction Closing Date , as reported by NASDAQ . At the closing of the EPD Transaction , former shareholders of Mylan Inc. owned approximately 78% of Mylan N.V. 's ordinary shares and certain affiliates of Abbott (the "Abbott Shareholders") owned approximately 22% of Mylan N.V. 's ordinary shares. On the EPD Transaction Closing Date , Mylan N.V. , Abbott and the Abbott Shareholders entered into a shareholder agreement. Following an underwritten public offering of the Abbott Shareholders of a portion of the Mylan N.V. ordinary shares held by them, which offering closed on April 6, 2015, the Abbott Shareholders collectively owned approximately 13% of Mylan N.V. 's outstanding ordinary shares. The Company and Abbott engage in commercial transactions for the supply of products. In addition, Abbott provides certain transitional services to Mylan. The Company believes that these transactions are conducted on commercially reasonable terms.
In accordance with U.S. GAAP, Mylan N.V. used the acquisition method of accounting to account for the EPD Transaction with Mylan Inc. being treated as the accounting acquirer. Under the purchase method of accounting, the assets acquired and liabilities assumed in the EPD Transaction were recorded at their respective estimated fair values at the EPD Transaction Closing Date . The purchase price was finalized during the fourth quarter of 2015. The allocation of the $6.31 billion purchase price (as valued on the EPD Transaction Closing Date ) to the assets acquired and liabilities assumed for the
107
Table of Contents
(In millions) |
| ||
Accounts receivable | $ | 443.8 | |
Inventories | 198.5 | | |
Other current assets | 43.0 | | |
Property, plant and equipment | 140.8 | | |
Identified intangible assets | 4,843.0 | | |
Goodwill | 1,341.0 | | |
Other assets | 41.0 | | |
Total assets acquired | 7,051.1 | | |
Current liabilities | (268.9 | ) | |
Deferred tax liabilities | (421.9 | ) | |
Other non-current liabilities | (54.5 | ) | |
Net assets acquired | $ | 6,305.8 | |
The identified intangible assets of $4.84 billion are comprised of $4.52 billion of product rights and licenses that have weighted average useful lives of 13 years and $320 million of contractual rights that have weighted average useful lives ranging from two to five years . Significant assumptions utilized in the valuation of identified intangible assets were based on company specific information and projections which are not observable in the market and are thus considered Level 3 measurements as defined by U.S. GAAP. The goodwill of $1.34 billion arising from the acquisition primarily relates to the expected synergies of the combined company and the value of the employee workforce. A majority of the goodwill was assigned to the North America segment. Goodwill of $486.4 million is currently expected to be deductible for income tax purposes. Acquisition related costs of approximately $86.1 million were incurred during the year ended December 31, 2015, which were recorded as a component of SG&A in the Consolidated Statements of Operations .
The operating results of the acquired EPD Business have been included in the Company's Consolidated Statements of Operations since February 27, 2015 . The revenues of the acquired EPD Business for the period from the acquisition date to December 31, 2015 were $1.47 billion and the net loss, net of tax, was $62.4 million . The net loss, net of tax, includes the effects of the purchase accounting adjustments and acquisition related costs.
Unaudited Pro Forma Financial Results
| Year Ended December 31, | ||||||||||
(Unaudited, in millions, except per share amounts) | 2016 |
| 2015 |
| 2014 | ||||||
Total revenues | $ | 12,376.0 | |
| $ | 11,930.0 | |
| $ | 9,704.6 | |
Net earnings attributable to Mylan N.V. ordinary shareholders | $ | 560.6 | |
| $ | 604.1 | |
| $ | 694.0 | |
Earnings per ordinary share attributable to Mylan N.V. ordinary shareholders: |
|
|
|
|
| ||||||
Basic | $ | 1.06 | |
| $ | 1.17 | |
| $ | 1.37 | |
Diluted | $ | 1.05 | |
| $ | 1.11 | |
| $ | 1.37 | |
Weighted average ordinary shares outstanding: |
|
|
|
|
| ||||||
Basic | 528.7 | |
| 516.9 | |
| 483.7 | | |||
Diluted | 536.2 | |
| 542.1 | |
| 508.0 | | |||
108
Table of Contents
Other Transactions
On January 9, 2017, the Company announced that it has agreed to acquire the global rights to Cold-EEZE® brand cold remedy line from ProPhase Labs, Inc. for approximately $50 million in cash. The closing of the transaction is subject to the approval of the shareholders of ProPhase Labs, Inc and other customary closing conditions. On February 14, 2017, the Company entered into a joint development and marketing agreement for a respiratory product that will result in approximately $50 million in expense in the first quarter of 2017.
During the year ended December 31, 2016, the Company entered into an agreement to acquire a marketed pharmaceutical product for an upfront payment of approximately $57.9 million , which is included in investing activities in the Consolidated Statements of Cash Flows. The Company accounted for this transaction as an asset acquisition and is amortizing the product over a weighted useful life of five years .
In December 2015, the Company entered into an agreement to acquire certain European intellectual property rights and marketing authorizations. The purchase price was $202.5 million including approximately $2.5 million of transaction costs. The Company accounted for this transaction as an asset acquisition. The Company paid $10 million at the closing of the transaction, which is included in investing in the Consolidated Statements of Cash Flows . The Company paid approximately $165 million during 2016, which is also included in investing in the Consolidated Statements of Cash Flows, and the remaining $25 million is expected to be paid during the first quarter of 2017, subject to certain timing conditions. The asset is being amortized over a useful life of five years .
On April 3, 2015 , the Company and Stichting Preferred Shares Mylan (the "Foundation") entered into a call option agreement (the "Call Option Agreement"). Pursuant to the terms of the Call Option Agreement, Mylan N.V. granted the Foundation a call option (the "Option"), permitting the Foundation to acquire from time-to-time Mylan N.V. preferred shares up to a maximum number equal to the total number of Mylan N.V. ordinary shares issued at such time to the extent such shares are not held by the Foundation. The exercise price of the Option is €0.01 per preferred share. On April 21, 2015, the Company received a letter from the President and Chief Executive Officer of Teva Pharmaceutical Industries Ltd. ("Teva"), containing a non-binding expression of interest from Teva to acquire Mylan for $82 per Mylan ordinary share. On July 23, 2015, in response to Teva's unsolicited expression of interest in acquiring Mylan, the Foundation exercised the Option and acquired 488,388,431 Mylan preferred shares pursuant to the terms of the Call Option Agreement. In compliance with the current statutory arrangement, 25% of the nominal value of the preferred shares, approximately $1.3 million , was paid to Mylan in cash upon issuance. Each Mylan ordinary share and preferred share is entitled to one vote on each matter properly brought before a general meeting of shareholders. On July 27, 2015, Teva announced its entry into an agreement to acquire the Generic Drug Unit of Allergan plc and the withdrawal of its unsolicited, non-binding expression of interest to acquire Mylan. On September 19, 2015, the Foundation requested the redemption of the Mylan preferred shares issued on July 23, 2015, informing Mylan that it was reasonably convinced that the influences that might adversely affect or threaten the strategy, mission, independence, continuity and/or identity of Mylan and its business in a manner that is contrary to the interest of Mylan, its business, and its stakeholders had been sufficiently addressed. Mylan ordinary shareholders approved the redemption of the preferred shares on January 7, 2016 at an extraordinary general meeting of shareholders, and on March 17, 2016 the redemption of the Mylan preferred shares became effective. The Foundation will continue to have the right to exercise the Option in the future in response to a new threat to the interests of Mylan, its businesses and its stakeholders from time to time.
During 2015, the Company entered into agreements with multiple counterparties to acquire certain marketed pharmaceutical products for upfront payments totaling approximately $360.8 million , which were paid during the year ended December 31, 2015 and are included in investing activities in the Consolidated Statements of Cash Flows . The Company will be subject to potential future sales and other contingent milestone payments under the terms of one of the agreements.
4. | Balance Sheet Components |
Selected balance sheet components consist of the following:
Accounts receivable, net
(In millions) | December 31, 2016 |
| December 31, 2015 | ||||
Trade receivables, net | $ | 3,015.4 | |
| $ | 2,434.0 | |
Other receivables | 295.5 | |
| 255.1 | | ||
Accounts receivable, net | $ | 3,310.9 | |
| $ | 2,689.1 | |
109
Table of Contents
Trade receivables, net includes certain sales allowances totaling $2.05 billion and $1.84 billion at December 31, 2016 and 2015 , respectively. See Note 2 Summary of Significant Accounting Policies for further discussion of such allowances. Total allowances for doubtful accounts were $59.0 million and $33.6 million at December 31, 2016 and 2015 , respectively. Mylan performs ongoing credit evaluations of its customers and generally does not require collateral. Approximately 45% and 42% of the accounts receivable balances represent amounts due from three customers at December 31, 2016 and 2015 , respectively.
Inventories
(In millions) | December 31, 2016 |
| December 31, 2015 | ||||
Raw materials | $ | 783.4 | |
| $ | 592.4 | |
Work in process | 436.0 | |
| 387.0 | | ||
Finished goods | 1,237.0 | |
| 971.6 | | ||
Inventories | $ | 2,456.4 | |
| $ | 1,951.0 | |
Inventory reserves totaled $174.6 million and $157.3 million at December 31, 2016 and 2015 , respectively. Included as a component of cost of sales is expense related to the net realizable value of inventories of $195.7 million , $221.4 million and $182.5 million for the years ended December 31, 2016 , 2015 and 2014, respectively.
Prepaid and other current assets
(In millions) | December 31, 2016 |
| December 31, 2015 | ||||
Prepaid expenses | $ | 138.3 | |
| $ | 137.3 | |
Restricted cash | 148.1 | |
| 106.6 | | ||
Available-for-sale securities | 83.7 | |
| 54.0 | | ||
Fair value of financial instruments | 62.2 | |
| 64.7 | | ||
Momenta collaboration prepaid expenses | 30.8 | |
| - | | ||
Trading securities | 29.6 | |
| 22.8 | | ||
Other current assets | 263.7 | |
| 211.2 | | ||
Prepaid expenses and other current assets | $ | 756.4 | |
| $ | 596.6 | |
Prepaid expenses consists primarily of prepaid rent, insurance and other individually insignificant items. At December 31, 2016, restricted cash includes $50 million paid into escrow for contingent consideration related to the acquisition of the Topicals Business.
Property, plant and equipment, net
(In millions) | December 31, 2016 |
| December 31, 2015 | ||||
Machinery and equipment | $ | 2,227.9 | |
| $ | 1,928.4 | |
Buildings and improvements | 1,106.5 | |
| 950.6 | | ||
Construction in progress | 328.8 | |
| 290.5 | | ||
Land and improvements | 144.7 | |
| 124.5 | | ||
Gross property, plant and equipment | 3,807.9 | |
| 3,294.0 | | ||
Accumulated depreciation | 1,485.7 | |
| 1,310.1 | | ||
Property, plant and equipment, net | $ | 2,322.2 | |
| $ | 1,983.9 | |
Capitalized software costs included on our Consolidated Balance Sheets were $145.4 million and $130.0 million , net of accumulated depreciation, at December 31, 2016 and 2015 , respectively. The Company periodically reviews the original estimated useful lives of assets and makes adjustments when appropriate. Depreciation expense was approximately $259.4 million , $186.1 million and $172.8 million for the years ended December 31, 2016 , 2015 and 2014 , respectively.
110
Table of Contents
Other assets
(In millions) | December 31, 2016 |
| December 31, 2015 | ||||
Equity method investments, clean energy investments | $ | 320.6 | |
| $ | 379.3 | |
Equity method investments, Sagent Agila | 75.8 | |
| 96.2 | | ||
Restricted cash | - | |
| 100.0 | | ||
Other long-term assets | 172.2 | |
| 176.0 | | ||
Other assets | $ | 568.6 | |
| $ | 751.5 | |
During the year ended December 31, 2016, restricted cash of $100 million principally related to amounts deposited in escrow for potential contingent consideration payments related to the Company's acquisition of Agila Specialties ("Agila") was reclassified to prepaid expenses and other current assets or released from restrictions, in conjunction with the Strides Settlement, as discussed in Note 14 Commitments .
Trade accounts payable
(In millions) | December 31, 2016 |
| December 31, 2015 | ||||
Trade accounts payable | $ | 939.5 | |
| $ | 717.5 | |
Other payables | 408.6 | |
| 392.1 | | ||
Trade accounts payable | $ | 1,348.1 | |
| $ | 1,109.6 | |
Other current liabilities
(In millions) | December 31, 2016 |
| December 31, 2015 | ||||
Accrued sales allowances | $ | 809.0 | |
| $ | 681.8 | |
Payroll and employee benefit plan accruals | 409.8 | |
| 367.9 | | ||
Legal and professional accruals, including litigation accruals | 720.4 | |
| 122.6 | | ||
Contingent consideration | 256.9 | |
| 35.0 | | ||
Restructuring | 138.6 | |
| 14.8 | | ||
Compulsory acquisition proceeding | 70.2 | |
| - | | ||
Equity method investments, clean energy investments | 64.7 | |
| 62.3 | | ||
Accrued interest | 41.0 | |
| 25.1 | | ||
Fair value of financial instruments | 15.3 | |
| 19.8 | | ||
Other | 732.6 | |
| 512.6 | | ||
Other current liabilities | $ | 3,258.5 | |
| $ | 1,841.9 | |
Included in legal and professional accruals, including litigation accruals at December 31, 2016 was $465 million for a settlement with the U.S. Department of Justice and other government agencies related to the classification of the EpiPen® Auto-Injector and EpiPen Jr® Auto-Injector (collectively, " EpiPen® Auto-Injector ") for purposes of the Medicaid Drug Rebate Program (the "Medicaid Drug Rebate Program Settlement") and approximately $96.5 million related to the Modafinil antitrust litigation matter, as discussed further in Note 18 Litigation .
At the close of the Meda transaction, the Company recorded a current liability of approximately $431.0 million related to the purchase of the non-tendered shares of Meda pursuant to the compulsory acquisition proceeding. In conjunction with the November Offer, Meda shareholders, holding approximately 19 million of the outstanding non-tendered shares, tendered their shares to the Company and in the fourth quarter of 2016, the Company paid approximately $330.3 million for the tendered Meda shares. At December 31, 2016 , the Company's current liability associated with the compulsory acquisition proceeding was approximately $70.2 million . Included in other current liabilities at December 31, 2016 was approximately $316.9 million of accrued expenses assumed from Meda. Refer to Note 16 Restructuring for further information regarding the $138.6 million recorded related to restructuring costs at December 31, 2016.
111
Table of Contents
Other long-term obligations
(In millions) | December 31, 2016 |
| December 31, 2015 | ||||
Employee benefit liabilities | $ | 396.7 | |
| $ | 118.1 | |
Equity method investments, clean energy investments | 302.3 | |
| 357.0 | | ||
Contingent consideration | 307.7 | |
| 491.4 | | ||
Tax contingencies | 239.3 | |
| 247.2 | | ||
Other | 112.6 | |
| 152.3 | | ||
Other long-term obligations | $ | 1,358.6 | |
| $ | 1,366.0 | |
5. | Equity Method Investments |
(In millions) | December 31, 2016 |
| December 31, 2015 | ||||
Clean energy investments: |
|
|
| ||||
Other assets | $ | 320.6 | |
| $ | 379.3 | |
Total liabilities | 367.0 | |
| 419.3 | | ||
Included in other current liabilities | 64.7 | |
| 62.3 | | ||
Included in other long-term obligations | 302.3 | |
| 357.0 | | ||
Sagent Agila: |
|
|
| ||||
Other assets | $ | 75.8 | |
| $ | 96.2 | |
Summarized financial information, in the aggregate, of the Company's equity method investments on a 100% basis as of December 31, 2016 and 2015 and for the years ended December 31, 2016 , 2015 and 2014 are as follows:
(In millions) | December 31, 2016 |
| December 31, 2015 | ||||
Current assets | $ | 75.6 | |
| $ | 97.6 | |
Noncurrent assets | 12.3 | |
| 14.6 | | ||
Total assets | 87.9 | |
| 112.2 | | ||
Current liabilities | 50.7 | |
| 74.9 | | ||
Noncurrent liabilities | 2.6 | |
| 2.6 | | ||
Total liabilities | 53.3 | |
| 77.5 | | ||
Net assets | $ | 34.6 | |
| $ | 34.7 | |
(In millions) | Year Ended December 31, | ||||||||||
| 2016 |
| 2015 |
| 2014 | ||||||
Total revenues | $ | 589.4 | |
| $ | 774.6 | |
| $ | 536.8 | |
Gross (loss) profit | (13.2 | ) |
| (11.3 | ) |
| (7.8 | ) | |||
Operating and non-operating expense | 22.2 | |
| 25.6 | |
| 16.9 | | |||
Net loss | $ | (35.4 | ) |
| $ | (36.9 | ) |
| $ | (24.7 | ) |
112
Table of Contents
The Company's net losses from equity method investments includes amortization expense related to the excess of the cost basis of the Company's investment to the underlying assets of each individual investee. For the years ended December 31, 2016 , 2015 and 2014 , the Company's share of the net loss of the equity method investments was $112.8 million , $105.1 million and $91.4 million , respectively, which was recognized as a component of other expense, net in the Consolidated Statements of Operations. The Company recognizes the income tax credits and benefits from the clean energy investments as part of its provision for income taxes.
6. | Goodwill and Other Intangible Assets |
The changes in the carrying amount of goodwill for the years ended December 31, 2016 and 2015 are as follows:
(In millions) | North America |
| Europe |
| Rest of World |
| Total | ||||||||
Balance at December 31, 2014: |
|
|
|
|
|
|
| ||||||||
Goodwill | $ | 1,815.9 | |
| $ | 1,123.5 | |
| $ | 1,494.9 | |
| $ | 4,434.3 | |
Accumulated impairment losses | (385.0 | ) |
| - | |
| - | |
| (385.0 | ) | ||||
| 1,430.9 | |
| 1,123.5 | |
| 1,494.9 | |
| 4,049.3 | | ||||
Acquisitions (1) | 1,450.3 | |
| 15.7 | |
| 192.2 | |
| 1,658.2 | | ||||
Reclassifications | - | |
| 10.1 | |
| (10.1 | ) |
| - | | ||||
Foreign currency translation | (87.5 | ) |
| (148.8 | ) |
| (91.1 | ) |
| (327.4 | ) | ||||
| 2,793.7 | |
| 1,000.5 | |
| 1,585.9 | |
| 5,380.1 | | ||||
Balance at December 31, 2015: |
|
|
|
|
|
|
| ||||||||
Goodwill | 3,178.7 | |
| 1,000.5 | |
| 1,585.9 | |
| 5,765.1 | | ||||
Accumulated impairment losses | (385.0 | ) |
| - | |
| - | |
| (385.0 | ) | ||||
| 2,793.7 | |
| 1,000.5 | |
| 1,585.9 | |
| 5,380.1 | | ||||
Acquisitions and measurement period adjustments (1) | 818.6 | |
| 2,993.0 | |
| 190.6 | |
| 4,002.2 | | ||||
Foreign currency translation | (6.9 | ) |
| (134.4 | ) |
| (9.1 | ) |
| (150.4 | ) | ||||
| 3,605.4 | |
| 3,859.1 | |
| 1,767.4 | |
| 9,231.9 | | ||||
Balance at December 31, 2016: |
|
|
|
|
|
|
| ||||||||
Goodwill | 3,990.4 | |
| 3,859.1 | |
| 1,767.4 | |
| 9,616.9 | | ||||
Accumulated impairment losses | (385.0 | ) |
| - | |
| - | |
| (385.0 | ) | ||||
| $ | 3,605.4 | |
| $ | 3,859.1 | |
| $ | 1,767.4 | |
| $ | 9,231.9 | |
____________
(1) | In 2015, includes goodwill related to the acquisition of the EPD Business and Jai Pharma Limited totaling approximately $1.34 billion and $317.2 million , respectively. In 2016, includes measurement period adjustments related to the acquisition of Jai Pharma Limited and the recognition of goodwill related to the acquisitions of Meda and the Topicals Business totaling approximately $6.7 million , $3.68 billion and $318.6 million , respectively. |
113
Table of Contents
(In millions) | Weighted Average Life (Years) |
| Original Cost |
| Accumulated Amortization |
| Net Book Value | ||||||
December 31, 2016 |
|
|
|
|
|
|
| ||||||
Amortized intangible assets: |
|
|
|
|
|
|
| ||||||
Product rights and licenses | 15 |
| $ | 16,968.4 | |
| $ | 3,585.7 | |
| $ | 13,382.7 | |
Patents and technologies | 20 |
| 116.6 | |
| 108.5 | |
| 8.1 | | |||
Other (1) | 6 |
| 465.9 | |
| 330.0 | |
| 135.9 | | |||
|
|
| 17,550.9 | |
| 4,024.2 | |
| 13,526.7 | | |||
In-process research and development |
|
| 921.1 | |
| - | |
| 921.1 | | |||
|
|
| $ | 18,472.0 | |
| $ | 4,024.2 | |
| $ | 14,447.8 | |
December 31, 2015 |
|
|
|
|
|
|
| ||||||
Amortized intangible assets: |
|
|
|
|
|
|
| ||||||
Product rights and licenses | 11 |
| $ | 8,848.6 | |
| $ | 2,652.7 | |
| $ | 6,195.9 | |
Patents and technologies | 20 |
| 116.6 | |
| 103.8 | |
| 12.8 | | |||
Other (1) | 6 |
| 465.3 | |
| 189.8 | |
| 275.5 | | |||
|
|
| 9,430.5 | |
| 2,946.3 | |
| 6,484.2 | | |||
In-process research and development |
|
| 737.7 | |
| - | |
| 737.7 | | |||
|
|
| $ | 10,168.2 | |
| $ | 2,946.3 | |
| $ | 7,221.9 | |
____________
(1) | Other intangibles consist principally of customer lists, contractual rights and other contracts. |
During the year ended December 31, 2016, the Company acquired product rights and licenses from Meda and the Topicals Business totaling approximately $8.06 billion and $454.0 million , respectively. The Company also acquired IPR&D totaling approximately $275.0 million from the Topicals Business. During the years ended December 31, 2016 and 2015 , approximately $32.6 million and $59.4 million , respectively, was reclassified from acquired IPR&D to product rights and licenses. During the years ended December 31, 2016 and 2015, the Company acquired approximately $341 million and $425 million , respectively, for products rights and licenses related to certain marketed pharmaceutical products with multiple counterparties, as further described in Note 3 Acquisitions and Other Transactions .
114
Table of Contents
(In millions) | December 31, 2016 |
| December 31, 2015 | ||||
Central Nervous System and Anesthesia | $ | 2,172.0 | |
| $ | 949.8 | |
Dermatology | 2,070.2 | |
| 52.9 | | ||
Gastroenterology | 1,906.2 | |
| 1,289.9 | | ||
Diabetes and Metabolism | 1,395.7 | |
| 720.1 | | ||
Cardiovascular | 1,718.0 | |
| 1,105.5 | | ||
Respiratory and Allergy | 1,691.0 | |
| 209.1 | | ||
Infectious Disease | 490.6 | |
| 368.7 | | ||
Oncology | 413.4 | |
| 169.3 | | ||
Women's Healthcare | 371.4 | |
| 432.4 | | ||
Immunology | 284.9 | |
| 322.7 | | ||
Other (1) | 869.3 | |
| 575.5 | | ||
| $ | 13,382.7 | |
| $ | 6,195.9 | |
(1) | Other consists of numerous therapeutic classes, none of which individually exceeds 5% of total product rights and licenses. |
| Year ended December 31, | ||||||||||
(In millions) | 2016 |
| 2015 |
| 2014 | ||||||
Intangible asset amortization expense | $ | 1,195.3 | |
| $ | 814.7 | |
| $ | 366.1 | |
Intangible asset impairment charges | 68.3 | |
| 31.3 | |
| 27.7 | | |||
Total intangible asset amortization expense (including impairment charges) | $ | 1,263.6 | |
| $ | 846.0 | |
| $ | 393.8 | |
Indefinite-lived intangibles, such as the Company's IPR&D assets, are tested at least annually for impairment, but they may be tested whenever certain impairment indicators are present. Impairment is determined to exist when the fair value is less than the carrying value of the assets being tested. In addition, the Company monitors long-lived intangible assets for potential triggering events or changes in circumstances that would indicate that the carrying amount of the asset may not be recoverable. During the year ended December 31, 2016 , the Company recorded impairment charges on certain product rights and licenses and IPR&D assets of approximately $18.4 million and $49.9 million , respectively, which were recorded as components of amortization expense. During the years ended December 31, 2016 and 2015, the Company revised its estimated useful lives on certain intangible assets. During the year ended December 31, 2014 , the Company recorded impairment charges of approximately $10 million related to product rights and licenses, which was recorded as a component of amortization expense.
The Company performed its annual impairment review of certain IPR&D assets during the third and fourth quarters of 2016 . This review of IPR&D assets principally related to assets acquired as part of the Jai Pharma Limited acquisition in 2015, the Agila acquisition in December 2013, the respiratory delivery platform acquisition in December 2011 and the Bioniche Pharma acquisition in September 2010. The impairment charges recorded resulted from the Company's estimate of the fair value of the assets, which was based upon updated forecasts and commercial development plans, compared with the assigned fair values at the acquisition date. The fair value was determined based upon detailed valuations employing the income approach which utilized Level 3 inputs, as defined in Note 7 Financial Instruments and Risk Management . The fair value of IPR&D was calculated as the present value of the estimated future net cash flows using a market rate of return. The assumptions inherent in the estimated future cash flows include, among other things, the impact of changes to the development programs, the projected development and regulatory time frames and the current competitive environment. Discount rates ranging between 8.5% and 11.9% were utilized in the valuations performed during the third and fourth quarters of 2016 .
115
Table of Contents
Discount rates ranging between 9.8% and 11.8% were utilized in valuation during the third and fourth quarters of 2015 . Changes to any of the Company's assumptions may result in a future reduction to the estimated fair value of the IPR&D asset.
(In millions) |
| ||
2017 | $ | 1,243 | |
2018 | 1,206 | | |
2019 | 1,106 | | |
2020 | 1,014 | | |
2021 | 923 | | |
7. | Financial Instruments and Risk Management |
The Company is exposed to certain financial risks relating to its ongoing business operations. The primary financial risks that are managed by using derivative instruments are foreign currency risk, interest rate risk and equity risk.
Foreign Currency Risk Management
In order to manage foreign currency risk, the Company enters into foreign exchange forward contracts to mitigate risk associated with changes in spot exchange rates of mainly non-functional currency denominated assets or liabilities. The foreign exchange forward contracts are measured at fair value and reported as current assets or current liabilities on the Consolidated Balance Sheets . Any gains or losses on the foreign exchange forward contracts are recognized in earnings in the period incurred in the Consolidated Statements of Operations.
During 2016, in order to economically hedge the foreign currency exposure associated with the expected payment of the Swedish krona-denominated cash portion of the purchase price of the Offer, the Company entered into a series of non-designated foreign exchange forward and option contracts with a total notional amount of 45.2kr billion . During the year ended December 31, 2016, the Company recognized losses of $128.6 million for the changes in fair value related to these contracts which is included in other expense, net in the Consolidated Statements of Operations . These contracts were settled in 2016. As of December 31, 2016, the Company has not hedged the foreign currency risk associated with the remaining liability for the compulsory acquisition proceeding of approximately $70.2 million .
The Company has also entered into forward contracts to hedge forecasted foreign currency denominated sales from certain international subsidiaries. These contracts are designated as cash flow hedges to manage foreign currency transaction risk and are measured at fair value and reported as current assets or current liabilities on the Consolidated Balance Sheets . Any changes in fair value are included in earnings or deferred through accumulated other comprehensive earnings ("AOCE"), depending on the nature and effectiveness of the offset.
Following the acquisition of Meda, the Company designated certain Euro borrowings as a hedge of its investment in certain Euro-functional currency subsidiaries in order to manage the foreign currency translation risk. The notional amount of the net investment hedges was €288 million and consisted primarily of Euro denominated debt which had a maturity date in August 2017. Borrowings designated as net investment hedges are marked to market using the current spot exchange rate as of the end of the period, with gains and losses included in the foreign currency translation component of AOCE until the sale or substantial liquidation of the underlying net investments. In the fourth quarter of 2016, the Company repaid the related Euro borrowings, and as such, the hedging designation was terminated. The Company recorded no ineffectiveness from its net investment hedges for the year ended December 31, 2016.
During the fourth quarter of 2016, the Company issued approximately €3.0 billion of Euro Notes, as defined in Note 8 Debt . During the year ended December 31, 2016, the Company recognized approximately $32.0 million of mark-to-market gains in other expense, net in the Consolidated Statements of Operations, related to the Euro Notes. During this time, the Company was partially managing the related foreign exchange risk of the Euro Notes through certain Euro denominated financial assets. In the first quarter of 2017, the Euro Notes were designated as a hedge of its investment in certain Euro-functional currency subsidiaries in order to manage the foreign currency translation risk.
116
Table of Contents
Interest Rate Risk Management
The Company enters into interest rate swaps in order to manage interest rate risk associated with the Company's fixed- and floating-rate debt. These derivative instruments are measured at fair value and reported as current assets or current liabilities on the Consolidated Balance Sheets .
Cash Flow Hedging Relationships
The Company's interest rate swaps designated as cash flow hedges fix the interest rate on a portion of the Company's variable-rate debt or hedge part of the Company's interest rate exposure associated with the variability in the future cash flows attributable to changes in interest rates. Any changes in fair value are included in earnings or deferred through AOCE, depending on the nature and effectiveness of the offset. Any ineffectiveness in a cash flow hedging relationship is recognized immediately in earnings in the Consolidated Statements of Operations .
Following the acquisition of Meda, the Company designated certain interest rate swaps with a notional amount of €750 million as cash flow hedges. The maturity date of these swaps was June 2017. In the fourth quarter of 2016, the Company repaid the related debt instrument and terminated these swaps.
In anticipation of issuing fixed-rate debt, the Company may use treasury rate locks or forward starting interest rate swaps that are designated as cash flow hedges. In September 2015, the Company entered into a series of forward starting swaps to hedge against changes in interest rates related to future debt issuances. These swaps were designated as cash flow hedges of expected future issuances of long-term bonds. The Company executed $500 million of notional value swaps with an effective date of June 2016 and an additional $500 million of notional value swaps with an effective date of November 2016. Both sets of swaps had a maturity of ten years . As discussed further in Note 8 Debt , during the second quarter of 2016, the Company issued $2.25 billion in an aggregate principal amount of 3.950% Senior Notes due 2026 and the Company terminated these swaps. As a result of this termination, the Company recorded losses of $64.9 million in AOCE, which are being amortized over the life of the 3.950% Senior Notes due 2026. In addition, during the second quarter of 2016, approximately $2.1 million of hedge ineffectiveness related to these forward starting swaps was recorded in interest expense on the Consolidated Statements of Operations .
In August 2014, the Company entered into a series of forward starting swaps to hedge against changes in interest rates that could impact future debt issuances. These swaps were designed as cash flow hedges of expected future issuances of long-term bonds. The Company executed $575 million of notional value swaps with an effective date of September 2015. These swaps had a maturity of ten years . In September 2015, the Company terminated these swaps, and as a result of this termination, the Company has recognized losses, net of tax, of approximately $22.4 million , which were recorded in AOCE. During the fourth quarter of 2015, the Company issued $500 million aggregate principal amount of 3.000% Senior Notes due December 2018 and $500 million aggregate principal amount of 3.750% Senior Notes due December 2020. The Company recognized approximately $11.8 million of the loss, net of tax, previously recorded to AOCE in other expense, net during the fourth quarter of 2015. The remaining loss, net of tax, of approximately $10.6 million will be amortized over the remaining lives of the 3.000% Senior Notes due December 2018 and 3.750% Senior Notes due December 2020.
In April 2013, the Company entered into a series of forward starting swaps to hedge against changes in interest rates that could impact future debt issuances. These swaps were designated as cash flow hedges of expected future issuances of long-term bonds. The Company executed $1 billion of notional value swaps with an effective date of August 2015. These swaps had a maturity of ten years . In August 2015, the Company terminated these swaps. As a result of this termination, the Company incurred losses, net of tax, of approximately $32.9 million , which were recorded in AOCE in the third quarter of 2015. During the fourth quarter of 2015, the balance in AOCE was recognized in other expense, net as the forecasted transaction was no longer probable of occurring.
In December 2014, the Company terminated certain forward starting swaps designated as cash flow hedges of expected future issuances of long-term bonds. As a result of this termination, the Company has recognized a loss of approximately $14.6 million during the year ended December 31, 2014.
Fair Value Hedging Relationships
The Company's interest rate swaps designated as fair value hedges convert the fixed rate on a portion of the Company's fixed-rate senior notes to a variable rate. Any changes in the fair value of these derivative instruments, as well as the offsetting change in fair value of the portion of the fixed-rate debt being hedged, is included in interest expense. In November 2014, in conjunction with the redemption of the Company's 6.000% Senior Notes due 2018, the Company's counterparties
117
Table of Contents
exercised their right to terminate certain swaps that had been designated as a fair value hedge on a portion of the Company's 6.000% Senior Notes due 2018. As a result, during the year ended December 31, 2014, the Company received a payment of approximately $15 million related to the swap termination, which was recognized in other expense, net .
In June 2013, the Company entered into interest rate swaps with a notional value of $500 million that were designated as hedges of the Company's 1.800% Senior Notes due 2016. In October 2014, the Company terminated these fair value swaps. In December 2013, the Company entered into interest rate swaps with a notional value of $750 million that were designated as hedges of the Company's 3.125% Senior Notes due 2023. The variable rate was 1.30% at December 31, 2016 . The total notional amount of the Company's interest rate swaps on fixed-rate debt was $750 million as of December 31, 2016 and 2015 .
Equity Risk Management
In connection with the consummation of the EPD Transaction, Mylan Inc. and Mylan N.V. executed a supplemental indenture that amended the indenture governing the Cash Convertible Notes so that, among other things, all relevant determinations for purposes of the cash conversion rights to which holders were entitled from time-to-time in accordance with such indenture were made by reference to the Mylan N.V. ordinary shares. As adjusted in connection with the consummation of the EPD Transaction, holders could convert their Cash Convertible Notes subject to certain conversion provisions determined by a) the market price of Mylan N.V. 's ordinary shares, b) specified distributions to common shareholders, c) a fundamental change, as defined in the indenture governing the Cash Convertible Notes, or d) certain time periods specified in the indenture governing the Cash Convertible Notes. The conversion feature could only be settled in cash and, therefore, it was bifurcated from the Cash Convertible Notes and treated as a separate derivative instrument. In order to offset the cash flow risk associated with the cash conversion feature, the Company entered into a convertible note hedge with certain counterparties. In connection with the consummation of the EPD Transaction, the terms of the convertible note hedge were adjusted so that the cash settlement value would be based on Mylan N.V. ordinary shares. Both the cash conversion feature and the purchased convertible note hedge were measured at fair value with gains and losses recorded in the Company's Consolidated Statements of Operations . The Company's convertible note hedge on its Cash Convertible Notes, which was entered into in order to offset the cash flow risk associated with the cash conversion feature of the Cash Convertible Notes, was settled in conjunction with the maturity and full redemption of the Cash Convertible Notes on September 15, 2015.
Also, in conjunction with the issuance of the Cash Convertible Notes, Mylan Inc. entered into several warrant transactions with certain counterparties. In connection with the consummation of the EPD Transaction, the terms of the warrants were also adjusted so that the Company was able settle the obligations under the warrant transaction by delivering Mylan N.V. ordinary shares. Settlement of the warrants occurred during the second quarter of 2016. The warrants met the definition of derivatives; however, because these instruments had been determined to be indexed to the Company's own ordinary shares, and were recorded in shareholders' equity in the Company's Consolidated Balance Sheets , the instruments were exempt from the scope of U.S. GAAP guidance regarding accounting for derivative instruments and hedging activities and were not subject to the fair value provisions set forth therein.
The Company regularly reviews the creditworthiness of its financial counterparties and does not expect to incur a significant loss from failure of any counterparties to perform under any agreements. The Company is not subject to any obligations to post collateral under derivative instrument contracts. Certain derivative instrument contracts entered into by the Company are governed by master agreements, which contain credit-risk-related contingent features that would allow the counterparties to terminate the contracts early and request immediate payment should the Company trigger an event of default on other specified borrowings. The Company records all derivative instruments on a gross basis in the Consolidated Balance Sheets . Accordingly, there are no offsetting amounts that net assets against liabilities.
Fair Values of Derivative Instruments
Derivatives Designated as Hedging Instruments
| Asset Derivatives | ||||||||||
| December 31, 2016 |
| December 31, 2015 | ||||||||
(In millions) | Balance Sheet Location |
| Fair Value |
| Balance Sheet Location |
| Fair Value | ||||
Interest rate swaps | Prepaid expenses and other current assets |
| $ | 26.2 | |
| Prepaid expenses and other current assets |
| $ | 36.3 | |
Foreign currency forward contracts | Prepaid expenses and other current assets |
| 21.9 | |
| Prepaid expenses and other current assets |
| 8.4 | | ||
Total |
| $ | 48.1 | |
|
|
| $ | 44.7 | | |
118
Table of Contents
| Liability Derivatives | ||||||||||
| December 31, 2016 |
| December 31, 2015 | ||||||||
(In millions) | Balance Sheet Location |
| Fair Value |
| Balance Sheet Location |
| Fair Value | ||||
Interest rate swaps | Other current liabilities |
| $ | - | |
| Other current liabilities |
| $ | 10.5 | |
Total |
| $ | - | |
|
|
| $ | 10.5 | | |
Fair Values of Derivative Instruments
Derivatives Not Designated as Hedging Instruments
| Asset Derivatives | ||||||||||
| December 31, 2016 |
| December 31, 2015 | ||||||||
(In millions) | Balance Sheet Location |
| Fair Value |
| Balance Sheet Location |
| Fair Value | ||||
Foreign currency forward contracts | Prepaid expenses and other current assets |
| $ | 14.0 | |
| Prepaid expenses and other current assets |
| $ | 20.0 | |
Total |
| $ | 14.0 | |
|
|
| $ | 20.0 | | |
| Liability Derivatives | ||||||||||
| December 31, 2016 |
| December 31, 2015 | ||||||||
(In millions) | Balance Sheet Location |
| Fair Value |
| Balance Sheet Location |
| Fair Value | ||||
Foreign currency forward contracts | Other current liabilities |
| $ | 15.3 | |
| Other current liabilities |
| $ | 9.3 | |
Total |
| $ | 15.3 | |
|
|
| $ | 9.3 | | |
The Effect of Derivative Instruments on the Consolidated Statements of Operations
Derivatives in Fair Value Hedging Relationships
| Location of (Loss) or Gain Recognized in Earnings on Derivatives | Amount of (Loss) or Gain Recognized in Earnings on Derivatives | ||||||||||
| Year Ended December 31, | |||||||||||
(In millions) | 2016 |
| 2015 |
| 2014 | |||||||
Interest rate swaps | Interest expense | $ | (10.0 | ) |
| $ | 5.9 | |
| $ | 35.6 | |
Total | $ | (10.0 | ) |
| $ | 5.9 | |
| $ | 35.6 | | |
| Location of Gain or (Loss) Recognized in Earnings on Hedged Items | Amount of Gain or (Loss) Recognized in Earnings on Hedging Items | ||||||||||
| Year Ended December 31, | |||||||||||
(In millions) | 2016 |
| 2015 |
| 2014 | |||||||
2023 Senior Notes (3.125% coupon) | Interest expense | $ | 10.0 | |
| $ | (5.9 | ) |
| $ | (45.7 | ) |
2016 Senior Notes (1.800% coupon) | Interest expense | - | |
| - | |
| (0.9 | ) | |||
2018 Senior Notes (6.000% coupon) | Other expense, net | - | |
| - | |
| 15.0 | | |||
2018 Senior Notes (6.000% coupon) | Interest expense | - | |
| - | |
| 4.6 | | |||
Total | $ | 10.0 | |
| $ | (5.9 | ) |
| $ | (27.0 | ) | |
119
Table of Contents
The Effect of Derivative Instruments on the Consolidated Statements of Comprehensive Earnings
Derivatives in Net Investment Hedging Relationships
| Amount of Loss Recognized in AOCE (Net of Tax) on Derivatives (Effective Portion) | ||||||||||
| Year Ended December 31, | ||||||||||
(In millions) | 2016 |
| 2015 |
| 2014 | ||||||
Foreign currency borrowings and forward contracts | $ | (1.4 | ) |
| $ | - | |
| $ | - | |
Total | $ | (1.4 | ) |
| $ | - | |
| $ | - | |
The Effect of Derivative Instruments on the Consolidated Statements of Comprehensive Earnings
Derivatives in Cash Flow Hedging Relationships
| Amount of (Loss) or Gain Recognized in AOCE (Net of Tax) on Derivatives (Effective Portion) | ||||||||||
| Year Ended December 31, | ||||||||||
(In millions) | 2016 |
| 2015 |
| 2014 | ||||||
Foreign currency forward contracts | $ | (27.5 | ) |
| $ | (44.5 | ) |
| $ | (26.8 | ) |
Interest rate swaps | (38.7 | ) |
| 13.5 | |
| (135.1 | ) | |||
Total | $ | (66.2 | ) |
| $ | (31.0 | ) |
| $ | (161.9 | ) |
The Effect of Derivative Instruments on the Consolidated Statements of Operations
Derivatives in Cash Flow Hedging Relationships
| Location of Loss Reclassified from AOCE into Earnings (Effective Portion) | Amount of Loss Reclassified from AOCE into Earnings (Effective Portion) | ||||||||||
| Year Ended December 31, | |||||||||||
(In millions) | 2016 |
| 2015 |
| 2014 | |||||||
Foreign currency forward contracts | Net sales | $ | (44.3 | ) |
| $ | (40.3 | ) |
| $ | (47.9 | ) |
Interest rate swaps | Interest expense | (8.7 | ) |
| (0.8 | ) |
| (0.6 | ) | |||
Total | $ | (53.0 | ) |
| $ | (41.1 | ) |
| $ | (48.5 | ) | |
| Location of Gain Excluded from the Assessment of Hedge Effectiveness | Amount of Gain Excluded from the Assessment of Hedge Effectiveness | ||||||||||
| Year Ended December 31, | |||||||||||
(In millions) | 2016 |
| 2015 |
| 2014 | |||||||
Foreign currency forward contracts | Other expense, net | $ | 33.5 | |
| $ | 45.1 | |
| $ | 82.3 | |
Total | $ | 33.5 | |
| $ | 45.1 | |
| $ | 82.3 | | |
At December 31, 2016 , the Company expects that approximately $27.8 million of pre-tax net losses on cash flow hedges will be reclassified from AOCE into earnings during the next twelve months .
120
Table of Contents
The Effect of Derivative Instruments on the Consolidated Statements of Operations
Derivatives Not Designated as Hedging Instruments
| Location of (Loss) or Gain Recognized in Earnings on Derivatives | Amount of (Loss) or Gain Recognized in Earnings on Derivatives | ||||||||||
| Year Ended December 31, | |||||||||||
(In millions) | 2016 |
| 2015 |
| 2014 | |||||||
Foreign currency option and forward contracts | Other expense, net | $ | (104.5 | ) |
| $ | 41.7 | |
| $ | (78.3 | ) |
Interest rate swaps | Other expense, net | - | |
| (71.2 | ) |
| - | | |||
Cash conversion feature of Cash Convertible Notes | Other expense, net | - | |
| 1,853.5 | |
| (550.2 | ) | |||
Purchased cash convertible note hedge | Other expense, net | - | |
| (1,853.5 | ) |
| 550.2 | | |||
Total | $ | (104.5 | ) |
| $ | (29.5 | ) |
| $ | (78.3 | ) | |
Fair Value Measurement
Fair value is based on the price that would be received from the sale of an identical asset or paid to transfer an identical liability in an orderly transaction between market participants at the measurement date. In order to increase consistency and comparability in fair value measurements, a fair value hierarchy has been established that prioritizes observable and unobservable inputs used to measure fair value into three broad levels, which are described below:
Level 1: | Quoted prices (unadjusted) in active markets that are accessible at the measurement date for identical assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs. |
Level 2: | Observable market-based inputs other than quoted prices in active markets for identical assets or liabilities. |
Level 3: | Unobservable inputs are used when little or no market data is available. The fair value hierarchy gives the lowest priority to Level 3 inputs. |
In determining fair value, the Company utilizes valuation techniques that maximize the use of observable inputs and minimize the use of unobservable inputs to the extent possible, as well as considers counterparty credit risk in its assessment of fair value.
121
Table of Contents
Financial assets and liabilities carried at fair value are classified in the tables below in one of the three categories described above:
| December 31, 2016 | ||||||||||||||
(In millions) | Level 1 |
| Level 2 |
| Level 3 |
| Total | ||||||||
Recurring fair value measurements |
|
|
|
|
|
|
| ||||||||
Financial Assets | |||||||||||||||
Cash equivalents: |
|
|
|
|
|
|
| ||||||||
Money market funds | $ | 433.7 | |
| $ | - | |
| $ | - | |
| $ | 433.7 | |
Total cash equivalents | 433.7 | |
| - | |
| - | |
| 433.7 | | ||||
Trading securities: |
|
|
|
|
|
|
| ||||||||
Equity securities - exchange traded funds | 29.6 | |
| - | |
| - | |
| 29.6 | | ||||
Total trading securities | 29.6 | |
| - | |
| - | |
| 29.6 | | ||||
Available-for-sale fixed income investments: |
|
|
|
|
|
|
| ||||||||
Corporate bonds | - | |
| 17.5 | |
| - | |
| 17.5 | | ||||
U.S. Treasuries | - | |
| 6.0 | |
| - | |
| 6.0 | | ||||
Agency mortgage-backed securities | - | |
| 4.0 | |
| - | |
| 4.0 | | ||||
Asset backed securities | - | |
| 1.6 | |
| - | |
| 1.6 | | ||||
Other | - | |
| 2.3 | |
| - | |
| 2.3 | | ||||
Total available-for-sale fixed income investments | - | |
| 31.4 | |
| - | |
| 31.4 | | ||||
Available-for-sale equity securities: |
|
|
|
|
|
|
| ||||||||
Marketable securities | 52.3 | |
| - | |
| - | |
| 52.3 | | ||||
Total available-for-sale equity securities | 52.3 | |
| - | |
| - | |
| 52.3 | | ||||
Foreign exchange derivative assets | - | |
| 35.9 | |
| - | |
| 35.9 | | ||||
Interest rate swap derivative assets | - | |
| 26.2 | |
| - | |
| 26.2 | | ||||
Total assets at recurring fair value measurement | $ | 515.6 | |
| $ | 93.5 | |
| $ | - | |
| $ | 609.1 | |
Financial Liabilities | |||||||||||||||
Foreign exchange derivative liabilities | $ | - | |
| $ | 15.3 | |
| $ | - | |
| $ | 15.3 | |
Contingent consideration | - | |
| - | |
| 564.6 | |
| 564.6 | | ||||
Total liabilities at recurring fair value measurement | $ | - | |
| $ | 15.3 | |
| $ | 564.6 | |
| $ | 579.9 | |
122
Table of Contents
| December 31, 2015 | ||||||||||||||
(In millions) | Level 1 |
| Level 2 |
| Level 3 |
| Total | ||||||||
Recurring fair value measurements |
|
|
|
|
|
|
| ||||||||
Financial Assets | |||||||||||||||
Cash equivalents: |
|
|
|
|
|
|
| ||||||||
Money market funds | $ | 923.3 | |
| $ | - | |
| $ | - | |
| $ | 923.3 | |
Total cash equivalents | 923.3 | |
| - | |
| - | |
| 923.3 | | ||||
Trading securities: |
|
|
|
|
|
|
| ||||||||
Equity securities - exchange traded funds | 22.8 | |
| - | |
| - | |
| 22.8 | | ||||
Total trading securities | 22.8 | |
| - | |
| - | |
| 22.8 | | ||||
Available-for-sale fixed income investments: |
|
|
|
|
|
|
| ||||||||
Corporate bonds | - | |
| 15.7 | |
| - | |
| 15.7 | | ||||
U.S. Treasuries | - | |
| 4.7 | |
| - | |
| 4.7 | | ||||
Agency mortgage-backed securities | - | |
| 3.9 | |
| - | |
| 3.9 | | ||||
Asset backed securities | - | |
| 2.3 | |
| - | |
| 2.3 | | ||||
Other | - | |
| 1.4 | |
| - | |
| 1.4 | | ||||
Total available-for-sale fixed income investments | - | |
| 28.0 | |
| - | |
| 28.0 | | ||||
Available-for-sale equity securities: |
|
|
|
|
|
|
| ||||||||
Marketable securities | 26.0 | |
| - | |
| - | |
| 26.0 | | ||||
Total available-for-sale equity securities | 26.0 | |
| - | |
| - | |
| 26.0 | | ||||
Foreign exchange derivative assets | - | |
| 28.4 | |
| - | |
| 28.4 | | ||||
Interest rate swap derivative assets | - | |
| 36.3 | |
| - | |
| 36.3 | | ||||
Total assets at recurring fair value measurement | $ | 972.1 | |
| $ | 92.7 | |
| $ | - | |
| $ | 1,064.8 | |
Financial Liabilities | |||||||||||||||
Foreign exchange derivative liabilities | $ | - | |
| $ | 9.3 | |
| $ | - | |
| $ | 9.3 | |
Interest rate swap derivative liabilities | - | |
| 10.5 | |
| - | |
| 10.5 | | ||||
Contingent consideration | - | |
| - | |
| 526.4 | |
| 526.4 | | ||||
Total liabilities at recurring fair value measurement | $ | - | |
| $ | 19.8 | |
| $ | 526.4 | |
| $ | 546.2 | |
For financial assets and liabilities that utilize Level 2 inputs, the Company utilizes both direct and indirect observable price quotes, including the LIBOR yield curve, foreign exchange forward prices, and bank price quotes. For the years ended December 31, 2016 and 2015 , there were no transfers between Level 1 and 2 of the fair value hierarchy. Below is a summary of valuation techniques for Level 1 and Level 2 financial assets and liabilities:
• | Cash equivalents - valued at observable net asset value prices. |
• | Trading securities - valued at the active quoted market price from broker or dealer quotations or transparent pricing sources at the reporting date. |
• | Available-for-sale fixed income investments - valued at the quoted market price from broker or dealer quotations or transparent pricing sources at the reporting date. |
• | Available-for-sale equity securities - valued using quoted stock prices from public exchanges at the reporting date. |
• | Interest rate swap derivative assets and liabilities - valued using the LIBOR/EURIBOR yield curves at the reporting date. Counterparties to these contracts are highly rated financial institutions. |
• | Foreign exchange derivative assets and liabilities - valued using quoted forward foreign exchange prices and spot rates at the reporting date. Counterparties to these contracts are highly rated financial institutions. |
123
Table of Contents
Contingent Consideration
The fair value measurement of contingent consideration is determined using Level 3 inputs. The Company's contingent consideration represents a component of the total purchase consideration for the acquisitions of the respiratory delivery platform, Agila , Jai Pharma Limited , the Topicals Business and certain other acquisitions. The measurement is calculated using unobservable inputs based on the Company's own assumptions. For the respiratory delivery platform, Jai Pharma Limited, the Topicals Business and certain other acquisitions, significant unobservable inputs in the valuation include the probability and timing of future development and commercial milestones and future profit sharing payments. When valuing the contingent consideration related to the respiratory delivery platform and Jai Pharma Limited, the value of the obligations are derived from a probability assessment based on expectations of when certain milestones or profit sharing payments occur which are discounted using a market rate of return. At December 31, 2016 and 2015 , discount rates ranging from 0.9% to 9.8% were utilized in such valuations. Significant changes in unobservable inputs could result in material changes to the contingent consideration liability.
In conjunction with the acquisition of Agila on December 4, 2013 , the Company recorded estimated contingent consideration totaling $250 million as part of the purchase price. During the third quarter of 2014, the Company entered into an agreement with Strides Arcolab Limited ("Strides Arcolab") to settle a portion of the contingent consideration for $150 million , for which the Company accrued $230 million at the acquisition date. As a result of this agreement, the Company recognized a gain of $80 million during the year ended December 31, 2014, which is included in litigation settlements and other contingencies, net in the Consolidated Statements of Operations . On November 1, 2016, the Company and Strides Arcolab agreed on a settlement of substantially all outstanding regulatory, warranty and indemnity claims (the "Strides Settlement") related to the acquisition of Agila. As a result of the settlement, the Company received approximately $80 million of cash in the fourth quarter of 2016, which was previously classified as restricted cash. Approximately $110 million will be paid to either settle these pre-acquisition claims or be remitted to Strides. As such, in addition to the $20 million of contingent consideration recorded upon acquisition, the Company recorded expense of approximately $90 million , of which $74.8 million represented additional contingent consideration, which is included in litigation settlements and other contingencies, net in the Consolidated Statements of Operations for the year ended December 31, 2016.
(In millions) | Current Portion (1) |
| Long-Term Portion (2) |
| Total Contingent Consideration | ||||||
Balance at December 31, 2014 | $ | 20.0 | |
| $ | 450.0 | |
| $ | 470.0 | |
Acquisitions | - | |
| 18.0 | |
| 18.0 | | |||
Reclassifications | 15.0 | |
| (15.0 | ) |
| - | | |||
Accretion | - | |
| 38.4 | |
| 38.4 | | |||
Balance at December 31, 2015 | $ | 35.0 | |
| $ | 491.4 | |
| $ | 526.4 | |
Acquisitions | 21.6 | |
| 1.2 | |
| 22.8 | | |||
Payments | (44.4 | ) |
| (0.5 | ) |
| (44.9 | ) | |||
Reclassifications | 169.8 | |
| (169.8 | ) |
| - | | |||
Accretion | 0.1 | |
| 41.7 | |
| 41.8 | | |||
Fair value loss (gain) (3) | 74.8 | |
| (55.9 | ) |
| 18.9 | | |||
Foreign currency translation | - | |
| (0.4 | ) |
| (0.4 | ) | |||
Balance at December 31, 2016 | $ | 256.9 | |
| $ | 307.7 | |
| $ | 564.6 | |
(1) | Included in other current liabilities on the Consolidated Balance Sheets. |
(2) | Included in other long-term obligations on the Consolidated Balance Sheets. |
(3) | Included in litigation settlements and other contingencies, net in the Consolidated Statements of Operations. |
2015 Changes to Contingent Consideration: Total contingent consideration increased $18.0 million in 2015 due to the acquisition of Jai Pharma Limited. During the year ended December 31, 2015, the Company reclassified $15.0 million of contingent consideration from other long-term obligations to other current liabilities representing milestone payments related to the respiratory delivery platform that were paid in 2016.
124
Table of Contents
2016 Changes to Contingent Consideration: During 2016, the Company recorded a fair value loss resulting in an additional $74.8 million of contingent consideration related to the Strides Settlement, of which approximately $28.3 million was paid in the fourth quarter of 2016. In addition, the Company recorded a fair value loss of $12.6 million related to the Jai Pharma Limited acquisition. Offsetting these items was a fair value gain of approximately $68.5 million related to the respiratory delivery platform contingent consideration. As part of the acquisition of the Topicals Business, the Company recorded contingent consideration of $16 million at the acquisition date. Additionally, the Company reclassified $169.8 million of contingent consideration from other long-term obligations to other current liabilities representing milestone and profit sharing payments related to the respiratory delivery platform, milestone payments related to Jai Pharma Limited and payments related to the Strides Settlement which are expected to be paid in 2017.
The Company expects to incur approximately $30 million to $35 million of non-cash accretion expense related to the increase in the net present value of the contingent consideration liability in 2017.
Although the Company has not elected the fair value option for financial assets and liabilities, any future transacted financial asset or liability will be evaluated for the fair value election.
Available-for-Sale Securities
The amortized cost and estimated fair value of available-for-sale securities, included in prepaid expenses and other current assets, were as follows:
(In millions) | Cost |
| Gross |
| Gross |
| Fair | ||||||||
December 31, 2016 |
|
|
|
|
|
|
| ||||||||
Debt securities | $ | 31.4 | |
| $ | - | |
| $ | - | |
| $ | 31.4 | |
Equity securities | 28.0 | |
| 24.6 | |
| (0.3 | ) |
| 52.3 | | ||||
| $ | 59.4 | |
| $ | 24.6 | |
| $ | (0.3 | ) |
| $ | 83.7 | |
December 31, 2015 |
|
|
|
|
|
|
| ||||||||
Debt securities | $ | 28.3 | |
| $ | - | |
| $ | (0.3 | ) |
| $ | 28.0 | |
Equity securities | 27.3 | |
| - | |
| (1.3 | ) |
| 26.0 | | ||||
| $ | 55.6 | |
| $ | - | |
| $ | (1.6 | ) |
| $ | 54.0 | |
Maturities of available-for-sale debt securities at fair value as of December 31, 2016 , were as follows:
(In millions) |
| ||
Mature within one year | $ | 1.8 | |
Mature in one to five years | 16.1 | | |
Mature in five years and later | 13.5 | | |
| $ | 31.4 | |
125
Table of Contents
8. | Debt |
(In millions) | Coupon |
| December 31, |
| December 31, | |||||
Current portion of long-term debt: |
|
|
|
|
| |||||
2016 Senior Notes (a) * | 1.800 | % |
| $ | - | |
| $ | 500.1 | |
2016 Senior Notes (b) * | 1.350 | % |
| - | |
| 499.9 | | ||
Meda Bank Loans (c) |
|
| 219.6 | |
| - | | |||
Other |
|
| 3.7 | |
| 1.6 | | |||
Deferred financing fees |
|
| - | |
| (2.9 | ) | |||
Current portion of long-term debt |
|
| $ | 223.3 | |
| $ | 998.7 | | |
|
|
|
|
|
| |||||
Non-current portion of long-term debt: |
|
|
|
|
| |||||
2016 Term Loans (d) ** |
|
| $ | 1,600.0 | |
| $ | - | | |
2015 Term Loans (e) * |
|
| - | |
| 1,600.0 | | |||
2014 Term Loan (f) * |
|
| - | |
| 800.0 | | |||
Meda Medium Term Notes (g) |
|
| 146.4 | |
| - | | |||
2018 Euro Senior Notes (h) ** |
|
| 526.0 | |
| - | | |||
2018 Senior Notes (i) * | 2.600 | % |
| 649.6 | |
| 649.3 | | ||
2018 Senior Notes (i) ** | 3.000 | % |
| 499.6 | |
| 499.4 | | ||
2019 Senior Notes (j) ** | 2.500 | % |
| 999.1 | |
| - | | ||
2019 Senior Notes (k) * | 2.550 | % |
| 499.5 | |
| 499.2 | | ||
2020 Euro Senior Notes (l) ** | 1.250 | % |
| 785.7 | |
| - | | ||
2020 Senior Notes (m) ** | 3.750 | % |
| 499.9 | |
| 499.8 | | ||
2021 Senior Notes (n) ** | 3.150 | % |
| 2,247.7 | |
| - | | ||
2023 Senior Notes (k) * | 3.125 | % |
| 775.3 | |
| 785.2 | | ||
2023 Senior Notes (o) * | 4.200 | % |
| 498.6 | |
| 498.4 | | ||
2024 Euro Senior Notes (p)** | 2.250 | % |
| 1,049.2 | |
| - | | ||
2026 Senior Notes (q) ** | 3.950 | % |
| 2,233.5 | |
| - | | ||
2028 Euro Senior Notes (r) ** | 3.125 | % |
| 781.1 | |
| - | | ||
2043 Senior Notes (s) * | 5.400 | % |
| 497.0 | |
| 497.0 | | ||
2046 Senior Notes (t) ** | 5.250 | % |
| 999.8 | |
| - | | ||
Other |
|
| 7.1 | |
| 2.7 | | |||
Deferred financing fees |
|
| (92.2 | ) |
| (35.4 | ) | |||
Total long-term debt |
|
| $ | 15,202.9 | |
| $ | 6,295.6 | | |
(a) | Instrument matured on June 24, 2016 , and the Company paid the principal amount of $500.0 million and final interest payment of $4.5 million upon maturity. |
(b) | Instrument matured on November 29, 2016, and the Company paid the principal amount of $500.0 million and final interest payment of $3.4 million upon maturity. |
(c) | Represents a bank loan of 2.0kr billion with AB Svensk Exportkredit (publ), as lender ("Svensk Exportkredit"), which matures in October 2017, and accordingly is included in current portion of long-term debt and other long-term obligations in the Consolidated Balance Sheets at December 31, 2016 . |
(d) | The 2016 Term Loans mature on November 22, 2019. |
(e) | The 2015 Term Loans were terminated and repaid in the fourth quarter of 2016 in conjunction with the effectiveness of the 2016 Term Loans. |
(f) | The 2014 Term Loan was terminated and repaid in the fourth quarter of 2016 in conjunction with the effectiveness of the 2016 Term Loans. |
(g) | Swedish medium term notes ("MTN") program with an upper limit of 7kr billion . Of the total amount outstanding of 1.33kr billion , 583kr million matures on April 5, 2018 and 750kr million matures on May 21, 2019. |
126
Table of Contents
(h) | Instrument bears interest at a rate of three-month EURIBOR plus 0.870% per annum, reset quarterly. |
(i) | Instrument is callable by the Company at any time at the greater of 100% of the principal amount and the sum of the present values of the remaining scheduled payments of principal and interest discounted at the U.S. Treasury rate plus 0.30% plus, in each case, accrued and unpaid interest. |
(j) | Instrument is callable by the Company at any time at the greater of 100% of the principal amount and the sum of the present values of the remaining scheduled payments of principal and interest discounted at the U.S. Treasury rate plus 0.25% plus, in each case, accrued and unpaid interest. |
(k) | Instrument is callable by the Company at any time at the greater of 100% of the principal amount and the sum of the present values of the remaining scheduled payments of principal and interest discounted at the U.S. Treasury rate plus 0.20% plus, in each case, accrued and unpaid interest. |
(l) | Instrument is callable by the Company at any time prior to the date that is one month prior to the instrument's maturity date at the greater of 100% of the principal amount and the sum of the present values of the remaining scheduled payments of principal and interest discounted to the redemption date on an annual basis, at a rate equal to the applicable Bund Rate (as defined in the Euro Notes Indenture (as defined herein)), plus 0.30% plus, in each case, accrued and unpaid interest. On or after such date, the instrument is callable by the Company at 100% of the principal amount plus accrued and unpaid interest. |
(m) | Instrument is callable by the Company at any time prior to the date that is one month prior to the instrument's maturity date at the greater of 100% of the principal amount and the sum of the present values of the remaining scheduled payments of principal and interest discounted at the U.S. Treasury rate plus 0.35% plus, in each case, accrued and unpaid interest. On or after such date, the instrument is callable by the Company at 100% of the principal amount plus accrued and unpaid interest. |
(n) | Instrument is callable by the Company at any time prior to the date that is one month prior to the instrument's maturity date at the greater of 100% of the principal amount and the sum of the present values of the remaining scheduled payments of principal and interest discounted at the U.S. Treasury rate plus 0.30% plus, in each case, accrued and unpaid interest. On or after such date, the instrument is callable by the Company at 100% of the principal amount plus accrued and unpaid interest. |
(o) | Instrument is callable by the Company at any time prior to August 29, 2023 at the greater of 100% of the principal amount and the sum of the present values of the remaining scheduled payments of principal and interest discounted at the U.S. Treasury rate plus 0.25% plus, in each case, accrued and unpaid interest. On or after such date, the instrument is callable by the Company at 100% of the principal amount plus accrued and unpaid interest. |
(p) | Instrument is callable by the Company at any time prior to the date that is two months prior to the instrument's maturity date at the greater of 100% of the principal amount and the sum of the present values of the remaining scheduled payments of principal and interest discounted to the redemption date on an annual basis, at a rate equal to the applicable Bund Rate (as defined in the Euro Notes Indenture), plus 0.35% plus, in each case, accrued and unpaid interest. On or after such date, the instrument is callable by the Company at 100% of the principal amount plus accrued and unpaid interest. |
(q) | Instrument is callable by the Company at any time prior to the date that is three months prior to the instrument's maturity date at the greater of 100% of the principal amount and the sum of the present values of the remaining scheduled payments of principal and interest discounted at the U.S. Treasury rate plus 0.35% plus, in each case, accrued and unpaid interest. On or after such date, the instrument is callable by the Company at 100% of the principal amount plus accrued and unpaid interest. |
(r) | Instrument is callable by the Company at any time prior to the date that is three months prior to the instrument's maturity date at the greater of 100% of the principal amount and the sum of the present values of the remaining scheduled payments of principal and interest discounted to the redemption date on an annual basis, at a rate equal to the applicable Bund Rate (as defined in the Euro Notes Indenture), plus 0.45% plus, in each case, accrued and unpaid interest. On or after such date, the instrument is callable by the Company at 100% of the principal amount plus accrued and unpaid interest. |
(s) | Instrument is callable by the Company at any time prior to May 29, 2043 at the greater of 100% of the principal amount and the sum of the present values of the remaining scheduled payments of principal and interest discounted at the U.S. Treasury rate plus 0.25% plus, in each case, accrued and unpaid interest. On or after such date, the instrument is callable by the Company at 100% of the principal amount plus accrued and unpaid interest. |
(t) | Instrument is callable by the Company at any time prior to the date that is six months prior to the instrument's maturity date at the greater of 100% of the principal amount and the sum of the present values of the remaining scheduled payments of principal and interest discounted at the U.S. Treasury rate plus 0.40% plus, in each case, accrued and unpaid interest. On or after such date, the instrument is callable by the Company at 100% of the principal amount plus accrued and unpaid interest. |
* | Instrument was issued by Mylan Inc. |
** | Instrument was issued by Mylan N.V. |
127
Table of Contents
Short-Term Borrowings
The Company's subsidiaries in India have working capital facilities with several banks. At December 31, 2016 , the working capital facilities had a weighted average interest rate of 8.0% on borrowings of approximately $46.4 million outstanding under such facilities. At December 31, 2015 , the Company had no amounts outstanding under such facilities.
Receivables Facility
Mylan Pharmaceuticals Inc. (" MPI "), a wholly owned subsidiary of the Company, has a $400 million accounts receivable securitization facility ("Receivables Facility"), which will expire in January 2018. Although from time-to-time, the available amount of the Receivables Facility may be less than $400 million based on accounts receivable concentration limits and other eligibility requirements. In January 2015, the Receivables Facility was amended and restated, and its maturity was extended through January 2018.
Under the terms of the Receivables Facility, our subsidiary, MPI , sells certain accounts receivable to Mylan Securitization LLC ("Mylan Securitization"), a wholly owned special purpose entity which in turn sells a percentage ownership interest in the receivables to financial institutions and commercial paper conduits sponsored by financial institutions. MPI is the servicer of the receivables under the Receivables Facility. Purchases under the Receivables Facility will be repaid as accounts receivable are collected, with new purchases being advanced as new accounts receivable are originated by MPI . Mylan Securitization's assets have been pledged to the agent in support of its obligations under the Receivables Facility. Any amounts outstanding under the facility will be recorded as a secured loan and the receivables underlying any borrowings will continue to be included in accounts receivable, net, in the Consolidated Balance Sheets of the Company.
The Receivables Facility contains requirements relating to the performance of the accounts receivable and covenants related to the Company. If we do not comply with the covenants under the Receivables Facility, our ability to use the Receivables Facility may be suspended and repayment of any outstanding balances under the Receivables Facility may be required.
As of December 31, 2016 and 2015, the Company had $1.13 billion and $914.2 million , respectively, of accounts receivable balances sold to Mylan Securitization and no short-term borrowings included in the Consolidated Balance Sheets . During the year ended December 31, 2015 , the Company paid approximately $1.5 million in upfront fees and other fees which were recorded as deferred financing costs in the Consolidated Balance Sheets .
2016 Activity
2016 Senior Revolving Credit Agreement
On November 22, 2016, the Company entered into a revolving credit agreement (the "2016 Senior Revolving Credit Agreement") among the Company, as borrower, Mylan Inc., as a guarantor (the "Guarantor"), certain lenders and issuing banks and Bank of America, N.A., as the administrative agent (in such capacity, the "Revolving Administrative Agent"). The 2016 Senior Revolving Credit Agreement contains a revolving credit facility (the " 2016 Senior Revolving Facility ") under which the Company may obtain extensions of credit in an aggregate principal amount not to exceed $2.0 billion , subject to the satisfaction of customary conditions, in U.S. Dollars or alternative currencies including Euro, Sterling, Yen and any other currency that is approved by the Revolving Administrative Agent and each lender under the 2016 Senior Revolving Facility . The 2016 Senior Revolving Facility includes a $200 million subfacility for the issuance of letters of credit and a $175 million sublimit for swingline borrowings. The swingline borrowings will be made available in U.S. Dollars only. The Company may seek additional commitments under the 2016 Senior Revolving Facility from lenders or other financial institutions designated by the Company up to an aggregate amount such that the Company would be in compliance with the financial covenant described below, after giving effect to such increase in the commitments and the application of proceeds therefrom. In determining pro forma compliance with the financial covenant described below, any indebtedness that is proposed to be incurred will be added to the Company's consolidated total indebtedness, and if such indebtedness is incurred in connection with an acquisition, the consolidated EBITDA of the acquired business for the trailing four quarters will be added to (or, if negative, subtracted from) the Company's consolidated EBITDA for the same period.
Proceeds from the 2016 Senior Revolving Facility will be used for working capital, capital expenditures and other lawful corporate purposes, including, without limitation, to repay outstanding obligations of the Company and its subsidiaries. The effectiveness of the 2016 Senior Revolving Credit Agreement was concurrent with, and contingent upon, the termination of the Revolving Credit Agreement, dated as of December 19, 2014 (as amended, restated, supplemented or otherwise modified from time to time, the " 2014 Revolving Credit Agreement "), among the Company, as guarantor, Mylan Inc., as borrower, the
128
Table of Contents
lenders and issuing banks from time to time party thereto and Bank of America, N.A., as administrative agent. The 2016 Senior Revolving Facility is guaranteed by (1) the Guarantor; provided that if the Guarantor is no longer a borrower in respect of third party indebtedness in excess of $500 million , the Guarantor shall be released from such guarantee at the option of the Company or the Guarantor and (2) each subsidiary of the Company that guarantees (or is otherwise a co-obligor of) third party indebtedness in excess of $500 million of the Company, or if the Guarantor is at such time a guarantor of the 2016 Senior Revolving Facility , indebtedness in excess of $500 million of the Guarantor. As of December 31, 2016, no subsidiary of the Company (other than the Guarantor) is required to provide a guarantee of the 2016 Senior Revolving Facility , but will automatically do so upon the occurrence of the above. The 2016 Senior Revolving Facility is unsecured.
Borrowings under the 2016 Senior Revolving Facility will bear interest at LIBOR (determined in accordance with the 2016 Senior Revolving Credit Agreement) plus 1.200% per annum, if the Company chooses to make LIBOR borrowings, or at a base rate (determined in accordance with the 2016 Senior Revolving Credit Agreement) plus 0.200% per annum. The 2016 Senior Revolving Facility has a facility fee, which currently accrues at 0.175% on the daily amount of the aggregate revolving commitments of the lenders. The applicable margins over LIBOR and the base rate for the revolver can fluctuate based on the long term unsecured senior, non-credit enhanced debt rating of the Company by S&P Global Ratings, Moody's Investors Service, Inc. and Fitch Ratings, Inc.
The 2016 Senior Revolving Credit Agreement contains customary affirmative covenants for facilities of this type, including, among others, covenants pertaining to the delivery of financial statements, notices of default and certain other material events, maintenance of corporate existence and rights, business, property and insurance and compliance with laws, as well as customary negative covenants for facilities of this type, including, among others, limitations on the incurrence of subsidiary indebtedness, liens, mergers and certain other fundamental changes, investments and loans, acquisitions, transactions with affiliates, payments of dividends and other restricted payments and changes in the Company's line of business. The 2016 Senior Revolving Credit Agreement contains a financial covenant requiring maintenance of a maximum ratio of 3.75 to 1.00 for consolidated total indebtedness as of the end of any quarter to consolidated EBITDA for the trailing four quarters. Following certain qualifying acquisitions (which includes our acquisition of Meda), at the Company's election, the maximum ratio in the financial covenant will be increased to 4.25 to 1.00 for the three full quarters following such qualifying acquisition. This financial covenant was first tested at the quarter ending December 31, 2016 , and the Company was in compliance.
The 2016 Senior Revolving Credit Agreement contains default provisions customary for facilities of this type, which are subject to customary grace periods and materiality thresholds, including, among others, defaults related to payment failures, failure to comply with covenants, material misrepresentations, defaults under other material indebtedness, the occurrence of a "change in control", bankruptcy and related events, material judgments, certain events related to pension plans and the invalidity or revocation of any loan document or any guarantee agreement of the Company or any subsidiary that becomes a guarantor as described above. If an event of default occurs under the 2016 Senior Revolving Credit Agreement, the lenders may, among other things, terminate their commitments and declare immediately payable all borrowings.
Amounts drawn on the 2016 Senior Revolving Facility become due and payable on November 22, 2021 and may be voluntarily prepaid without penalty or premium, other than customary breakage costs related to prepayments of LIBOR borrowings. At December 31, 2016, the Company had no amounts outstanding on the 2016 Senior Revolving Facility .
Termination of 2014 Revolving Credit Agreement
In conjunction with the effectiveness of the 2016 Senior Revolving Credit Agreement in the fourth quarter of 2016, the 2014 Revolving Credit Agreement was terminated. The Company had no amounts outstanding on the 2014 Revolving Credit Agreement at the time of termination.
2016 Senior Term Credit Agreement
On November 22, 2016, the Company entered into a term loan credit agreement (the "2016 Senior Term Credit Agreement") among the Company, as borrower, the Guarantor, as a guarantor, certain lenders and Goldman Sachs Bank USA, as administrative agent (in such capacity, the "Term Administrative Agent") pursuant to which the Company borrowed $2.0 billion in term loans denominated in U.S. Dollars (the "2016 Term Loans"). The proceeds of the 2016 Term Loans were used to repay outstanding obligations under, and thereby terminate, the facilities agreement, dated as of December 17, 2014 (as amended, restated, supplemented or otherwise modified from time to time, the "Meda Credit Agreement"), among Meda, as borrower, the lenders from time to time party thereto and Danske Bank A/S, as agent. The effectiveness of the 2016 Senior Term Credit Agreement was concurrent with, and contingent upon, the termination of (i) the 2014 Term Credit Agreement (as defined below), and (ii) the 2015 Term Credit Agreement (as defined below).
129
Table of Contents
The 2016 Senior Term Credit Agreement is guaranteed by (1) the Guarantor; provided that if the Guarantor is no longer a borrower in respect of third party indebtedness in excess of $500 million , the Guarantor shall be released from such guarantee at the option of the Company or the Guarantor and (2) each subsidiary of the Company that guarantees (or is otherwise a co-obligor of) third party indebtedness in excess of $500 million of the Company, or if the Guarantor is at such time a guarantor of the 2016 Term Loans, indebtedness in excess of $500 million of the Guarantor. As of December 31, 2016, no subsidiary of the Company (other than the Guarantor) is required to provide a guarantee of the 2016 Term Loans, but will automatically do so upon the occurrence of the above. The 2016 Terms Loans are unsecured.
The 2016 Term Loans currently bear interest at LIBOR (determined in accordance with the 2016 Senior Term Credit Agreement) plus 1.375% per annum, if the Company chooses to make LIBOR borrowings, or at a base rate (determined in accordance with the 2016 Senior Term Credit Agreement) plus 0.375% per annum. The applicable margins over LIBOR and the base rate for the 2016 Term Loans can fluctuate based on the long term unsecured senior, non-credit enhanced debt rating of the Company by S&P Global Ratings, Moody's Investors Service, Inc. and Fitch Ratings, Inc. At December 31, 2016 , the weighted average interest rate of the 2016 Term Loans was approximately 2.124% .
The 2016 Senior Term Credit Agreement contains customary affirmative covenants for facilities of this type, including, among others, covenants pertaining to the delivery of financial statements, notices of default and certain other material events, maintenance of corporate existence and rights, business, property and insurance, compliance with laws and repayment of indebtedness and termination of commitments under the Meda Credit Agreement within five business days of closing, as well as customary negative covenants for facilities of this type, including, among others, limitations on the incurrence of subsidiary indebtedness, liens, mergers and certain other fundamental changes, investments and loans, acquisitions, transactions with affiliates, payments of dividends and other restricted payments and changes in the Company's line of business. The 2016 Senior Term Credit Agreement contains a financial covenant requiring maintenance of a maximum ratio of 3.75 to 1.00 for consolidated total indebtedness as of the end of any quarter to consolidated EBITDA for the trailing four quarters. Following certain qualifying acquisitions (which includes our acquisition of Meda), at the Company's election, the maximum ratio in the financial covenant will be increased to 4.25 to 1.00 for the three full quarters following such qualifying acquisition. This financial covenant was first tested at the quarter ending December 31, 2016, and the Company was in compliance.
The 2016 Senior Term Credit Agreement contains default provisions customary for facilities of this type, which are subject to customary grace periods and materiality thresholds, including, among others, defaults related to payment failures, failure to comply with covenants, material misrepresentations, defaults under other material indebtedness, the occurrence of a change in control, bankruptcy and related events, material judgments, certain events related to pension plans and the invalidity or revocation of any loan document or any guarantee agreement of the Company or any subsidiary that becomes a guarantor as described above. If an event of default occurs under the 2016 Senior Term Credit Agreement, the lenders may, among other things, terminate their commitments and declare immediately payable all borrowings.
The 2016 Term Loans mature on November 22, 2019 and have no required amortization payments. The entire principal amount on the 2016 Term Loans will be due and payable on November 22, 2019. The 2016 Term Loans may be voluntarily prepaid without penalty or premium, other than customary breakage costs related to prepayments of LIBOR borrowings. The Company voluntarily prepaid $400 million of the aggregate principal amount of the 2016 Term Loans in the fourth quarter, and at December 31, 2016, the Company had an aggregate principal amount of $1.6 billion outstanding under the 2016 Term Loans. During the year ended December 31, 2016 , the Company incurred fees of approximately $6.4 million related to the 2016 Term Loans which were recorded as deferred financing fees in the Consolidated Balance Sheets.
Termination of 2015 Term Credit Agreement
On July 15, 2015 , the Company entered into a term credit agreement (as amended, restated, supplemented or otherwise modified from time to time, the "2015 Term Credit Agreement") among the Company, as guarantor, Mylan Inc., as borrower, certain lenders and PNC Bank, National Association as the administrative agent. In conjunction with the effectiveness of the 2016 Senior Term Credit Agreement in the fourth quarter of 2016, the 2015 Term Credit Agreement was terminated and the Company repaid the $1.6 billion aggregate principal amount outstanding.
Termination of 2014 Term Credit Agreement
On December 19, 2014, the Company entered into a term credit agreement (as amended, restated, supplemented or otherwise modified from time to time, the "2014 Term Credit Agreement") among the Company, as guarantor, Mylan Inc., as borrower, certain lenders and Bank of America, N.A., as the administrative agent. In conjunction with the effectiveness of the
130
Table of Contents
2016 Senior Term Credit Agreement in the fourth quarter of 2016, the 2014 Term Credit Agreement was terminated and the Company repaid the $800 million aggregate principal amount outstanding.
Issuance of Euro Notes
On November 22, 2016, the Company completed its offering of €500 million aggregate principal amount of the Company's Floating Rate Senior Notes due 2018 (the "Floating Rate Euro Notes"), €750 million aggregate principal amount of the Company's 1.250% Senior Notes due 2020 (the "2020 Euro Notes"), €1.0 billion aggregate principal amount of the Company's 2.250% Senior Notes due 2024 (the "2024 Euro Notes") and €750 million aggregate principal amount of the Company's 3.125% Senior Notes due 2028 (the "2028 Euro Notes," together with the 2020 Euro Notes and the 2024 Euro Notes, the "Fixed Rate Euro Notes"), issued pursuant to the indenture dated November 22, 2016 (the "Euro Notes Indenture"), among the Company, Mylan Inc. (the "Guarantor") and Citibank, N.A., London Branch, as trustee, paying agent, transfer agent, registrar and calculation agent. The Floating Rate Euro Notes and the Fixed Rate Euro Notes, together, are referred to as the "Euro Notes."
The Euro Notes were issued in a private offering exempt from the registration requirements of the Securities Act of 1933, as amended (the "Securities Act"), to persons outside of the United States pursuant to Regulation S under the Securities Act.
The Euro Notes are the Company's senior unsecured indebtedness and are guaranteed on a senior unsecured basis by the Guarantor. In addition, if a subsidiary of the Company becomes a guarantor or an obligor in respect of certain indebtedness, such subsidiary will guarantee the Euro Notes on the terms and subject to the conditions in the Euro Notes Indenture.
The Floating Rate Euro Notes bear interest at a rate per annum, reset quarterly, equal to the sum of (i) three-month EURIBOR (as defined in the Euro Notes Indenture) plus (ii) 0.870% , as determined by the calculation agent for the Floating Rate Euro Notes pursuant to the Euro Notes Indenture; provided, however, that the minimum interest rate for the Floating Rate Euro Notes is zero . Interest on the Floating Rate Euro Notes is payable quarterly in arrears on each February 22, May 22, August 22 and November 22, commencing on February 22, 2017. The Floating Rate Euro Notes will mature on November 22, 2018. The 2020 Euro Notes bear interest at a rate of 1.250% per annum, accruing from November 22, 2016. Interest on the 2020 Notes is payable annually in arrears on November 23, commencing on November 23, 2017. The 2020 Euro Notes will mature on November 23, 2020, subject to earlier repurchase or redemption in accordance with the terms of the Euro Notes Indenture. The 2024 Euro Notes bear interest at a rate of 2.250% per annum, accruing from November 22, 2016. Interest on the 2024 Euro Notes is payable annually in arrears on November 22, commencing on November 22, 2017. The 2024 Euro Notes will mature on November 22, 2024, subject to earlier repurchase or redemption in accordance with the terms of the Euro Notes Indenture. The 2028 Euro Notes bear interest at a rate of 3.125% per annum, accruing from November 22, 2016. Interest on the 2028 Euro Notes is payable annually in arrears on November 22, commencing on November 22, 2017. The 2028 Euro Notes will mature on November 22, 2028, subject to earlier repurchase or redemption in accordance with the terms of the Euro Notes Indenture.
At any time and from time to time prior to the date that is one month prior to their maturity date in the case of the 2020 Euro Notes, the date that is two months prior to their maturity date in the case of the 2024 Euro Notes and the date that is three months prior to their maturity date in the case of the 2028 Euro Notes, the Company may redeem some or all of the Fixed Rate Euro Notes of the applicable series, upon not less than 30 nor more than 60 days' prior notice, at a price equal to the greater of (1) 100% of the aggregate principal amount of any Fixed Rate Notes being redeemed, and (2) the sum of the present values of the remaining scheduled payments of principal and interest on the Fixed Rate Euro Notes being redeemed that would be due to their maturity date, in each case, not including unpaid interest accrued to, but excluding, the redemption date, discounted to the redemption date on an annual basis (ACTUAL/ACTUAL (ICMA), as defined in the rulebook of the International Capital Market Association), at a rate equal to the applicable Bund Rate (as defined in the Euro Notes Indenture) plus 30 basis points with respect to the 2020 Euro Notes, 35 basis points with respect to the 2024 Euro Notes and 45 basis points with respect to the 2028 Euro Notes, plus, in each case, unpaid interest on the Fixed Rate Euro Notes being redeemed accrued to, but excluding, the redemption date. The Floating Rate Euro Notes cannot be redeemed at the option of the Company.
If the Company experiences certain change of control events with respect to a series of Euro Notes, it must offer to purchase all Euro Notes of such series at a purchase price equal to 101% of the principal amount of such Euro Notes, plus accrued and unpaid interest, if any, to, but excluding, the date of purchase.
The Euro Notes Indenture contains covenants that, among other things, restrict the Company's ability and the ability of certain of its subsidiaries to enter into sale and leaseback transactions; create liens; and consolidate, merge or sell all or substantially all of the Company's assets. The Euro Notes Indenture also provides for customary events of default (subject in
131
Table of Contents
certain cases to customary grace and cure periods), which include nonpayment, breach of covenants, payment defaults or acceleration of other indebtedness, failure to pay certain judgments and certain events of bankruptcy and insolvency. These covenants and events of default are subject to a number of important qualifications, limitations and exceptions that are described in the Euro Notes Indenture. If an event of default with respect to the Euro Notes of a series occurs under the Euro Notes Indenture, the principal amount of all of the Euro Notes of such series then outstanding, plus accrued and unpaid interest, if any, to the date of acceleration, may become immediately due and payable.
The Company utilized the net proceeds from this offering to repay or otherwise refinance the Company's or any of the Company's subsidiaries indebtedness (the "Refinancing"), to pay fees and expenses associated with the Refinancing and for general corporate purposes. At December 31, 2016 , the outstanding balance of the Floating Rate Euro Notes, 2020 Euro Notes, 2024 Euro Notes and 2028 Euro Notes was $526.0 million , $785.7 million , $1.05 billion and $781.1 million , respectively, converted at the December 31, 2016 EUR to USD spot exchange rate. At December 31, 2016, discounts on the 2020 Euro Notes, 2024 Euro Notes and 2028 Euro Notes were approximately $3.3 million , $2.8 million and $7.9 million , respectively, converted at the December 31, 2016 EURO to USD spot exchange rate. During the year ended December 31, 2016 , the Company recorded mark-to-market gains, included in other expense, net on the Consolidated Statements of Operations, related to the Floating Rate Euro Notes, 2020 Euro Notes, 2024 Euro Notes and 2028 Euro Notes of approximately $5.3 million , $8.0 million , $10.7 million and $8.0 million , respectively. During the year ended December 31, 2016 , the Company incurred approximately $15.6 million in financing fees related to the Euro Notes, which were recorded as deferred financing fees in the Consolidated Balance Sheets .
Meda Borrowings
Upon settlement of the Offer on August 5, 2016, Meda became a controlled subsidiary of Mylan. Meda is party to certain debt obligations, all of which remained outstanding following the settlement of the Offer. In conjunction with the effectiveness of the 2016 Term Loans, the Meda Credit Agreement was terminated. As a result of the termination, the Company repaid 16.9kr billion ( $1.8 billion ) of borrowings outstanding thereunder. In addition, during the year ended December 31, 2016 , the Company repaid approximately $567 million of Meda's bank loans. At December 31, 2016 , Meda's borrowings include a bilateral bank loan of 2kr billion , a Swedish MTN program with an upper limit of 7kr billion and a Swedish commercial paper program with an upper limit of 4kr billion .
Bank Loans
The settlement of the Offer constituted a Change of Control (as defined in the Loan Agreement referred to below) under the Loan Agreement, dated as of September 17, 2014 (the "Loan Agreement"), between Meda, as borrower, and Svensk Exportkredit, as lender. As of December 31, 2016 , there was 2kr billion ( $219.6 million ) aggregate principal amount of loans outstanding under the Loan Agreement. In accordance with the terms of the Loan Agreement, Meda notified Svensk Exportkredit of the Change of Control. No agreement to amend the terms of the Loan Agreement was reached within 30 days of Svensk Exportkredit's receipt of notice from Meda of the Change of Control. As a result, Svensk Exportkredit was permitted to cancel its commitment and demand repayment of the loans in accordance with the terms of the Loan Agreement, but Svensk Exportkredit did not exercise such put rights. The Loan Agreement contains customary affirmative covenants, including among others, covenants pertaining to notices of default and certain material events, maintenance of authorizations and compliance with laws, as well as customary negative covenants, including limitations on disposals, liens, mergers and certain other corporate reconstructions and changes in Meda's lines of business.
On December 22, 2016, Meda entered into the Amendment and Waiver Agreement (the "Amendment") to the Loan Agreement. The Amendment provides for (i) the deletion of the covenant limiting indebtedness of Meda's subsidiaries and the covenant requiring Meda to deliver its consolidated quarterly and annual financial statements to Svensk Exportkredit; (ii) the modification of the covenant limiting asset dispositions by Meda and its subsidiaries to permit any dispositions other than those that could reasonably be expected to jeopardize Meda's ability, or the Company's ability pursuant to the guarantee described below, to fulfill its obligations under the Loan Agreement; (iii) the deletion of the financial maintenance covenants applicable to Meda; (iv) the waiver of compliance by Meda with the financial maintenance covenants applicable to Meda and the related reporting requirements for the fiscal quarter ending September 30, 2016; (v) the waiver of any put rights (including those described above) arising in connection with the Company's acquisition of a majority of the issued share capital in Meda or any action taken in connection therewith; (vi) the modification of the change of control definition to provide that a change of control will occur under the Loan Agreement if (a) the Company fails to, directly or indirectly, own all or substantially all of the issued share capital or votes in Meda or (b) any person (other than Stichting Preferred Shares Mylan) acquires more than 50% of the issued share capital or votes in the Company; (vii) the modification of the covenant limiting mergers by Meda and its subsidiaries to permit mergers with the Company and its subsidiaries; and (viii) the modification of the cross default and
132
Table of Contents
insolvency default provisions in the Loan Agreement to conform with the cross default and insolvency default provisions in the Company's 2016 Senior Revolving Credit Agreement and 2016 Senior Term Credit Agreement.
Concurrent with, and as a condition to, the effectiveness of the Amendment, the Company and Meda entered into the Guarantee Agreement, dated as of December 22, 2016 (the "Guarantee"), among Meda, the Company and Svensk Exportkredit. Under the Guarantee, the Company guarantees the payment and performance of all obligations, including repayment of the 2kr billion loan, of Meda under the Loan Agreement (collectively, the "Obligations"). The Guarantee and the obligations of the Company thereunder will automatically terminate upon the payment in full of the Obligations.
MTN Program
On December 20, 2016, the Company issued a guarantee (the "MTN Guarantee") in favor of each of the holders of the 2013/2018 583kr million floating rate notes and 2014/2019 750kr million floating rate notes (collectively, the "Meda MTN") issued by Meda. Under the MTN Guarantee, the Company guarantees the fulfillment of Meda's obligations to the holders of the Meda MTN according to the terms and conditions of the Meda MTN, including payment of interest in accordance with the terms and conditions of the Meda MTN and the repayment of the principal on the respective maturity date of the Meda MTN. The MTN Guarantee and the obligations of the Company thereunder will automatically terminate upon the payment in full of the Meda MTN.
The MTN program contains covenants that, among other things, restrict Meda's ability and the ability of certain of Meda's subsidiaries to substantially change the general nature of its business; create liens to secure debt securities or other publicly traded debt; or sell or dispose of Meda's assets to the extent such sales or disposition could jeopardize Meda's ability to fulfill its obligations under the MTN program; and require Meda to maintain the listing of the loans under the MTN program on Nasdaq Stockholm. As long as the loans under the MTN program are listed on Nasdaq Stockholm, Meda is required to comply with certain Nasdaq Stockholm disclosure requirements. The MTN program also provides for customary events of default (subject in certain cases to customary grace and cure periods), which include nonpayment, breach of covenants, payment defaults or acceleration of other indebtedness, failure to pay certain judgments and certain events of bankruptcy and insolvency. These covenants and events of default are subject to a number of important qualifications, limitations and exceptions that are described in the general terms and conditions of the MTN program. If an event of default with respect to the loans under the MTN program occurs, the principal amount of all of the loans under the MTN program then outstanding, plus accrued and unpaid interest, if any, to the date of acceleration, may become immediately due and payable.
Issuance of June 2016 Senior Notes
During 2016, in anticipation of the completion of the Offer, Mylan N.V. issued $1.00 billion aggregate principal amount of 2.500% Senior Notes due 2019, $2.25 billion aggregate principal amount of 3.150% Senior Notes due 2021, $2.25 billion aggregate principal amount of 3.950% Senior Notes due 2026 and $1.00 billion aggregate principal amount of 5.250% Senior Notes due 2046 (collectively, the "June 2016 Senior Notes") in a private offering exempt from the registration requirements of the Securities Act, to qualified institutional buyers in accordance with Rule 144A and to persons outside of the U.S. pursuant to Regulation S under the Securities Act. The June 2016 Senior Notes were issued pursuant to an indenture, dated as of June 9, 2016 (the "June 2016 Indenture"), among the Company, the Guarantor, and The Bank of New York Mellon, as trustee. The June 2016 Senior Notes were guaranteed by the Guarantor upon issuance. In addition, the Company entered into a registration rights agreement, dated as of June 9, 2016, pursuant to which the Company and Mylan Inc. were required to use commercially reasonable efforts to file a registration statement with respect to an offer to exchange each series of the June 2016 Senior Notes for new notes with the same aggregate principal amount and terms identical in all material respects. In December 2016, Mylan N.V. and Mylan Inc. filed a registration statement with the SEC with respect to an offer to exchange these notes for registered notes with the same aggregate principal amount and terms substantially identical in all material respects, which was declared effective on January 3, 2017. The exchange offer expired on January 31, 2017 and settled on February 3, 2017.
The June 2016 Indenture contains covenants that, among other things, restrict the Company's ability and the ability of certain of its subsidiaries to enter into sale and leaseback transactions; create liens; consolidate, merge or sell all or substantially all of the Company's assets; and with respect to such subsidiaries only, guarantee certain of our or our other subsidiaries' outstanding obligations or incur certain obligations without also guaranteeing our obligations under the June 2016 Senior Notes on a senior basis. The June 2016 Indenture also provides for customary events of default (subject in certain cases to customary grace and cure periods), which include nonpayment, breach of covenants, payment defaults or acceleration of other indebtedness, failure to pay certain judgments and certain events of bankruptcy and insolvency. These covenants and events of default are subject to a number of important qualifications, limitations and exceptions that are described in the June 2016 Indenture. If an event of default with respect to the June 2016 Senior Notes of a series occurs under the June 2016 Indenture,
133
Table of Contents
the principal amount of all of the June 2016 Senior Notes of such series then outstanding, plus accrued and unpaid interest, if any, to the date of acceleration, may become immediately due and payable.
The 2.500% Senior Notes due 2019 mature on June 7, 2019, subject to earlier repurchase or redemption in accordance with the terms of the June 2016 Indenture. The 2.500% Senior Notes due 2019 bear interest at a rate of 2.500% per annum, accruing from June 9, 2016. Interest on the 2.500% Senior Notes due 2019 is payable semi-annually in arrears on June 7 and December 7 of each year, commencing on December 7, 2016. The 3.150% Senior Notes due 2021 mature on June 15, 2021, subject to earlier repurchase or redemption in accordance with the terms of the June 2016 Indenture. The 3.150% Senior Notes due 2021 bear interest at a rate of 3.150% per annum, accruing from June 9, 2016. Interest on the 3.150% Senior Notes due 2021 is payable semi-annually in arrears on June 15 and December 15 of each year, commencing on December 15, 2016. The 3.950% Senior Notes due 2026 mature on June 15, 2026, subject to earlier repurchase or redemption in accordance with the terms of the June 2016 Indenture. The 3.950% Senior Notes due 2026 bear interest at a rate of 3.950% per annum, accruing from June 9, 2016. Interest on the 3.950% Senior Notes due 2026 is payable semi-annually in arrears on June 15 and December 15 of each year, commencing on December 15, 2016. The 5.250% Senior Notes due 2046 mature on June 15, 2046, subject to earlier repurchase or redemption in accordance with the terms of the June 2016 Indenture. The 5.250% Senior Notes due 2046 bear interest at a rate of 5.250% per annum, accruing from June 9, 2016. Interest of the 5.250% Senior Notes due 2046 is payable semi-annually in arrears on June 15 and December 15 of each year, commencing on December 15, 2016.
At December 31, 2016 , the outstanding balance of the 2.500% Senior Notes due 2019, 3.150% Senior Notes due 2021, 3.950% Senior Notes due 2026 and 5.250% Senior Notes due 2046 was $999.1 million , $2.25 billion , $2.23 billion and $999.8 million , respectively, which includes discounts of $0.9 million , $2.4 million , $16.5 million and $0.2 million , respectively. During the year ended December 31, 2016 , the Company incurred approximately $48.7 million in financing fees, which were recorded as deferred financing costs in the Consolidated Balance Sheets .
2016 Bridge Credit Agreement
In connection with the Offer , on February 10, 2016 , the Company entered into a Bridge Credit Agreement (the "2016 Bridge Credit Agreement"), among the Company, as borrower, Mylan Inc. , as guarantor, Deutsche Bank AG Cayman Islands Branch, as administrative agent and a lender, Goldman Sachs Bank USA, as a lender, Goldman Sachs Lending Partners LLC, as a lender, and other lenders party thereto from time to time. The Company incurred total financing and ticking fees of approximately $45.2 million related to the 2016 Bridge Credit Agreement. During the first quarter of 2016, the Company wrote off approximately $3.0 million of financing fees related to the Tranche B Loans (as defined in the 2016 Bridge Credit Agreement) in conjunction with the termination of the Tranche B Loans. The remaining commitments under the 2016 Bridge Credit Agreement were permanently terminated in their entirety in connection with the completion of the offering of the June 2016 Senior Notes. As a result of the termination of the 2016 Bridge Credit Agreement, the Company expensed the remaining $30.2 million of unamortized financing fees related to the 2016 Bridge Credit Agreement to other expense, net in the Consolidated Statements of Operations during the year ended December 31, 2016.
2015 Activity
Issuance of December 2015 Senior Notes
In December 2015, the Company issued $500 million aggregate principal amount of 3.000% Senior Notes due December 2018 and $500 million aggregate principal amount of 3.750% Senior Notes due December 2020 (collectively, the "December 2015 Senior Notes") in a private offering exempt from the registration requirements of the Securities Act, to qualified institutional buyers in accordance with Rule 144A and to persons outside of the U.S. pursuant to Regulation S under the Securities Act. The December 2015 Senior Notes were issued pursuant to an indenture dated December 9, 2015 (the "December 2015 Indenture") entered into among the Company, the Guarantor and The Bank of New York Mellon as trustee. Interest payments on the December 2015 Senior Notes are due semi-annually in arrears on June 15th and December 15th of each year commencing on June 15, 2016. The December 2015 Senior Notes were guaranteed by the Guarantor upon issuance. In connection with the offering of the December 2015 Senior Notes, the Company entered into a registration rights agreement pursuant to which the Company and Mylan Inc. were required to use commercially reasonable efforts to file a registration statement with respect to an offer to exchange each series of the December 2015 Senior Notes for new notes with the same aggregate principal amount and terms substantially identical in all material respects and to cause the exchange offer registration statement to be declared effective by the SEC and to consummate the exchange offer not later than 365 days following the date of issuance of the December 2015 Senior Notes. In December 2016, Mylan N.V. and Mylan Inc. filed a registration statement with the SEC with respect to an offer to exchange these notes for registered notes with the same aggregate principal amount and terms substantially identical in all material respects, which was declared effective on January 3, 2017. The exchange offer expired on January 31, 2017 and settled on February 3, 2017.
134
Table of Contents
The Company may redeem the 3.000% Senior Notes due in 2018 at any time prior to the maturity date and the 3.750% Senior Notes due in 2020 at any time that is one month prior to the maturity date at a redemption price equal to the greater of 100% of the aggregate principal amount of the notes and the sum of the present values of the remaining scheduled payments of principal and interest on the notes being redeemed, discounted to the redemption date on a semi-annual basis (assuming a 360-day year consisting of twelve 30-day months) at a treasury rate plus 30 basis points with respect to the 3.000% Senior Notes due 2018 and 35 basis points with respect to the 3.750% Senior Notes due 2020, plus accrued and unpaid interest up to, but excluding the redemption date. If the Company experiences certain change of control events with respect to a series of December 2015 Senior Notes, it must offer to purchase all notes of such series at a purchase price equal to 101% of the principal amount of such notes, plus accrued but unpaid interest, if any, to (but not including) the date of purchase.
The December 2015 Indenture contains covenants that, among other things, restrict the Company's ability and the ability of certain of its subsidiaries to enter into sale and leaseback transactions; create liens; and consolidate, merge or sell all or substantially all of the Company's assets. The December 2015 Indenture also provides for customary events of default (subject in certain cases to customary grace and cure periods), which include nonpayment, breach of covenants, payment defaults or acceleration of other indebtedness, failure to pay certain judgments and certain events of bankruptcy and insolvency. These covenants and events of default are subject to a number of important qualifications, limitations and exceptions that are described in the December 2015 Indenture. If an event of default with respect to the Notes of a series occurs under the December 2015 Indenture, the principal amount of all of the Notes of such series then outstanding, plus accrued but unpaid interest to the date of acceleration, may become immediately due and payable.
The net proceeds from the offering were primarily used to repay amounts outstanding under the 2014 Revolving Credit Agreement and the Receivables Facility. In addition, the offering was used to finance a portion of the repurchase of our ordinary shares pursuant to the Share Repurchase Program. At December 31, 2015, the outstanding balance under the 3.000% Senior Notes due 2018 and 3.750% Senior notes due 2020 was $499.4 million and $499.8 million , respectively, which includes discounts of $0.6 million and $0.2 million , respectively. During the year ended December 31, 2015, the Company incurred approximately $4.7 million of fees, which were recorded as deferred financing costs in the Consolidated Balance Sheets .
July 2020 Senior Notes Redemption
On June 15, 2015 , the Company announced its intention to redeem all of its outstanding 7.875% Senior Notes due 2020 (the " July 2020 Senior Notes ") on July 15, 2015 at a redemption price of 103.938% of the principal amount, together with accrued and unpaid interest at the redemption date. On July 15, 2015 , the Company utilized a portion of the proceeds borrowed under the 2015 Term Credit Agreement to complete its redemption of the July 2020 Senior Notes for a total of approximately $1.08 billion , including a $39.4 million redemption premium and approximately $39.4 million of accrued interest. In addition, the Company expensed approximately $11.1 million of previously recorded deferred financing fees offset by the write-off of the remaining unamortized premium of approximately $9.7 million related to the July 2020 Senior Notes .
Senior Notes Consent Solicitation
During the first quarter of 2015, Mylan Inc. and Mylan N.V. completed consent solicitations relating to Mylan Inc. 's 3.750% Cash Convertible Notes due 2015, 7.875% Senior Notes due 2020, 3.125% Senior Notes due 2023, 1.800% Senior Notes due 2016, 2.600% Senior Notes due 2018, 1.350% Senior Notes due 2016, 2.550% Senior Notes due 2019, 4.200% Senior Notes due 2023 and 5.400% Senior Notes due 2043 (collectively, the "Senior Notes"). The consent solicitations modified the reporting covenants set forth in the indentures governing the Senior Notes so that, subject to certain conditions, the reports, information and other documents required to be filed with the SEC and furnished to holders of the Senior Notes may, at the option of Mylan Inc. , be filed by and be those of any direct or indirect parent entity, rather than Mylan Inc. During the year ended December 31, 2015, the Company incurred approximately $21.8 million of fees, which were recorded as deferred financing costs in the Consolidated Balance Sheets .
2015 Bridge Credit Agreement
On April 24, 2015 , the Company entered into a bridge credit agreement, which was amended on April 29, 2015 and on August 6, 2015 (the "Bridge Credit Agreement"), among the Company, as borrower, Mylan Inc., as guarantor, the lenders party thereto from time to time and Goldman Sachs Bank USA, as the administrative agent, in connection with the Company's previously announced offer (the "Perrigo Offer") to acquire all of the issued and outstanding ordinary shares of Perrigo Company plc. The Company announced on November 13, 2015 that the conditions to the Perrigo Offer had not been satisfied and the Perrigo Offer had lapsed in accordance with its terms. As such, the commitments under the Bridge Credit Agreement terminated. During the year ended December 31, 2015, the Company paid approximately $99.6 million in commitment and other fees under the Bridge Credit Agreement, which were expensed in the Consolidated Statement of Operations.
135
Table of Contents
Fair Value
At December 31, 2016 and December 31, 2015 , the fair value of the Senior Notes and Euro Notes was approximately $13.2 billion and $4.8 billion , respectively. The fair values of the Senior Notes and Euro Notes were valued at quoted market prices from broker or dealer quotations and were classified as Level 2 in the fair value hierarchy. Based on quoted market rates of interest and maturity schedules for similar debt issues, the fair values of the Company's 2016 Term Loan and Meda borrowings determined based on Level 2 inputs, approximate their carrying values at December 31, 2016 and December 31, 2015 .
Mandatory minimum repayments remaining on the outstanding long-term debt at December 31, 2016 , excluding the discounts and premiums, are as follows for each of the periods ending December 31:
(In millions) | Total | ||
2017 | $ | 220 | |
2018 | 1,740 | | |
2019 | 3,182 | | |
2020 | 1,289 | | |
2021 | 2,250 | | |
Thereafter | 6,841 | | |
Total | $ | 15,522 | |
9. | Comprehensive Earnings |
Accumulated other comprehensive loss , as reflected on the Consolidated Balance Sheets , is comprised of the following:
(In millions) | December 31, 2016 |
| December 31, 2015 | ||||
Accumulated other comprehensive loss: |
|
|
| ||||
Net unrealized gain (loss) on marketable securities, net of tax | $ | 14.5 | |
| $ | (1.0 | ) |
Net unrecognized losses and prior service cost related to defined benefit plans, net of tax | (0.5 | ) |
| (14.9 | ) | ||
Net unrecognized losses on derivatives in cash flow hedging relationships, net of tax | (38.6 | ) |
| (18.1 | ) | ||
Net unrecognized losses on derivatives in net investment hedging relationships, net of tax | (1.4 | ) |
| - | | ||
Foreign currency translation adjustment | (2,237.7 | ) |
| (1,730.3 | ) | ||
| $ | (2,263.7 | ) |
| $ | (1,764.3 | ) |
136
Table of Contents
Components of accumulated other comprehensive (loss) earnings, before tax, consist of the following:
| Year Ended December 31, 2016 | ||||||||||||||||||||||||||||
Gains and Losses on Derivatives in Cash Flow Hedging Relationships |
| Gains and Losses on Net Investment Hedges |
| Gains and Losses on Marketable Securities |
| Defined Pension Plan Items |
| Foreign Currency Translation Adjustment |
| Totals | |||||||||||||||||||
(In millions) | Foreign Currency Forward Contracts |
| Interest Rate Swaps |
| Total |
|
|
|
|
|
|
|
|
|
| ||||||||||||||
Balance at December 31, 2015, net of tax |
|
|
|
| $ | (18.1 | ) |
| $ | - | |
| $ | (1.0 | ) |
| $ | (14.9 | ) |
| $ | (1,730.3 | ) |
| $ | (1,764.3 | ) | ||
Other comprehensive (loss) earnings before reclassifications, before tax |
|
|
|
| (84.2 | ) |
| (1.8 | ) |
| 24.6 | |
| 20.0 | |
| (507.4 | ) |
| (548.8 | ) | ||||||||
Amounts reclassified from accumulated other comprehensive (loss) earnings, before tax: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||||
Loss on foreign exchange forward contracts classified as cash flow hedges, included in net sales | 44.3 | |
|
|
| 44.3 | |
|
|
|
|
|
|
|
|
| 44.3 | | |||||||||||
Loss on interest rate swaps classified as cash flow hedges, included in interest expense |
|
| 8.7 | |
| 8.7 | |
|
|
|
|
|
|
|
|
| 8.7 | | |||||||||||
Amortization of prior service costs included in SG&A |
|
|
|
|
|
|
|
|
|
| 0.3 | |
|
|
| 0.3 | | ||||||||||||
Amortization of actuarial loss included in SG&A |
|
|
|
|
|
|
|
|
|
| 1.1 | |
|
|
| 1.1 | | ||||||||||||
Net other comprehensive (loss) earnings, before tax |
|
|
|
| (31.2 | ) |
| (1.8 | ) |
| 24.6 | |
| 21.4 | |
| (507.4 | ) |
| (494.4 | ) | ||||||||
Income tax (benefit) provision |
|
|
|
| (10.7 | ) |
| (0.4 | ) |
| 9.1 | |
| 7.0 | |
| - | |
| 5.0 | | ||||||||
Balance at December 31, 2016, net of tax |
|
|
|
| $ | (38.6 | ) |
| $ | (1.4 | ) |
| $ | 14.5 | |
| $ | (0.5 | ) |
| $ | (2,237.7 | ) |
| $ | (2,263.7 | ) | ||
137
Table of Contents
| Year Ended December 31, 2015 | ||||||||||||||||||||||||
Gains and Losses on Derivatives in Cash Flow Hedging Relationships |
| Gains and Losses on Marketable Securities |
| Defined Pension Plan Items |
| Foreign Currency Translation Adjustment |
| Totals | |||||||||||||||||
(In millions) | Foreign Currency Forward Contracts |
| Interest Rate Swaps |
| Total |
|
|
|
|
|
|
|
| ||||||||||||
Balance at December 31, 2014, net of tax |
|
|
|
| $ | (28.4 | ) |
| $ | 0.3 | |
| $ | (19.5 | ) |
| $ | (939.4 | ) |
| $ | (987.0 | ) | ||
Other comprehensive earnings (loss) before reclassifications, before tax |
|
|
|
| 129.0 | |
| (2.0 | ) |
| 0.6 | |
| (790.9 | ) |
| (663.3 | ) | |||||||
Amounts reclassified from accumulated other comprehensive (loss) earnings, before tax: |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Loss on foreign exchange forward contracts classified as cash flow hedges, included in net sales | (40.3 | ) |
|
|
| (40.3 | ) |
|
|
|
|
|
|
| (40.3 | ) | |||||||||
Loss on interest rate swaps classified as cash flow hedges, included in interest expense |
|
| (0.8 | ) |
| (0.8 | ) |
|
|
|
|
|
|
| (0.8 | ) | |||||||||
Loss on interest rate swaps classified as cash flow hedges, included in other expense, net |
|
| (71.2 | ) |
| (71.2 | ) |
|
|
|
|
|
|
| (71.2 | ) | |||||||||
Amortization of prior service costs included in SG&A |
|
|
|
|
|
|
|
| 0.3 | |
|
|
| 0.3 | | ||||||||||
Amortization of actuarial gain included in SG&A |
|
|
|
|
|
|
|
| 2.2 | |
|
|
| 2.2 | | ||||||||||
Net other comprehensive earnings (loss), before tax |
|
|
|
| 16.7 | |
| (2.0 | ) |
| 3.1 | |
| (790.9 | ) |
| (773.1 | ) | |||||||
Income tax provision (benefit) |
|
|
|
| 6.4 | |
| (0.7 | ) |
| (1.5 | ) |
| - | |
| 4.2 | | |||||||
Balance at December 31, 2015, net of tax |
|
|
|
| $ | (18.1 | ) |
| $ | (1.0 | ) |
| $ | (14.9 | ) |
| $ | (1,730.3 | ) |
| $ | (1,764.3 | ) | ||
138
Table of Contents
| Year Ended December 31, 2014 | ||||||||||||||||||||||||
| Gains and Losses on Derivatives in Cash Flow Hedging Relationships |
| Gains and Losses on Marketable Securities |
| Defined Pension Plan Items |
| Foreign Currency Translation Adjustment |
| Totals | ||||||||||||||||
(In millions) | Foreign Currency Forward Contracts |
| Interest Rate Swaps |
| Total |
|
|
|
|
|
|
|
| ||||||||||||
Balance at December 31, 2013, net of tax |
|
|
|
| $ | 84.8 | |
| $ | 0.3 | |
| $ | (8.7 | ) |
| $ | (316.5 | ) |
| $ | (240.1 | ) | ||
Other comprehensive loss before reclassifications, before tax |
|
|
|
| (231.1 | ) |
| - | |
| (12.8 | ) |
| (622.9 | ) |
| (866.8 | ) | |||||||
Amounts reclassified from accumulated other comprehensive loss, before tax: |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Loss on foreign exchange forward contracts classified as cash flow hedges, included in net sales | (47.9 | ) |
|
|
| (47.9 | ) |
|
|
|
|
|
|
| (47.9 | ) | |||||||||
Loss on interest rate swaps classified as cash flow hedges, included in interest expense |
|
| (0.6 | ) |
| (0.6 | ) |
|
|
|
|
|
|
| (0.6 | ) | |||||||||
Amortization of prior service costs included in SG&A |
|
|
|
|
|
|
|
| (0.3 | ) |
|
|
| (0.3 | ) | ||||||||||
Amortization of actuarial gain included in SG&A |
|
|
|
|
|
|
|
| (0.7 | ) |
|
|
| (0.7 | ) | ||||||||||
Amounts reclassified from accumulated other comprehensive loss, before tax |
|
|
|
| (48.5 | ) |
| - | |
| (1.0 | ) |
| - | |
| (49.5 | ) | |||||||
Net other comprehensive loss, before tax |
|
|
|
| (182.6 | ) |
| - | |
| (11.8 | ) |
| (622.9 | ) |
| (817.3 | ) | |||||||
Income tax benefit |
|
|
|
| (69.4 | ) |
| - | |
| (1.0 | ) |
| - | |
| (70.4 | ) | |||||||
Balance at December 31, 2014, net of tax |
|
|
|
| $ | (28.4 | ) |
| $ | 0.3 | |
| $ | (19.5 | ) |
| $ | (939.4 | ) |
| $ | (987.0 | ) | ||
139
Table of Contents
10. | Income Taxes |
Income tax provision consisted of the following components:
| Year Ended December 31, | ||||||||||
(In millions) | 2016 |
| 2015 |
| 2014 | ||||||
U.S. Federal: |
|
|
|
|
| ||||||
Current | $ | 86.8 | |
| $ | 13.7 | |
| $ | 218.1 | |
Deferred | (303.8 | ) |
| (35.8 | ) |
| (147.5 | ) | |||
| (217.0 | ) |
| (22.1 | ) |
| 70.6 | | |||
U.S. State: |
|
|
|
|
| ||||||
Current | 13.8 | |
| 8.1 | |
| 33.8 | | |||
Deferred | (14.8 | ) |
| (11.9 | ) |
| (1.6 | ) | |||
| (1.0 | ) |
| (3.8 | ) |
| 32.2 | | |||
Non-U.S.: |
|
|
|
|
| ||||||
Current | 150.6 | |
| 161.8 | |
| 104.6 | | |||
Deferred | (290.9 | ) |
| (68.2 | ) |
| (166.0 | ) | |||
| (140.3 | ) |
| 93.6 | |
| (61.4 | ) | |||
Income tax provision | $ | (358.3 | ) |
| $ | 67.7 | |
| $ | 41.4 | |
Earnings before income taxes and noncontrolling interest: |
|
|
|
|
| ||||||
United Kingdom | $ | (129.4 | ) |
| $ | (189.6 | ) |
| $ | 14.2 | |
United States | (187.4 | ) |
| 474.4 | |
| 679.2 | | |||
Foreign - Other | 438.5 | |
| 630.6 | |
| 281.1 | | |||
Total earnings before income taxes and noncontrolling interest | $ | 121.7 | |
| $ | 915.4 | |
| $ | 974.5 | |
For all periods presented, the allocation of earnings before income taxes and noncontrolling interest between U.S. and non-U.S. operations includes intercompany interest allocations between certain domestic and foreign subsidiaries. These amounts are eliminated on a consolidated basis.
Temporary differences and carryforwards that result in deferred tax assets and liabilities were as follows:
(In millions) | December 31, 2016 |
| December 31, 2015 | ||||
Deferred tax assets: |
|
|
| ||||
Employee benefits | $ | 265.2 | |
| $ | 202.4 | |
Litigation reserves | 239.9 | |
| 20.7 | | ||
Accounts receivable allowances | 396.0 | |
| 224.9 | | ||
Tax credit and loss carryforwards | 634.0 | |
| 463.7 | | ||
Intangible assets | 96.7 | |
| 65.3 | | ||
Other | 283.5 | |
| 168.7 | | ||
| 1,915.3 | |
| 1,145.7 | | ||
Less: Valuation allowance | (460.7 | ) |
| (355.7 | ) | ||
Total deferred tax assets | 1,454.6 | |
| 790.0 | | ||
Deferred tax liabilities: |
|
|
| ||||
Plant and equipment | 183.9 | |
| 184.4 | | ||
Intangible assets and goodwill | 2,601.6 | |
| 827.0 | | ||
Other | 42.3 | |
| 39.1 | | ||
Total deferred tax liabilities | 2,827.8 | |
| 1,050.5 | | ||
Deferred tax liabilities, net | $ | (1,373.2 | ) |
| $ | (260.5 | ) |
140
Table of Contents
For those foreign subsidiaries whose investments are permanent in duration, income and foreign withholding taxes have not been provided on the amount by which the investment in those subsidiaries, as recorded for financial reporting, exceeds the tax basis. This amount may become taxable upon a repatriation of assets from the subsidiary or a sale or liquidation of the subsidiary. The amount of such temporary differences is approximately $1.8 billion at December 31, 2016 . Determination of the amount of any unrecognized deferred income tax liability on this temporary difference is not practicable as such determination involves material uncertainties about the potential extent and timing of any distributions, the availability and complexity of calculating foreign tax credits, and the potential indirect tax consequences of such distributions, including withholding taxes. No deferred taxes have been recorded on the instances whereby the Company's investment in foreign subsidiaries is currently greater for tax purposes than for U.S. GAAP purposes, as management has no current plans that would cause that temporary difference to reverse in the foreseeable future.
Prior to the EPD Transaction, the statutory income tax rate applicable to Mylan Inc. in the U.S. was 35% . Since the EPD Transaction the statutory income tax rate applicable to Mylan N.V. in the United Kingdom (the "U.K.") has been 20% for the years ending December 31, 2016 and 2015 . A reconciliation of the statutory tax rate to the effective tax rate is as follows:
| Year Ended December 31, | |||||||
| 2016 |
| 2015 |
| 2014 | |||
Statutory tax rate | 20.0 | % |
| 20.0 | % |
| 35.0 | % |
|
|
|
|
|
| |||
United States Operations |
|
|
|
|
| |||
Clean energy and research credits | (85.9 | )% |
| (13.0 | )% |
| (9.6 | )% |
U.S. rate differential | (36.9 | )% |
| 4.6 | % |
| - | % |
Other U.S. items | 3.5 | % |
| - | % |
| (2.2 | )% |
Foreign tax credits, net | - | % |
| - | % |
| (0.6 | )% |
State income taxes and credits | (3.7 | )% |
| - | % |
| 2.2 | % |
Other Foreign Operations |
|
|
|
|
| |||
Luxembourg | (54.1 | )% |
| 1.7 | % |
| 11.6 | % |
Luxembourg - U.S. Branch | (28.8 | )% |
| (11.2 | )% |
| - | % |
Gibraltar | (49.2 | )% |
| (4.9 | )% |
| (21.3 | )% |
India | (13.0 | )% |
| (0.6 | )% |
| (0.1 | )% |
Ireland | (7.3 | )% |
| (1.9 | )% |
| (1.3 | )% |
Other | (5.2 | )% |
| 1.7 | % |
| (0.8 | )% |
Uncertain tax positions | 0.8 | % |
| (0.3 | )% |
| 1.6 | % |
Valuation allowance | 79.9 | % |
| 6.5 | % |
| 0.9 | % |
Merger of foreign subsidiaries | (123.5 | )% |
| - | % |
| (15.2 | )% |
Other foreign items | 9.0 | % |
| 4.8 | % |
| 4.0 | % |
Effective tax rate | (294.4 | )% |
| 7.4 | % |
| 4.2 | % |
The Company's jurisdictional location of earnings is a component of the effective tax rate each year, and the rate impact of this component is also influenced by the level of such earnings as compared to the Company's total earnings. The jurisdictional mix of earnings can vary as a result of operating fluctuations in the normal course of business and as a result of the extent and location of other income and expense items, such as internal restructurings, and gains and losses on strategic business decisions.
During 2016, the Company merged its wholly owned subsidiary, Jai Pharma Limited, into Mylan Laboratories Limited. The merger resulted in the recognition of a deferred tax asset of approximately $150 million for the tax deductible goodwill in excess of the book goodwill with a corresponding benefit to income tax provision for the year ended December 31, 2016.
During 2014, the Company merged its wholly owned subsidiaries, Agila Specialties Private Limited and Onco Therapies Limited, into Mylan Laboratories Limited . The merger resulted in the recognition of a deferred tax asset of approximately $156.0 million for the tax deductible goodwill in excess of the book goodwill with a corresponding benefit to income tax provision for the year ended December 31, 2014.
141
Table of Contents
Valuation Allowance
A valuation allowance is provided when it is more likely than not that some portion or all of the deferred tax assets will not be realized. At December 31, 2016 , a valuation allowance has been applied to certain foreign and state deferred tax assets in the amount of $460.7 million . The valuation allowance increased by $156.2 million during 2016 .
Net Operating Losses
As of December 31, 2016 , the Company has U.S. federal net operating loss carryforwards of $28 million , and U.S. state income tax loss carryforwards of approximately $2.5 billion . The Company also has non-U.S. net operating loss carryforwards of approximately $1.5 billion , of which $916 million can be carried forward indefinitely, with the remaining $633 million expiring in years 2016 through 2036. Most of the net operating losses have a full valuation allowance.
The Company has $82.0 million of capital loss carryforwards expiring in 2019 through 2022. A full valuation allowance is recorded against these losses. The Company also has $38 million of foreign, U.S. and U.S. state credit carryovers, expiring in various amounts through 2036.
Tax Examinations
The Company is subject to income taxes in a number of jurisdictions. As a result, a certain degree of estimation is required in recording the assets and liabilities related to income taxes. The Company's tax positions are subject to audit by the local taxing authorities in each tax jurisdiction. These tax audits and examinations can involve complex issues, interpretations and judgments and the resolution of matters may span multiple years, particularly if subject to litigations or the negotiation of a settlement. As of December 31, 2016, the Company has two separate matters undergoing U.S. Tax Court proceedings. In addition, the Company has certain other ongoing tax matters in jurisdictions outside of the U.S. It is reasonably possible that one or all of these matters are resolved during 2017.
Although the Company believes that adequate provisions have been made for these uncertain tax positions, the Company's assessment of uncertain tax positions is based on estimates and assumptions that the Company believes are reasonable but the estimates for unrecognized tax benefits and potential tax benefits may not be representative of actual outcomes, and variations from such estimates could materially affect the Company's financial condition, results of operations or cash flows in the period of resolution, settlement or when the statues of limitations expire.
Mylan is subject to ongoing U.S. Internal Revenue Service ("IRS") examinations and is a voluntary participant in the IRS Compliance Assurance Process. The years 2015 and 2016 are the open years under examination. The years 2012, 2013 and 2014 have one issue open and a Tax Court petition has been filed regarding the matter. Years 2007 through 2011 are currently scheduled for U.S. Tax Court proceedings in 2017. Tax and interest continue to be accrued related to uncertain tax positions.
The Company's major state taxing jurisdictions remain open from fiscal year 2007 through 2016, with several state audits currently in progress. The Company's major international taxing jurisdictions remain open from 2008 through 2016, some of which are indemnified by Merck KGaA and Strides Arcolab for tax assessments.
Accounting for Uncertainty in Income Taxes
The impact of an uncertain tax position that is more likely than not of being sustained upon audit by the relevant taxing authority must be recognized at the largest amount that is more likely than not to be sustained. No portion of an uncertain tax position will be recognized if the position has less than a 50% likelihood of being sustained.
As of December 31, 2016 and 2015 , the Company's Consolidated Balance Sheets reflect net liabilities for unrecognized tax benefits of $190.9 million and $174.1 million , of which $146.0 million and $128.1 million , respectively, would affect the Company's effective tax rate if recognized. Accrued interest and penalties included in the Consolidated Balance Sheets were $79.8 million and $73.1 million as of December 31, 2016 and 2015 , respectively. For the years ended December 31, 2016 , 2015 and 2014 , Mylan recognized $6.9 million , $0.8 million and $2.2 million , respectively, for interest expense related to uncertain tax positions. Interest expense and penalties related to income taxes are included in the tax provision.
142
Table of Contents
A reconciliation of the unrecognized tax benefits is as follows:
| Year Ended December 31, | ||||||||||
(In millions) | 2016 |
| 2015 |
| 2014 | ||||||
Unrecognized tax benefit - beginning of year | $ | 174.1 | |
| $ | 191.2 | |
| $ | 174.7 | |
Additions for current year tax positions | 2.1 | |
| 1.2 | |
| 21.9 | | |||
Additions for prior year tax positions | - | |
| - | |
| 6.3 | | |||
Reductions for prior year tax positions | (1.8 | ) |
| (9.0 | ) |
| (5.1 | ) | |||
Settlements | - | |
| (1.5 | ) |
| (1.5 | ) | |||
Reductions due to expirations of statute of limitations | (7.7 | ) |
| (7.8 | ) |
| (5.1 | ) | |||
Addition due to acquisition | 24.2 | |
| - | |
| - | | |||
Unrecognized tax benefit - end of year | $ | 190.9 | |
| $ | 174.1 | |
| $ | 191.2 | |
The Company believes that it is reasonably possible that the amount of unrecognized tax benefits will decrease in the next twelve months by approximately $100 million , involving federal and state tax audits and settlements, possible resolution of U.S. Tax Court proceedings and expirations of certain state and foreign statutes of limitations. The Company does not anticipate significant increases to the reserve within the next twelve months .
11. | Share-Based Incentive Plan |
The Company's shareholders have approved the 2003 Long-Term Incentive Plan (as amended, the "2003 Plan"). Under the 2003 Plan, 55,300,000 ordinary shares are reserved for issuance to key employees, consultants, independent contractors and non-employee directors of the Company through a variety of incentive awards, including: stock options, stock appreciation rights ("SAR"), restricted ordinary shares and units, performance awards ("PSU"), other stock-based awards and short-term cash awards. Stock option awards are granted with an exercise price equal to the fair market value of the ordinary shares underlying the options at the date of the grant, generally become exercisable over periods ranging from three to four years , and generally expire in ten years . Since approval of the 2003 Plan, no further grants of stock options have been made under any other previous plan.
In February 2014, Mylan 's Compensation Committee and the independent members of the Board of Directors adopted the One-Time Special Performance-Based Five-Year Realizable Value Incentive Program (the " 2014 Program ") under the 2003 Plan. Under the 2014 Program , certain key employees received a one-time, performance-based incentive award (the "Awards") either in the form of a grant of SAR or PSU. The initial Awards were granted in February 2014 and contain a five -year cliff-vesting feature based on the achievement of various performance targets, external market conditions and the employee's continued services. Additional Awards were granted in 2016 and are subject to the same performance conditions as the Awards granted in February 2014 and with a service vesting condition of between two and six years. The market condition was met on June 10, 2015 and is therefore no longer applicable to any of the Awards.
143
Table of Contents
The following table summarizes stock option and SAR (together, "stock awards") activity:
| Number of Shares Under Stock Awards |
| Weighted Average Exercise Price per Share | |||
Outstanding at December 31, 2013 | 13,563,881 | |
| $ | 22.05 | |
Granted | 6,226,185 | |
| 52.37 | | |
Exercised | (2,720,048 | ) |
| 20.25 | | |
Forfeited | (862,241 | ) |
| 38.28 | | |
Outstanding at December 31, 2014 | 16,207,777 | |
| $ | 33.21 | |
Granted | 937,873 | |
| 54.92 | | |
Exercised | (5,092,660 | ) |
| 22.48 | | |
Forfeited | (220,491 | ) |
| 46.36 | | |
Converted | (4,100,000 | ) |
| 53.33 | | |
Outstanding at December 31, 2015 | 7,732,499 | |
| $ | 31.85 | |
Granted | 876,397 | |
| 45.51 | | |
Exercised | (612,477 | ) |
| 23.13 | | |
Forfeited | (296,978 | ) |
| 50.70 | | |
Outstanding at December 31, 2016 | 7,699,441 | |
| $ | 33.38 | |
Vested and expected to vest at December 31, 2016 | 7,405,805 | |
| $ | 32.80 | |
Exercisable at December 31, 2016 | 5,672,524 | |
| $ | 28.10 | |
As of December 31, 2016 , stock awards outstanding, stock awards vested and expected to vest, and stock awards exercisable had average remaining contractual terms of 5.8 years , 5.7 years and 4.8 years , respectively. Also at December 31, 2016 , stock awards outstanding, stock awards vested and expected to vest and stock awards exercisable had aggregate intrinsic values of $74.0 million , $73.9 million and $73.2 million , respectively. During the year ended December 31, 2015, the Company recorded additional share-based compensation expense of approximately $21.8 million related to the accelerated vesting of equity awards as a result of the EPD Transaction.
A summary of the status of the Company's nonvested restricted ordinary shares and restricted stock unit awards, including PSUs (collectively, "restricted stock awards") as of December 31, 2016 and the changes during the year ended December 31, 2016 are presented below:
| Number of Restricted Stock Awards |
| Weighted Average Grant-Date Fair Value Per Share | |||
Nonvested at December 31, 2015 | 4,474,436 | |
| $ | 40.70 | |
Granted | 2,660,186 | |
| 45.05 | | |
Released | (1,088,088 | ) |
| 41.96 | | |
Forfeited | (378,704 | ) |
| 42.76 | | |
Nonvested at December 31, 2016 | 5,667,830 | |
| $ | 42.46 | |
Of the 2,660,186 restricted stock awards granted during the year ended December 31, 2016 , 1,368,088 vest ratably in five years or less and are not subject to market or performance conditions. Of the remaining restricted stock awards granted, 525,221 are subject to market conditions and will cliff vest in three years or less, 64,819 are subject to performance conditions and will cliff vest in less than three years and 110,756 are not subject to market or performance conditions and will cliff vest in three years or less. The remaining 543,442 restricted stock awards were granted under the 2014 Program and are subject to the performance condition and will cliff vest over various periods between two and six years . An additional 47,860 PSUs were granted and released as a result of exceeding certain performance targets during the year.
As of December 31, 2016 , the Company had $144.5 million of total unrecognized compensation expense, net of estimated forfeitures, related to all of its stock-based awards, which will be recognized over the remaining weighted average
144
Table of Contents
vesting period of 2.3 years . The total intrinsic value of stock-based awards exercised and restricted stock awards released during the years ended December 31, 2016 and 2015 was $60.7 million and $260.1 million , respectively.
2003 Plan
With respect to options granted under the Company's 2003 Plan, the fair value of each option grant was estimated at the date of grant using the Black-Scholes option pricing model. Black-Scholes utilizes assumptions related to volatility, the risk-free interest rate, the dividend yield and employee exercise behavior. Expected volatilities utilized in the model are based mainly on the implied volatility of the Company's stock price and other factors. The risk-free interest rate is derived from the U.S. Treasury yield curve in effect at the time of grant. The model incorporates exercise and post-vesting forfeiture assumptions based on an analysis of historical data. The expected lives of the grants are derived from historical and other factors.
The assumptions used for options granted under the 2003 Plan are as follows:
| Year Ended December 31, | ||||
| 2016 |
| 2015 |
| 2014 |
Volatility | 38.1% |
| 33.7% |
| 31.6% |
Risk-free interest rate | 1.4% |
| 1.7% |
| 1.9% |
Expected term (years) | 6.3 |
| 6.3 |
| 6.3 |
Forfeiture rate | 5.5% |
| 5.5% |
| 5.5% |
Weighted average grant date fair value per option | $17.90 |
| $20.18 |
| $17.44 |
2014 Program
Under the 2014 Program , approximately 4.4 million SARs and 1.5 million PSUs were granted in February 2014. The fair value of the Awards was determined using a Monte Carlo simulation as both the SARs and PSUs contain the same performance and market conditions. The Monte Carlo simulation involves a series of random trials that result in different future stock price paths over the contractual life of the SAR or PSU based on appropriate probability distributions. Conditions are imposed on each Monte Carlo simulation to determine the extent to which the performance conditions would have been met, and therefore the extent to which the Awards would have vested, for the particular stock price path. The market condition was met on June 10, 2015. In determining the fair value of the performance-based SARs and PSUs, the Company considered the achievement of the market condition in determining the estimated fair value. The Restricted Ordinary Shares and PSUs remain subject to the achievement of the performance condition and the employee's continued service. Subsequent to the initial grant under the 2014 Program , approximately 300,000 awards have been forfeited.
On June 10, 2015, 4.1 million shares of the Company's performance-based SARs were converted into 1.1 million restricted ordinary shares (the "Restricted Ordinary Shares") pursuant to the terms of the 2014 Program . In addition, the maximum number of the Company's PSUs granted in February 2014 under the 2014 Program that could vest was fixed at 1.4 million units. Each SAR or PSU is equal to one ordinary share with the maximum value of each Award upon vesting subject to varying limitations.
| Year Ended December 31, |
| 2014 |
Volatility | 31.6% |
Risk-free interest rate | 1.9% |
Expected term (years) | 6.3 |
Forfeiture rate | 5.5% |
Weighted average grant date fair value per SAR | $9.43 |
Weighted average grant date fair value per PSU | $34.58 |
145
Table of Contents
12. | Employee Benefit Plans |
Defined Benefit Plans
The Company sponsors various defined benefit pension plans in several countries. Benefits provided generally depend on length of service, pay grade and remuneration levels. The Company maintains two fully frozen defined benefit pension plans in the U.S., and employees in the U.S. and Puerto Rico are provided retirement benefits through defined contribution plans rather than through a defined benefit plan. The Company assumed net unfunded pension and other postretirement liabilities, primarily in Germany and the U.S., of approximately $322.3 million as a result of the Meda transaction.
The Company also sponsors other postretirement benefit plans. There are plans that provide for postretirement supplemental medical coverage. Benefits from these plans are paid to certain employees and their spouses and dependents who meet various minimum age and service requirements. In addition, there are plans that provide for life insurance benefits and postretirement medical coverage for certain officers and management employees.
Accounting for Defined Benefit Pension and Other Postretirement Plans
The Company recognizes on its balance sheet an asset or liability equal to the over- or under-funded benefit obligation of each defined benefit pension and other postretirement plan. Actuarial gains or losses and prior service costs or credits that arise during the period are not recognized as components of net periodic benefit cost but are recognized, net of tax, as a component of other comprehensive income.
| Pension Benefits |
| Other Postretirement Benefits | ||||||||||||
| December 31, |
| December 31, | ||||||||||||
(In millions) | 2016 |
| 2015 |
| 2016 |
| 2015 | ||||||||
Unrecognized actuarial (gains) losses | $ | (9.1 | ) |
| $ | 13.5 | |
| $ | 5.9 | |
| $ | 5.6 | |
Unrecognized prior service costs | 2.0 | |
| 3.0 | |
| - | |
| - | | ||||
Total | $ | (7.1 | ) |
| $ | 16.5 | |
| $ | 5.9 | |
| $ | 5.6 | |
(In millions) | Pension Benefits |
| Other Postretirement Benefits | ||||
Unrecognized actuarial (gain)/loss | $ | (24.1 | ) |
| $ | 0.7 | |
Amortization of actuarial (gain)/loss | (1.1 | ) |
| (0.3 | ) | ||
Unrecognized prior service costs | (0.6 | ) |
| - | | ||
Amortization of prior service costs | (0.4 | ) |
| - | | ||
Impact of foreign currency translation | 0.4 | |
| - | | ||
Net change | $ | (25.8 | ) |
| $ | 0.4 | |
Components of net periodic benefit cost, change in projected benefit obligation, change in plan assets, funded status, fair value of plan assets, assumptions used to determine net periodic benefit cost, funding policy and estimated future benefit payments are summarized below for the Company's pension plans and other postretirement plans.
146
Table of Contents
Net Periodic Benefit Cost
| Pension Benefits |
| Other Postretirement Benefits | ||||||||||||||||||||
| December 31, |
| December 31, | ||||||||||||||||||||
(In millions) | 2016 |
| 2015 |
| 2014 |
| 2016 |
| 2015 |
| 2014 | ||||||||||||
Service cost | $ | 17.4 | |
| $ | 11.9 | |
| $ | 5.0 | |
| $ | 0.6 | |
| $ | 0.6 | |
| $ | 0.5 | |
Interest cost | 8.2 | |
| 4.0 | |
| 2.6 | |
| 1.3 | |
| 1.1 | |
| 1.0 | | ||||||
Expected return on plan assets | (10.9 | ) |
| (6.4 | ) |
| (1.7 | ) |
| - | |
| - | |
| - | | ||||||
Plan curtailment, settlement and termination | (2.4 | ) |
| 1.1 | |
| 0.2 | |
| - | |
| - | |
| - | | ||||||
Amortization of prior service costs | 0.3 | |
| 0.3 | |
| 0.3 | |
| - | |
| - | |
| - | | ||||||
Recognized net actuarial losses | 0.8 | |
| 0.9 | |
| 0.6 | |
| 0.3 | |
| 0.3 | |
| 0.1 | | ||||||
Net periodic benefit cost | $ | 13.4 | |
| $ | 11.8 | |
| $ | 7.0 | |
| $ | 2.2 | |
| $ | 2.0 | |
| $ | 1.6 | |
Change in Projected Benefit Obligation, Change in Plan Assets and Funded Status
The table below presents components of the change in projected benefit obligation, change in plan assets and funded status at December 31, 2016 and 2015 .
| Pension Benefits |
| Other Postretirement Benefits | ||||||||||||
(In millions) | 2016 |
| 2015 |
| 2016 |
| 2015 | ||||||||
Change in Projected Benefit Obligation |
|
|
|
|
|
|
| ||||||||
Projected benefit obligation, beginning of year | $ | 234.4 | |
| $ | 75.7 | |
| $ | 26.0 | |
| $ | 26.2 | |
Service cost | 17.4 | |
| 11.9 | |
| 0.6 | |
| 0.6 | | ||||
Interest cost | 8.2 | |
| 4.0 | |
| 1.3 | |
| 1.1 | | ||||
Participant contributions | 1.1 | |
| 0.8 | |
| 0.2 | |
| 0.1 | | ||||
Transferred liabilities | 2.1 | |
| 0.4 | |
| - | |
| - | | ||||
Acquisitions | 441.2 | |
| 166.1 | |
| 11.1 | |
| - | | ||||
Plan settlements and terminations | (7.9 | ) |
| (2.5 | ) |
| - | |
| - | | ||||
Actuarial losses (gains) | (28.6 | ) |
| (5.9 | ) |
| 0.7 | |
| (0.1 | ) | ||||
Benefits paid | (17.8 | ) |
| (8.9 | ) |
| (1.9 | ) |
| (1.9 | ) | ||||
Impact of foreign currency translation | (17.2 | ) |
| (7.2 | ) |
| - | |
| - | | ||||
Projected benefit obligation, end of year | $ | 632.9 | |
| $ | 234.4 | |
| $ | 38.0 | |
| $ | 26.0 | |
|
|
|
|
|
|
|
| ||||||||
Change in Plan Assets |
|
|
|
|
|
|
| ||||||||
Fair value of plan assets, beginning of year | $ | 162.0 | |
| $ | 33.2 | |
| $ | - | |
| $ | - | |
Actual return on plan assets | 6.1 | |
| (0.8 | ) |
| - | |
| - | | ||||
Company contributions | 17.4 | |
| 11.3 | |
| 1.8 | |
| 1.7 | | ||||
Participant contributions | 1.1 | |
| 0.8 | |
| 0.1 | |
| 0.1 | | ||||
Acquisitions | 128.6 | |
| 131.7 | |
| - | |
| - | | ||||
Transferred assets | 2.1 | |
| 0.4 | |
| - | |
| - | | ||||
Plan settlements | (4.0 | ) |
| (2.7 | ) |
| - | |
| - | | ||||
Benefits paid | (17.8 | ) |
| (8.9 | ) |
| (1.9 | ) |
| (1.9 | ) | ||||
Other | (0.8 | ) |
| - | |
| - | |
| 0.1 | | ||||
Impact of foreign currency translation | (3.0 | ) |
| (3.0 | ) |
| - | |
| - | | ||||
Fair value of plan assets, end of year | 291.7 | |
| 162.0 | |
| - | |
| - | | ||||
Funded status of plans | $ | (341.2 | ) |
| $ | (72.4 | ) |
| $ | (38.0 | ) |
| $ | (26.0 | ) |
147
Table of Contents
| Pension Benefits |
| Other Postretirement Benefits | ||||||||||||
| December 31, |
| December 31, | ||||||||||||
(In millions) | 2016 |
| 2015 |
| 2016 |
| 2015 | ||||||||
Noncurrent assets | $ | 3.4 | |
| $ | 0.5 | |
| $ | - | |
| $ | - | |
Current liabilities | (9.8 | ) |
| (2.8 | ) |
| (1.9 | ) |
| (1.2 | ) | ||||
Noncurrent liabilities | (334.8 | ) |
| (70.1 | ) |
| (36.1 | ) |
| (24.8 | ) | ||||
Net accrued benefit costs | $ | (341.2 | ) |
| $ | (72.4 | ) |
| $ | (38.0 | ) |
| $ | (26.0 | ) |
The projected benefit obligation is the actuarial present value of benefits attributable to employee service rendered to date, including the effects of estimated future pay increases. The accumulated benefit obligation is the actuarial present value of benefits attributable to employee service rendered to date, but does not include the effects of estimated future pay increases. The accumulated benefit obligation for the Company's pension plans was $569.6 million and $212.3 million at December 31, 2016 and 2015 , respectively.
| December 31, | ||||||
(In millions) | 2016 |
| 2015 | ||||
Plans with accumulated benefit obligation in excess of plan assets: |
|
|
| ||||
Accumulated benefit obligation | $ | 471.7 | |
| $ | 136.8 | |
Projected benefit obligation | 493.7 | |
| 147.3 | | ||
Fair value of plan assets | 164.3 | |
| 81.5 | | ||
Fair Value of Plan Assets
| December 31, 2016 | ||||||||||||||
(In millions) | Level 1 |
| Level 2 |
| Level 3 |
| Total | ||||||||
Cash and cash equivalents | $ | 0.8 | |
| $ | 0.3 | |
| $ | - | |
| $ | 1.1 | |
Equity securities | 94.5 | |
| 37.8 | |
| - | |
| 132.3 | | ||||
Fixed income securities | 59.5 | |
| 39.6 | |
| - | |
| 99.1 | | ||||
Assets held by insurance companies and other | 8.7 | |
| 19.2 | |
| 31.3 | |
| 59.2 | | ||||
Total | $ | 163.5 | |
| $ | 96.9 | |
| $ | 31.3 | |
| $ | 291.7 | |
| December 31, 2015 | ||||||||||||||
(In millions) | Level 1 |
| Level 2 |
| Level 3 |
| Total | ||||||||
Cash and cash equivalents | $ | 1.7 | |
| $ | - | |
| $ | - | |
| $ | 1.7 | |
Equity securities | 6.2 | |
| 78.7 | |
| - | |
| 84.9 | | ||||
Fixed income securities | 3.5 | |
| 51.1 | |
| - | |
| 54.6 | | ||||
Assets held by insurance companies and other | 8.6 | |
| 9.2 | |
| 3.0 | |
| 20.8 | | ||||
Total | $ | 20.0 | |
| $ | 139.0 | |
| $ | 3.0 | |
| $ | 162.0 | |
Risk tolerance on invested pension plan assets is established through careful consideration of plan liabilities, plan funded status and corporate financial condition. Investment risk is measured and monitored on an ongoing basis through annual
148
Table of Contents
liability measures, periodic asset/liability studies and investment portfolio reviews. The Company's investment strategy is to maintain, where possible, a diversified investment portfolio across several asset classes that, when combined with the Company's contributions to the plans, will ensure that required benefit obligations are met.
Assumptions
| Pension Benefits |
| Other Postretirement Benefits | ||||||||
| 2016 |
| 2015 |
| 2016 |
| 2015 | ||||
Discount rate | 2.2 | % |
| 2.1 | % |
| 4.2 | % |
| 4.3 | % |
Expected return on plan assets | 4.9 | % |
| 4.9 | % |
| - | % |
| - | % |
Rate of compensation increase | 2.8 | % |
| 5.5 | % |
| - | % |
| - | % |
| Pension Benefits |
| Other Postretirement Benefits | ||||||||||||||
| 2016 |
| 2015 |
| 2014 |
| 2016 |
| 2015 |
| 2014 | ||||||
Discount rate | 2.1 | % |
| 2.3 | % |
| 4.1 | % |
| 4.3 | % |
| 4.0 | % |
| 4.8 | % |
Expected return on plan assets | 4.9 | % |
| 4.5 | % |
| 5.6 | % |
| - | % |
| - | % |
| - | % |
Rate of compensation increase | 3.2 | % |
| 5.2 | % |
| 6.9 | % |
| - | % |
| - | % |
| - | % |
The assumptions for each plan are reviewed on an annual basis. The discount rate reflects the current rate at which the pension and other benefit liabilities could be effectively settled at the measurement date. In setting the discount rates, we utilize comparable corporate bond indices as an indication of interest rate movements and levels. Corporate bond indices were selected based on individual plan census data and duration. The expected return on plan assets was determined using historical market returns and long-term historical relationships between equities and fixed income securities. The Company compares the expected return on plan assets assumption to actual historic returns to ensure reasonableness. Current market factors such as inflation and interest rates are also evaluated.
(In millions) | Increase |
| Decrease | ||||
Increase (decrease) in the projected benefit obligation | $ | 1.7 | |
| $ | (1.4 | ) |
Increase in the aggregate of service and interest cost components of annual expense | 0.1 | |
| - | | ||
Estimated Future Benefit Payments
The Company's funding policy for its funded pension plans is based upon local statutory requirements. The Company's funding policy is subject to certain statutory regulations with respect to annual minimum and maximum company contributions. Plan benefits for the nonqualified plans are paid as they come due.
149
Table of Contents
(In millions) | Pension Benefits |
| Other Postretirement Benefits | ||||
2017 | $ | 28.5 | |
| $ | 1.9 | |
2018 | 29.5 | |
| 2.4 | | ||
2019 | 30.2 | |
| 2.1 | | ||
2020 | 33.6 | |
| 2.5 | | ||
2021 | 34.6 | |
| 2.0 | | ||
Thereafter | 184.2 | |
| 12.3 | | ||
Total | $ | 340.6 | |
| $ | 23.2 | |
Defined Contribution Plans
The Company sponsors defined contribution plans covering its employees in the U.S. and Puerto Rico, as well as certain employees in a number of countries outside the U.S. The Company's domestic defined contribution plans consist primarily of a 401(k) retirement plan with a profit sharing component for non-union represented employees (the "Profit Sharing 401(k) Plan") and a 401(k) retirement plan for union-represented employees. Profit sharing contributions are made at the discretion of the Board of Directors. The Company's non-domestic plans vary in form depending on local legal requirements. The Company's contributions are based upon employee contributions, service hours, or pre-determined amounts depending upon the plan. Obligations for contributions to defined contribution plans are recognized as expense in the Consolidated Statements of Operations when they are earned.
The Company adopted a 401(k) Restoration Plan (the "Restoration Plan"), which permits employees who earn compensation in excess of the limits imposed by Section 401(a)(17) of the Code to (i) defer a portion of base salary and bonus compensation, (ii) be credited with a Company matching contribution in respect of deferrals under the Restoration Plan, and (iii) be credited with Company non-elective contributions (to the extent so made by the Company), in each case, to the extent that participants otherwise would be able to defer or be credited with such amounts, as applicable, under the Profit Sharing 401(k) Plan if not for the limits on contributions and deferrals imposed by the Code.
The Company adopted an Income Deferral Plan (the "Income Deferral Plan"), which permits certain management or highly compensated employees who are designated by the plan administrator to participate in the Income Deferral Plan to elect to defer up to 50% of base salary and up to 100% of bonus compensation, in each case, in addition to any amounts that may be deferred by such participants under the Profit Sharing 401(k) Plan and the Restoration Plan. In addition, under the Income Deferral Plan, eligible participants may be granted employee deferral awards, which awards will be subject to the terms and conditions (including vesting) as determined by the plan administrator at the time such awards are granted.
Total employer contributions to defined contribution plans were approximately $95.6 million , $102.4 million and $87.4 million for the years ended December 31, 2016 , 2015 and 2014 , respectively.
Other Benefit Arrangements
The Company participated in a multi-employer pension plan under previous collective bargaining agreements. The PACE Industry Union-Management Pension Fund (the "Plan") provides defined benefits to certain retirees and certain production and maintenance employees at the Company's manufacturing facility in Morgantown, West Virginia who were covered by the previous collective bargaining agreements. Pursuant to a collective bargaining agreement entered into on April 16, 2012, the Company withdrew from the Plan effective May 10, 2012. In the fourth quarter of 2013, the Plan trustee notified the Company that its withdrawal liability was approximately $27 million , which was accrued by the Company at December 31, 2013. The withdrawal liability is being paid over a period of approximately nine years; payments began in March 2014 . The withdrawal liability was approximately $20.7 million and $23.2 million at December 31, 2016 and 2015 , respectively. The Employee Identification Number for this Plan is 11-6166763.
13. | Segment Information |
As a result of our acquisition of Meda on August 5, 2016 and the integration of our portfolio across our branded, generics and OTC platforms in all of our regions, effective October 1, 2016, the Company has expanded its reportable
150
Table of Contents
segments. The Company has three reportable segments on a geographic basis as follows: North America, Europe and Rest of World. Our North America segment is made up of our operations in the U.S. and Canada and includes the operations of our previously reported Specialty segment. Our Europe segment is made up of our operations in 35 countries within the region. Our Rest of World segment is primarily made up of our operations in India, Australia, Japan and New Zealand. Also included in our Rest of World segment are our operations in emerging markets, which includes countries in Africa (including South Africa) as well as Brazil and other countries throughout Asia and the Middle East. Comparative segment financial information has been recast for prior periods to conform to this revised segment reporting.
The Company's chief operating decision maker is the Chief Executive Officer, who evaluates the performance of its segments based on total revenues and segment profitability. Segment profitability represents segment gross profit less direct R&D expenses and direct SG&A expenses. Certain general and administrative and R&D expenses not allocated to the segments, net charges for litigation settlements and other contingencies, impairment charges and other expenses not directly attributable to the segments and certain intercompany transactions, including eliminations, are reported in Corporate/Other. Additionally, amortization of intangible assets and other purchase accounting related items, as well as certain other significant special items, are included in Corporate/Other. Items below the earnings from operations line on the Company's Consolidated Statements of Operations are not presented by segment, since they are excluded from the measure of segment profitability. The Company does not report depreciation expense, total assets and capital expenditures by segment, as such information is not used by the chief operating decision maker.
The accounting policies of the segments are the same as those described in Note 2 Summary of Significant Accounting Policies to Consolidated Financial Statements . Intersegment revenues are accounted for at current market values and are eliminated at the consolidated level.
Presented in the table below is segment information for the periods identified and a reconciliation of segment information to total consolidated information.
(In millions) | North America |
| Europe |
| Rest of World |
| Corporate / Other |
| Consolidated | ||||||||||
Year Ended December 31, 2016 |
|
|
|
|
|
|
|
|
| ||||||||||
Third party net sales | $ | 5,629.5 | |
| $ | 2,953.8 | |
| $ | 2,383.8 | |
| $ | - | |
| $ | 10,967.1 | |
Other revenue | 88.4 | |
| 12.6 | |
| 8.8 | |
| - | |
| 109.8 | | |||||
Intersegment revenue | 45.4 | |
| 106.3 | |
| 407.6 | |
| (559.3 | ) |
| - | | |||||
Total | $ | 5,763.3 | |
| $ | 3,072.7 | |
| $ | 2,800.2 | |
| $ | (559.3 | ) |
| $ | 11,076.9 | |
|
|
|
|
|
|
|
|
|
| ||||||||||
Segment profitability | $ | 2,921.2 | |
| $ | 669.4 | |
| $ | 423.5 | |
| $ | (3,312.5 | ) |
| $ | 701.6 | |
Year Ended December 31, 2015 |
|
|
|
|
|
|
|
|
| ||||||||||
Third party net sales | $ | 5,100.4 | |
| $ | 2,205.6 | |
| $ | 2,056.6 | |
| $ | - | |
| $ | 9,362.6 | |
Other revenue | 55.3 | |
| 5.3 | |
| 6.1 | |
| - | |
| 66.7 | | |||||
Intersegment revenue | 26.5 | |
| 109.9 | |
| 378.0 | |
| (514.4 | ) |
| - | | |||||
Total | $ | 5,182.2 | |
| $ | 2,320.8 | |
| $ | 2,440.7 | |
| $ | (514.4 | ) |
| $ | 9,429.3 | |
|
|
|
|
|
|
|
|
|
| ||||||||||
Segment profitability | $ | 2,720.8 | |
| $ | 421.5 | |
| $ | 320.7 | |
| $ | (2,002.1 | ) |
| $ | 1,460.9 | |
Year Ended December 31, 2014 |
|
|
|
|
|
|
|
|
| ||||||||||
Third party net sales | $ | 4,548.4 | |
| $ | 1,476.8 | |
| $ | 1,621.3 | |
| $ | - | |
| $ | 7,646.5 | |
Other revenue | 50.1 | |
| 7.5 | |
| 15.5 | |
| - | |
| 73.1 | | |||||
Intersegment revenue | 21.4 | |
| 130.3 | |
| 389.7 | |
| (541.4 | ) |
| - | | |||||
Total | $ | 4,619.9 | |
| $ | 1,614.6 | |
| $ | 2,026.5 | |
| $ | (541.4 | ) |
| $ | 7,719.6 | |
|
|
|
|
|
|
|
|
|
| ||||||||||
Segment profitability | $ | 2,376.3 | |
| $ | 130.1 | |
| $ | 200.3 | |
| $ | (1,354.1 | ) |
| $ | 1,352.6 | |
151
Table of Contents
During 2016, the Company refined its classifications for therapeutic franchises and prior year amounts have been reclassified to conform to the current year presentation. The Company's third party net sales are generated via the sale of products in the following therapeutic franchises:
| Year Ended December 31, | ||||||||||
(In millions) | 2016 |
| 2015 |
| 2014 | ||||||
Central Nervous System and Anesthesia | $ | 2,030.4 | |
| $ | 1,724.7 | |
| $ | 1,412.8 | |
Respiratory and Allergy | 1,813.1 | |
| 1,500.3 | |
| 1,317.5 | | |||
Infectious Disease | 1,303.0 | |
| 1,322.2 | |
| 1,106.8 | | |||
Cardiovascular | 1,165.1 | |
| 1,075.3 | |
| 895.8 | | |||
Gastroenterology | 1,029.3 | |
| 830.5 | |
| 420.0 | | |||
Diabetes and Metabolism | 972.0 | |
| 935.2 | |
| 792.1 | | |||
Oncology | 764.2 | |
| 633.3 | |
| 395.1 | | |||
Women's Healthcare | 593.5 | |
| 467.7 | |
| 364.0 | | |||
Dermatology | 369.5 | |
| 226.6 | |
| 238.6 | | |||
Immunology | 133.1 | |
| 118.1 | |
| 130.6 | | |||
Other (1) | 793.9 | |
| 528.7 | |
| 573.2 | | |||
| $ | 10,967.1 | |
| $ | 9,362.6 | |
| $ | 7,646.5 | |
____________
(1) | Other consists of numerous therapeutic franchises, none of which individually exceeds 5% of consolidated net sales. |
The following table represents the percentage of consolidated third party net sales to Mylan's major customers during the years ended December 31, 2016 , 2015 and 2014 .
| Percentage of Third Party Net Sales | |||||||
| 2016 |
| 2015 |
| 2014 | |||
McKesson Corporation | 16 | % |
| 15 | % |
| 19 | % |
AmerisourceBergen Corporation | 14 | % |
| 16 | % |
| 13 | % |
Cardinal Health, Inc. | 11 | % |
| 12 | % |
| 12 | % |
Sales by Country Information
Third party net sales by country are presented on the basis of geographic location of our subsidiaries:
| Year Ended December 31, | ||||||||||
(In millions) | 2016 |
| 2015 |
| 2014 | ||||||
United States | $ | 5,385.6 | |
| $ | 4,848.9 | |
| $ | 4,425.3 | |
India | 985.8 | |
| 1,033.4 | |
| 941.4 | | |||
The Netherlands (1) | 88.3 | |
| 66.5 | |
| 61.1 | | |||
Other countries (2) | 4,507.4 | |
| 3,413.8 | |
| 2,218.7 | | |||
| $ | 10,967.1 | |
| $ | 9,362.6 | |
| $ | 7,646.5 | |
____________
(1) | Mylan N.V. is domiciled in the Netherlands. |
(2) | No other country's net sales represents more than 10% of consolidated net sales for the years ended December 31, 2016 , 2015 and 2014 , respectively. |
152
Table of Contents
14. | Commitments |
Operating Leases
The Company leases certain property under various operating lease arrangements. These leases generally provide the Company with the option to renew the lease at the end of the lease term. For the years ended December 31, 2016 , 2015 and 2014 , the Company had operating lease expense of approximately $70.8 million , $57.1 million and $45.2 million , respectively.
Future minimum lease payments under operating lease commitments are as follows:
(In millions) |
| ||
December 31, |
| ||
2017 | $ | 75.6 | |
2018 | 63.5 | | |
2019 | 50.9 | | |
2020 | 33.6 | | |
2021 | 24.1 | | |
Thereafter | 48.1 | | |
| $ | 295.8 | |
Other Commitments
The Company has also entered into employment and other agreements with certain executives and other employees that provide for compensation, retirement and certain other benefits. These agreements provide for severance payments under certain circumstances. Additionally, the Company has split-dollar life insurance agreements with certain retired executives.
In the normal course of business, Mylan periodically enters into employment, legal settlement and other agreements which incorporate indemnification provisions. While the maximum amount to which Mylan may be exposed under such agreements cannot be reasonably estimated, the Company maintains insurance coverage, which management believes will effectively mitigate the Company's obligations under these indemnification provisions. No amounts have been recorded in the Consolidated Financial Statements with respect to the Company's obligations under such agreements.
153
Table of Contents
15. | Subsidiary Guarantors |
The following tables present condensed consolidating financial information for (a) Mylan N.V., the issuer of the June 2016 Senior Notes and December 2015 Senior Notes which are guaranteed on a senior unsecured basis by Mylan Inc.; (b) Mylan Inc., the issuer of the 1.800% Senior Notes due 2016, 1.350% Senior Notes due 2016, 2.600% Senior Notes due 2018, 2.550% Senior Notes due 2019, 3.125% Senior Notes due 2023, 4.200% Senior Notes due 2023 and 5.400% Senior Notes due 2043 (collectively, the "Mylan Inc. Senior Notes"), which are guaranteed on a senior unsecured basis by Mylan N.V.; and (c) all other subsidiaries of the Company on a combined basis, none of which guarantee the June 2016 Senior Notes, the December 2015 Senior Notes or guarantee the Mylan Inc. Senior Notes ("Non-Guarantor Subsidiaries"). The consolidating adjustments primarily relate to eliminations of investments in subsidiaries and intercompany balances and transactions. The condensed consolidating financial statements present investments in subsidiaries using the equity method of accounting.
The Company was incorporated on July 7, 2014 as an indirect wholly owned subsidiary of Mylan Inc. for the purpose of consummating the EPD Transaction. Upon consummation of the EPD Transaction, on February 27, 2015, Mylan Inc. became an indirect wholly owned subsidiary of the Company, and the Company fully and unconditionally guaranteed the Mylan Inc. Senior Notes. For periods prior to February 27, 2015, the parent entity was Mylan Inc.
The following financial information presents the related Condensed Consolidating Balance Sheet as of December 31, 2016 and 2015 and the related Condensed Consolidating Statements of Operations, Condensed Consolidating Statements of Comprehensive Earnings and Condensed Consolidating Statements of Cash Flows for each of the three years in the period ended December 31, 2016 . This condensed consolidating financial information has been prepared and presented in accordance with SEC Regulation S-X Rule 3-10 "Financial Statements of Guarantors and Issuers of Guaranteed Securities Registered or Being Registered."
154
Table of Contents
CONDENSED CONSOLIDATING BALANCE SHEET
As of December 31, 2016
(In millions) | Mylan N.V. |
| Mylan Inc. |
| Guarantor Subsidiaries |
| Non-Guarantor Subsidiaries |
| Eliminations |
| Consolidated | ||||||||||||
ASSETS |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Assets |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Current assets: |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Cash and cash equivalents | $ | 0.3 | |
| $ | 12.3 | |
| $ | - | |
| $ | 986.2 | |
| $ | - | |
| $ | 998.8 | |
Accounts receivable, net | - | |
| 12.3 | |
| - | |
| 3,298.6 | |
| - | |
| 3,310.9 | | ||||||
Inventories | - | |
| - | |
| - | |
| 2,456.4 | |
| - | |
| 2,456.4 | | ||||||
Intercompany receivables | 215.9 | |
| 416.0 | |
| - | |
| 10,506.6 | |
| (11,138.5 | ) |
| - | | ||||||
Prepaid expenses and other current assets | - | |
| 256.4 | |
| - | |
| 500.0 | |
| - | |
| 756.4 | | ||||||
Total current assets | 216.2 | |
| 697.0 | |
| - | |
| 17,747.8 | |
| (11,138.5 | ) |
| 7,522.5 | | ||||||
Property, plant and equipment, net | - | |
| 360.3 | |
| - | |
| 1,961.9 | |
| - | |
| 2,322.2 | | ||||||
Investments in subsidiaries | 15,606.2 | |
| 8,277.8 | |
| - | |
| - | |
| (23,884.0 | ) |
| - | | ||||||
Intercompany notes and interest receivable | 7,952.3 | |
| 9,817.3 | |
| - | |
| 16.7 | |
| (17,786.3 | ) |
| - | | ||||||
Intangible assets, net | - | |
| - | |
| - | |
| 14,447.8 | |
| - | |
| 14,447.8 | | ||||||
Goodwill | - | |
| 17.1 | |
| - | |
| 9,214.8 | |
| - | |
| 9,231.9 | | ||||||
Other assets | 5.2 | |
| 51.9 | |
| - | |
| 1,144.7 | |
| - | |
| 1,201.8 | | ||||||
Total assets | $ | 23,779.9 | |
| $ | 19,221.4 | |
| $ | - | |
| $ | 44,533.7 | |
| $ | (52,808.8 | ) |
| $ | 34,726.2 | |
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
LIABILITIES AND EQUITY |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Liabilities |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Current liabilities: |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Trade accounts payable | $ | 3.9 | |
| $ | 69.6 | |
| $ | - | |
| $ | 1,274.6 | |
| $ | - | |
| $ | 1,348.1 | |
Short-term borrowings | - | |
| - | |
| - | |
| 46.4 | |
| - | |
| 46.4 | | ||||||
Income taxes payable | - | |
| - | |
| - | |
| 97.7 | |
| - | |
| 97.7 | | ||||||
Current portion of long-term debt and other long-term obligations | - | |
| 0.2 | |
| - | |
| 289.8 | |
| - | |
| 290.0 | | ||||||
Intercompany payables | 416.0 | |
| 10,722.5 | |
| - | |
| - | |
| (11,138.5 | ) |
| - | | ||||||
Other current liabilities | 90.9 | |
| 388.8 | |
| - | |
| 2,778.8 | |
| - | |
| 3,258.5 | | ||||||
Total current liabilities | 510.8 | |
| 11,181.1 | |
| - | |
| 4,487.3 | |
| (11,138.5 | ) |
| 5,040.7 | | ||||||
Long-term debt | 12,151.5 | |
| 2,897.6 | |
| - | |
| 153.8 | |
| - | |
| 15,202.9 | | ||||||
Intercompany notes payable | - | |
| 3,870.9 | |
| - | |
| 13,915.4 | |
| (17,786.3 | ) |
| - | | ||||||
Other long-term obligations | - | |
| 58.1 | |
| - | |
| 3,306.9 | |
| - | |
| 3,365.0 | | ||||||
Total liabilities | 12,662.3 | |
| 18,007.7 | |
| - | |
| 21,863.4 | |
| (28,924.8 | ) |
| 23,608.6 | | ||||||
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Total equity | 11,117.6 | |
| 1,213.7 | |
| - | |
| 22,670.3 | |
| (23,884.0 | ) |
| 11,117.6 | | ||||||
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Total liabilities and equity | $ | 23,779.9 | |
| $ | 19,221.4 | |
| $ | - | |
| $ | 44,533.7 | |
| $ | (52,808.8 | ) |
| $ | 34,726.2 | |
155
Table of Contents
CONDENSED CONSOLIDATING BALANCE SHEET
As of December 31, 2015
(In millions) | Mylan N.V. |
| Mylan Inc. |
| Guarantor Subsidiaries |
| Non-Guarantor Subsidiaries |
| Eliminations |
| Consolidated | ||||||||||||
ASSETS |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Assets |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Current assets: |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Cash and cash equivalents | $ | - | |
| $ | 870.5 | |
| $ | - | |
| $ | 365.5 | |
| $ | - | |
| $ | 1,236.0 | |
Accounts receivable, net | - | |
| 14.4 | |
| - | |
| 2,674.7 | |
| - | |
| 2,689.1 | | ||||||
Inventories | - | |
| - | |
| - | |
| 1,951.0 | |
| - | |
| 1,951.0 | | ||||||
Intercompany receivables | 1,097.5 | |
| 283.2 | |
| - | |
| 8,936.4 | |
| (10,317.1 | ) |
| - | | ||||||
Other current assets | 0.3 | |
| 244.8 | |
| - | |
| 351.5 | |
| - | |
| 596.6 | | ||||||
Total current assets | 1,097.8 | |
| 1,412.9 | |
| - | |
| 14,279.1 | |
| (10,317.1 | ) |
| 6,472.7 | | ||||||
Property, plant and equipment, net | - | |
| 324.4 | |
| - | |
| 1,659.5 | |
| - | |
| 1,983.9 | | ||||||
Investments in subsidiaries | 9,947.7 | |
| 8,007.7 | |
| - | |
| - | |
| (17,955.4 | ) |
| - | | ||||||
Intercompany notes and interest receivable | - | |
| 9,704.4 | |
| - | |
| 18.7 | |
| (9,723.1 | ) |
| - | | ||||||
Intangible assets, net | - | |
| 0.5 | |
| - | |
| 7,221.4 | |
| - | |
| 7,221.9 | | ||||||
Goodwill | - | |
| 17.1 | |
| - | |
| 5,363.0 | |
| - | |
| 5,380.1 | | ||||||
Other assets | - | |
| 135.3 | |
| - | |
| 1,073.8 | |
| - | |
| 1,209.1 | | ||||||
Total assets | $ | 11,045.5 | |
| $ | 19,602.3 | |
| $ | - | |
| $ | 29,615.5 | |
| $ | (37,995.6 | ) |
| $ | 22,267.7 | |
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
LIABILITIES AND EQUITY |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Liabilities |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Current liabilities: |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Trade accounts payable | $ | - | |
| $ | 33.5 | |
| $ | - | |
| $ | 1,076.1 | |
| $ | - | |
| $ | 1,109.6 | |
Short-term borrowings | - | |
| - | |
| - | |
| 1.3 | |
| - | |
| 1.3 | | ||||||
Income taxes payable | - | |
| - | |
| - | |
| 92.4 | |
| - | |
| 92.4 | | ||||||
Current portion of long-term debt and other long-term obligations | - | |
| 1,010.1 | |
| - | |
| 66.9 | |
| - | |
| 1,077.0 | | ||||||
Intercompany payables | 283.2 | |
| 10,033.9 | |
| - | |
| - | |
| (10,317.1 | ) |
| - | | ||||||
Other current liabilities | 2.0 | |
| 320.1 | |
| - | |
| 1,519.8 | |
| - | |
| 1,841.9 | | ||||||
Total current liabilities | 285.2 | |
| 11,397.6 | |
| - | |
| 2,756.5 | |
| (10,317.1 | ) |
| 4,122.2 | | ||||||
Long-term debt | 994.5 | |
| 5,298.4 | |
| - | |
| 2.7 | |
| - | |
| 6,295.6 | | ||||||
Intercompany notes payable | - | |
| 18.7 | |
| - | |
| 9,704.4 | |
| (9,723.1 | ) |
| - | | ||||||
Other long-term obligations | - | |
| 122.2 | |
| - | |
| 1,961.9 | |
| - | |
| 2,084.1 | | ||||||
Total liabilities | 1,279.7 | |
| 16,836.9 | |
| - | |
| 14,425.5 | |
| (20,040.2 | ) |
| 12,501.9 | | ||||||
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Total equity | 9,765.8 | |
| 2,765.4 | |
| - | |
| 15,190.0 | |
| (17,955.4 | ) |
| 9,765.8 | | ||||||
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Total liabilities and equity | $ | 11,045.5 | |
| $ | 19,602.3 | |
| $ | - | |
| $ | 29,615.5 | |
| $ | (37,995.6 | ) |
| $ | 22,267.7 | |
156
Table of Contents
CONDENSED CONSOLIDATING STATEMENT OF OPERATIONS
Year Ended December 31, 2016
(In millions) | Mylan N.V. |
| Mylan Inc. |
| Guarantor Subsidiaries |
| Non-Guarantor Subsidiaries |
| Eliminations |
| Consolidated | ||||||||||||
Revenues: |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Net sales | $ | - | |
| $ | - | |
| $ | - | |
| $ | 10,967.1 | |
| $ | - | |
| $ | 10,967.1 | |
Other revenues | - | |
| - | |
| - | |
| 109.8 | |
| - | |
| 109.8 | | ||||||
Total revenues | - | |
| - | |
| - | |
| 11,076.9 | |
| - | |
| 11,076.9 | | ||||||
Cost of sales | - | |
| - | |
| - | |
| 6,379.9 | |
| - | |
| 6,379.9 | | ||||||
Gross profit | - | |
| - | |
| - | |
| 4,697.0 | |
| - | |
| 4,697.0 | | ||||||
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Research and development | - | |
| - | |
| - | |
| 826.8 | |
| - | |
| 826.8 | | ||||||
Selling, general and administrative | 71.6 | |
| 664.1 | |
| - | |
| 1,760.4 | |
| - | |
| 2,496.1 | | ||||||
Litigation settlements and other contingencies, net | - | |
| - | |
| - | |
| 672.5 | |
| - | |
| 672.5 | | ||||||
Total operating expenses | 71.6 | |
| 664.1 | |
| - | |
| 3,259.7 | |
| - | |
| 3,995.4 | | ||||||
Earnings from operations | (71.6 | ) |
| (664.1 | ) |
| - | |
| 1,437.3 | |
| - | |
| 701.6 | | ||||||
Interest expense | 198.4 | |
| 161.3 | |
| - | |
| 95.1 | |
| - | |
| 454.8 | | ||||||
Other (income) expense, net | (55.6 | ) |
| (193.2 | ) |
| - | |
| 373.9 | |
| - | |
| 125.1 | | ||||||
(Losses) earnings before income taxes and noncontrolling interest | (214.4 | ) |
| (632.2 | ) |
| - | |
| 968.3 | |
| - | |
| 121.7 | | ||||||
Income tax benefit | (19.5 | ) |
| (18.2 | ) |
| - | |
| (320.6 | ) |
| - | |
| (358.3 | ) | ||||||
Earnings (losses) of equity interest subsidiaries | 674.9 | |
| 1,360.2 | |
| - | |
| - | |
| (2,035.1 | ) |
| - | | ||||||
Net earnings | 480.0 | |
| 746.2 | |
| - | |
| 1,288.9 | |
| (2,035.1 | ) |
| 480.0 | | ||||||
Net earnings attributable to noncontrolling interest | - | |
| - | |
| - | |
| - | |
| - | |
| - | | ||||||
Net earnings attributable to Mylan N.V. ordinary shareholders | $ | 480.0 | |
| $ | 746.2 | |
| $ | - | |
| $ | 1,288.9 | |
| $ | (2,035.1 | ) |
| $ | 480.0 | |
157
Table of Contents
CONDENSED CONSOLIDATING STATEMENT OF OPERATIONS
Year Ended December 31, 2015
(In millions) | Mylan N.V. |
| Mylan Inc. |
| Guarantor Subsidiaries |
| Non-Guarantor Subsidiaries |
| Eliminations |
| Consolidated | ||||||||||||
Revenues: |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Net sales | $ | - | |
| $ | - | |
| $ | - | |
| $ | 9,362.6 | |
| $ | - | |
| $ | 9,362.6 | |
Other revenues | - | |
| - | |
| - | |
| 66.7 | |
| - | |
| 66.7 | | ||||||
Total revenues | - | |
| - | |
| - | |
| 9,429.3 | |
| - | |
| 9,429.3 | | ||||||
Cost of sales | - | |
| - | |
| - | |
| 5,213.2 | |
| - | |
| 5,213.2 | | ||||||
Gross profit | - | |
| - | |
| - | |
| 4,216.1 | |
| - | |
| 4,216.1 | | ||||||
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Research and development | - | |
| - | |
| - | |
| 671.9 | |
| - | |
| 671.9 | | ||||||
Selling, general and administrative | 106.1 | |
| 572.1 | |
| - | |
| 1,502.5 | |
| - | |
| 2,180.7 | | ||||||
Litigation settlements and other contingencies, net | - | |
| - | |
| - | |
| (97.4 | ) |
| - | |
| (97.4 | ) | ||||||
Total operating expenses | 106.1 | |
| 572.1 | |
| - | |
| 2,077.0 | |
| - | |
| 2,755.2 | | ||||||
Earnings from operations | (106.1 | ) |
| (572.1 | ) |
| - | |
| 2,139.1 | |
| - | |
| 1,460.9 | | ||||||
Interest expense | 58.3 | |
| 217.9 | |
| - | |
| 63.2 | |
| - | |
| 339.4 | | ||||||
Other expense, net | 41.1 | |
| - | |
| - | |
| 165.0 | |
| - | |
| 206.1 | | ||||||
(Losses) earnings before income taxes and noncontrolling interest | (205.5 | ) |
| (790.0 | ) |
| - | |
| 1,910.9 | |
| - | |
| 915.4 | | ||||||
Income tax (benefit) provision | - | |
| (23.2 | ) |
| - | |
| 90.9 | |
| - | |
| 67.7 | | ||||||
Earnings (losses) of equity interest subsidiaries | 1,053.2 | |
| 1,814.8 | |
| - | |
| - | |
| (2,868.0 | ) |
| - | | ||||||
Net earnings | 847.7 | |
| 1,048.0 | |
| - | |
| 1,820.0 | |
| (2,868.0 | ) |
| 847.7 | | ||||||
Net earnings attributable to noncontrolling interest | (0.1 | ) |
| - | |
| - | |
| (0.1 | ) |
| 0.1 | |
| (0.1 | ) | ||||||
Net earnings attributable to Mylan N.V. ordinary shareholders | $ | 847.6 | |
| $ | 1,048.0 | |
| $ | - | |
| $ | 1,819.9 | |
| $ | (2,867.9 | ) |
| $ | 847.6 | |
158
Table of Contents
CONDENSED CONSOLIDATING STATEMENT OF OPERATIONS
Year Ended December 31, 2014
(In millions) | Mylan Inc. |
| Guarantor Subsidiaries |
| Non-Guarantor Subsidiaries |
| Eliminations |
| Consolidated | ||||||||||
Revenues: |
|
|
|
|
|
|
|
|
| ||||||||||
Net sales | $ | - | |
| $ | - | |
| $ | 7,646.5 | |
| $ | - | |
| $ | 7,646.5 | |
Other revenues | - | |
| - | |
| 73.1 | |
| - | |
| 73.1 | | |||||
Total revenues | - | |
| - | |
| 7,719.6 | |
| - | |
| 7,719.6 | | |||||
Cost of sales | - | |
| - | |
| 4,191.6 | |
| - | |
| 4,191.6 | | |||||
Gross profit | - | |
| - | |
| 3,528.0 | |
| - | |
| 3,528.0 | | |||||
Operating expenses: |
|
|
|
|
|
|
|
|
| ||||||||||
Research and development | - | |
| - | |
| 581.8 | |
| - | |
| 581.8 | | |||||
Selling, general and administrative | 608.8 | |
| - | |
| 1,016.9 | |
| - | |
| 1,625.7 | | |||||
Litigation settlements and other contingencies, net | - | |
| - | |
| (32.1 | ) |
| - | |
| (32.1 | ) | |||||
Total operating expenses | 608.8 | |
| - | |
| 1,566.6 | |
| - | |
| 2,175.4 | | |||||
Earnings from operations | (608.8 | ) |
| - | |
| 1,961.4 | |
| - | |
| 1,352.6 | | |||||
Interest expense | 273.4 | |
| - | |
| 59.8 | |
| - | |
| 333.2 | | |||||
Other expense, net | - | |
| - | |
| 44.9 | |
| - | |
| 44.9 | | |||||
(Losses) earnings before income taxes and noncontrolling interest | (882.2 | ) |
| - | |
| 1,856.7 | |
| - | |
| 974.5 | | |||||
Income tax (benefit) provision | (39.7 | ) |
| - | |
| 81.1 | |
| - | |
| 41.4 | | |||||
Earnings (losses) of equity interest subsidiaries | 1,775.6 | |
| - | |
| - | |
| (1,775.6 | ) |
| - | | |||||
Net earnings | 933.1 | |
| - | |
| 1,775.6 | |
| (1,775.6 | ) |
| 933.1 | | |||||
Net earnings attributable to noncontrolling interest | - | |
| - | |
| (3.7 | ) |
| - | |
| (3.7 | ) | |||||
Net earnings attributable to Mylan N.V. ordinary shareholders | $ | 933.1 | |
| $ | - | |
| $ | 1,771.9 | |
| $ | (1,775.6 | ) |
| $ | 929.4 | |
159
Table of Contents
CONDENSED CONSOLIDATING STATEMENT OF COMPREHENSIVE EARNINGS
Year Ended December 31, 2016
(In millions) | Mylan N.V. |
| Mylan Inc. |
| Guarantor Subsidiaries |
| Non-Guarantor Subsidiaries |
| Eliminations |
| Consolidated | ||||||||||||
Net earnings | $ | 480.0 | |
| $ | 746.2 | |
| $ | - | |
| $ | 1,288.9 | |
| $ | (2,035.1 | ) |
| $ | 480.0 | |
Other comprehensive (loss) earnings, before tax: |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Foreign currency translation adjustment | (507.4 | ) |
| - | |
| - | |
| (507.4 | ) |
| 507.4 | |
| (507.4 | ) | ||||||
Change in unrecognized gain and prior service cost related to defined benefit plans | 21.4 | |
| (1.1 | ) |
| - | |
| 22.5 | |
| (21.4 | ) |
| 21.4 | | ||||||
Net unrecognized gain on derivatives in cash flow hedging relationships | (31.2 | ) |
| (47.7 | ) |
| - | |
| 16.5 | |
| 31.2 | |
| (31.2 | ) | ||||||
Net unrecognized loss on derivatives in net investment hedging relationships | (1.8 | ) |
| - | |
| - | |
| (1.8 | ) |
| 1.8 | |
| (1.8 | ) | ||||||
Net unrealized loss on marketable securities | 24.6 | |
| 24.6 | |
| - | |
| - | |
| (24.6 | ) |
| 24.6 | | ||||||
Other comprehensive (loss) earnings, before tax | (494.4 | ) |
| (24.2 | ) |
| - | |
| (470.2 | ) |
| 494.4 | |
| (494.4 | ) | ||||||
Income tax provision (benefit) | 5.0 | |
| (9.1 | ) |
| - | |
| 4.1 | |
| 5.0 | |
| 5.0 | | ||||||
Other comprehensive (loss) earnings, net of tax | (499.4 | ) |
| (15.1 | ) |
| - | |
| (474.3 | ) |
| 489.4 | |
| (499.4 | ) | ||||||
Comprehensive earnings | (19.4 | ) |
| 731.1 | |
| - | |
| 814.6 | |
| (1,545.7 | ) |
| (19.4 | ) | ||||||
Comprehensive earnings attributable to the noncontrolling interest | - | |
| - | |
| - | |
| - | |
| - | |
| - | | ||||||
Comprehensive earnings attributable to Mylan N.V. ordinary shareholders | $ | (19.4 | ) |
| $ | 731.1 | |
| $ | - | |
| $ | 814.6 | |
| $ | (1,545.7 | ) |
| $ | (19.4 | ) |
160
Table of Contents
CONDENSED CONSOLIDATING STATEMENT OF COMPREHENSIVE EARNINGS
Year Ended December 31, 2015
(In millions) | Mylan N.V. |
| Mylan Inc. |
| Guarantor Subsidiaries |
| Non-Guarantor Subsidiaries |
| Eliminations |
| Consolidated | ||||||||||||
Net earnings | $ | 847.7 | |
| $ | 1,048.0 | |
| $ | - | |
| $ | 1,820.0 | |
| $ | (2,868.0 | ) |
| $ | 847.7 | |
Other comprehensive (loss) earnings, before tax: |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Foreign currency translation adjustment | (790.9 | ) |
| - | |
| - | |
| (790.9 | ) |
| 790.9 | |
| (790.9 | ) | ||||||
Change in unrecognized gain and prior service cost related to defined benefit plans | 3.1 | |
| 0.4 | |
| - | |
| 2.7 | |
| (3.1 | ) |
| 3.1 | | ||||||
Net unrecognized gain (loss) on derivatives | 16.7 | |
| 23.4 | |
| - | |
| (6.7 | ) |
| (16.7 | ) |
| 16.7 | | ||||||
Net unrealized loss on marketable securities | (2.0 | ) |
| (1.3 | ) |
| - | |
| (0.7 | ) |
| 2.0 | |
| (2.0 | ) | ||||||
Other comprehensive (loss) earnings, before tax | (773.1 | ) |
| 22.5 | |
| - | |
| (795.6 | ) |
| 773.1 | |
| (773.1 | ) | ||||||
Income tax provision (benefit) | 4.2 | |
| 8.7 | |
| - | |
| (4.5 | ) |
| (4.2 | ) |
| 4.2 | | ||||||
Other comprehensive (loss) earnings, net of tax | (777.3 | ) |
| 13.8 | |
| - | |
| (791.1 | ) |
| 777.3 | |
| (777.3 | ) | ||||||
Comprehensive earnings | 70.4 | |
| 1,061.8 | |
| - | |
| 1,028.9 | |
| (2,090.7 | ) |
| 70.4 | | ||||||
Comprehensive earnings attributable to the noncontrolling interest | (0.1 | ) |
| - | |
| - | |
| (0.1 | ) |
| 0.1 | |
| (0.1 | ) | ||||||
Comprehensive earnings attributable to Mylan N.V. ordinary shareholders | $ | 70.3 | |
| $ | 1,061.8 | |
| $ | - | |
| $ | 1,028.8 | |
| $ | (2,090.6 | ) |
| $ | 70.3 | |
161
Table of Contents
CONDENSED CONSOLIDATING STATEMENT OF COMPREHENSIVE EARNINGS
Year Ended December 31, 2014
(In millions) | Mylan Inc. |
| Guarantor Subsidiaries |
| Non-Guarantor Subsidiaries |
| Eliminations |
| Consolidated | ||||||||||
Net earnings | $ | 933.1 | |
| $ | - | |
| $ | 1,775.6 | |
| $ | (1,775.6 | ) |
| $ | 933.1 | |
Other comprehensive (loss) earnings, before tax: |
|
|
|
|
|
|
|
|
| ||||||||||
Foreign currency translation adjustment | (622.9 | ) |
| - | |
| (622.9 | ) |
| 622.9 | |
| (622.9 | ) | |||||
Change in unrecognized loss and prior service cost related to defined benefit plans | (11.8 | ) |
| - | |
| (7.4 | ) |
| 7.4 | |
| (11.8 | ) | |||||
Net unrecognized (loss) gain on derivatives | (182.6 | ) |
| - | |
| 30.4 | |
| (30.4 | ) |
| (182.6 | ) | |||||
Other comprehensive (loss) earnings, before tax | (817.3 | ) |
| - | |
| (599.9 | ) |
| 599.9 | |
| (817.3 | ) | |||||
Income tax (benefit) provision | (70.4 | ) |
| - | |
| 10.0 | |
| (10.0 | ) |
| (70.4 | ) | |||||
Other comprehensive loss, net of tax | (746.9 | ) |
| - | |
| (609.9 | ) |
| 609.9 | |
| (746.9 | ) | |||||
Comprehensive earnings | 186.2 | |
| - | |
| 1,165.7 | |
| (1,165.7 | ) |
| 186.2 | | |||||
Comprehensive earnings attributable to the noncontrolling interest | (3.7 | ) |
| - | |
| (3.7 | ) |
| 3.7 | |
| (3.7 | ) | |||||
Comprehensive earnings attributable to Mylan N.V. ordinary shareholders | $ | 182.5 | |
| $ | - | |
| $ | 1,162.0 | |
| $ | (1,162.0 | ) |
| $ | 182.5 | |
162
Table of Contents
CONDENSED CONSOLIDATING STATEMENT OF CASH FLOWS
Year Ended December 31, 2016
(In millions) | Mylan N.V. |
| Mylan Inc. |
| Guarantor Subsidiaries |
| Non-Guarantor Subsidiaries |
| Eliminations |
| Consolidated | ||||||||||||
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Net cash (used in) provided by operating activities | $ | (0.3 | ) |
| $ | (518.3 | ) |
| $ | - | |
| $ | 2,565.8 | |
| $ | - | |
| $ | 2,047.2 | |
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Capital expenditures | - | |
| (94.4 | ) |
| - | |
| (296.0 | ) |
| - | |
| (390.4 | ) | ||||||
Change in restricted cash | - | |
| (49.5 | ) |
| - | |
| 106.6 | |
| - | |
| 57.1 | | ||||||
Cash paid for acquisitions, net | (5,608.8 | ) |
| (931.3 | ) |
| - | |
| 58.2 | |
| - | |
| (6,481.9 | ) | ||||||
Settlement of acquisition-related foreign currency derivatives | (128.6 | ) |
| - | |
| - | |
| - | |
| - | |
| (128.6 | ) | ||||||
Cash paid for deferred purchase price | - | |
| - | |
| - | |
| (308.0 | ) |
| - | |
| (308.0 | ) | ||||||
Purchase of marketable securities | - | |
| (4.3 | ) |
| - | |
| (25.9 | ) |
| - | |
| (30.2 | ) | ||||||
Proceeds from sale of marketable securities | - | |
| - | |
| - | |
| 21.5 | |
| - | |
| 21.5 | | ||||||
Investments in affiliates | - | |
| (49.6 | ) |
| - | |
| - | |
| 49.6 | |
| - | | ||||||
Dividends from affiliates | 135.6 | |
| - | |
| - | |
| - | |
| (135.6 | ) |
| - | | ||||||
Loans to affiliates | (14,073.5 | ) |
| (530.2 | ) |
| - | |
| (3,185.0 | ) |
| 17,788.7 | |
| - | | ||||||
Repayments of loans from affiliates | 8,539.6 | |
| 793.0 | |
| - | |
| 1,914.1 | |
| (11,246.7 | ) |
| - | | ||||||
Payments for product rights and other, net | - | |
| 3.3 | |
| - | |
| (363.5 | ) |
| - | |
| (360.2 | ) | ||||||
Net cash (used in) provided by investing activities | (11,135.7 | ) |
| (863.0 | ) |
| - | |
| (2,078.0 | ) |
| 6,456.0 | |
| (7,620.7 | ) | ||||||
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Payments of financing fees | (112.6 | ) |
| - | |
| - | |
| - | |
| - | |
| (112.6 | ) | ||||||
Purchase of ordinary shares | - | |
| - | |
| - | |
| - | |
| - | |
| - | | ||||||
Change in short-term borrowings, net | - | |
| - | |
| - | |
| 40.8 | |
| - | |
| 40.8 | | ||||||
Proceeds from convertible note hedge | - | |
| - | |
| - | |
| - | |
| - | |
| - | | ||||||
Proceeds from issuance of long-term debt | 11,652.6 | |
| - | |
| - | |
| 99.6 | |
| - | |
| 11,752.2 | | ||||||
Payments of long-term debt | (400.0 | ) |
| (3,400.0 | ) |
| - | |
| (2,496.3 | ) |
| - | |
| (6,296.3 | ) | ||||||
Proceeds from exercise of stock options | 13.8 | |
| - | |
| - | |
| - | |
| - | |
| 13.8 | | ||||||
Taxes paid related to net share settlement of equity awards | (17.5 | ) |
| - | |
| - | |
| - | |
| - | |
| (17.5 | ) | ||||||
Contingent consideration payments | - | |
| - | |
| - | |
| (35.5 | ) |
| - | |
| (35.5 | ) | ||||||
Capital contribution from affiliates | - | |
| - | |
| - | |
| 49.6 | |
| (49.6 | ) |
| - | | ||||||
Capital payments to affiliates | - | |
| - | |
| - | |
| (135.6 | ) |
| 135.6 | |
| - | | ||||||
Payments on borrowings from affiliates | - | |
| (3,021.9 | ) |
| - | |
| (8,224.9 | ) |
| 11,246.8 | |
| - | | ||||||
Proceeds from borrowings from affiliates | - | |
| 6,961.2 | |
| - | |
| 10,827.6 | |
| (17,788.8 | ) |
| - | | ||||||
Acquisition of noncontrolling interest | - | |
| - | |
| - | |
| (1.1 | ) |
| - | |
| (1.1 | ) | ||||||
Other items, net | - | |
| (16.2 | ) |
| - | |
| 17.0 | |
| - | |
| 0.8 | | ||||||
Net cash provided by (used in) financing activities | 11,136.3 | |
| 523.1 | |
| - | |
| 141.2 | |
| (6,456.0 | ) |
| 5,344.6 | | ||||||
Effect on cash of changes in exchange rates | - | |
| - | |
| - | |
| (8.3 | ) |
| - | |
| (8.3 | ) | ||||||
Net (decrease) increase in cash and cash equivalents | 0.3 | |
| (858.2 | ) |
| - | |
| 620.7 | |
| - | |
| (237.2 | ) | ||||||
Cash and cash equivalents - beginning of period | - | |
| 870.5 | |
| - | |
| 365.5 | |
| - | |
| 1,236.0 | | ||||||
Cash and cash equivalents - end of period | $ | 0.3 | |
| $ | 12.3 | |
| $ | - | |
| $ | 986.2 | |
| $ | - | |
| $ | 998.8 | |
Supplemental disclosures of cash flow information - |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Non-cash transactions: |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Contingent consideration | $ | - | |
| $ | - | |
| $ | - | |
| $ | 16.0 | |
| $ | - | |
| $ | 16.0 | |
Ordinary shares issued for acquisition | $ | 1,281.7 | |
| $ | - | |
| $ | - | |
| $ | - | |
| $ | - | |
| $ | 1,281.7 | |
163
Table of Contents
CONDENSED CONSOLIDATING STATEMENT OF CASH FLOWS
(In millions) | Mylan N.V. |
| Mylan Inc. |
| Guarantor Subsidiaries |
| Non-Guarantor Subsidiaries |
| Eliminations |
| Consolidated | ||||||||||||
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Net cash (used in) provided by operating activities | $ | (57.5 | ) |
| $ | (707.2 | ) |
| $ | - | |
| $ | 2,773.2 | |
| $ | - | |
| $ | 2,008.5 | |
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Capital expenditures | - | |
| (85.4 | ) |
| - | |
| (277.5 | ) |
| - | |
| (362.9 | ) | ||||||
Change in restricted cash | - | |
| (3.6 | ) |
| - | |
| 25.4 | |
| - | |
| 21.8 | | ||||||
Cash paid for acquisitions, net | - | |
| - | |
| - | |
| (693.1 | ) |
| - | |
| (693.1 | ) | ||||||
Purchase of marketable securities | - | |
| - | |
| - | |
| (62.1 | ) |
| - | |
| (62.1 | ) | ||||||
Proceeds from sale of marketable securities | - | |
| - | |
| - | |
| 33.1 | |
| - | |
| 33.1 | | ||||||
Investments in affiliates | - | |
| (607.9 | ) |
| - | |
| - | |
| 607.9 | |
| - | | ||||||
Loans to affiliates | (1,097.5 | ) |
| (5,856.4 | ) |
| - | |
| (7,682.2 | ) |
| 14,636.1 | |
| - | | ||||||
Repayments of loans from affiliates | - | |
| 358.5 | |
| - | |
| 1,198.5 | |
| (1,557.0 | ) |
| - | | ||||||
Payments for product rights and other, net | - | |
| (1.5 | ) |
| - | |
| (505.0 | ) |
| - | |
| (506.5 | ) | ||||||
Net cash (used in) provided by investing activities | (1,097.5 | ) |
| (6,196.3 | ) |
| - | |
| (7,962.9 | ) |
| 13,687.0 | |
| (1,569.7 | ) | ||||||
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Payments of financing fees | (104.4 | ) |
| (26.0 | ) |
| - | |
| - | |
| - | |
| (130.4 | ) | ||||||
Purchase of ordinary shares | (67.5 | ) |
| - | |
| - | |
| - | |
| - | |
| (67.5 | ) | ||||||
Change in short-term borrowings, net | - | |
| - | |
| - | |
| (329.2 | ) |
| - | |
| (329.2 | ) | ||||||
Proceeds from convertible note hedge | - | |
| 1,970.8 | |
| - | |
| - | |
| - | |
| 1,970.8 | | ||||||
Proceeds from issuance of long-term debt | 999.2 | |
| 2,540.0 | |
| - | |
| - | |
| - | |
| 3,539.2 | | ||||||
Payments of long-term debt | - | |
| (4,484.1 | ) |
| - | |
| - | |
| - | |
| (4,484.1 | ) | ||||||
Proceeds from exercise of stock options | 44.4 | |
| 53.3 | |
| - | |
| - | |
| - | |
| 97.7 | | ||||||
Taxes paid related to net share settlement of equity awards | - | |
| (25.9 | ) |
| - | |
| (5.9 | ) |
| - | |
| (31.8 | ) | ||||||
Capital contribution from affiliates | - | |
| - | |
| - | |
| 607.9 | |
| (607.9 | ) |
| - | | ||||||
Proceeds from borrowings from affiliates | 283.2 | |
| 8,779.7 | |
| - | |
| 5,573.2 | |
| (14,636.1 | ) |
| - | | ||||||
Payments on borrowings from affiliates | - | |
| (1,198.5 | ) |
| - | |
| (358.5 | ) |
| 1,557.0 | |
| - | | ||||||
Acquisition of noncontrolling interest | - | |
| - | |
| - | |
| (11.7 | ) |
| - | |
| (11.7 | ) | ||||||
Other items, net | - | |
| 51.8 | |
| - | |
| - | |
| - | |
| 51.8 | | ||||||
Net cash provided by (used in) financing activities | 1,154.9 | |
| 7,661.1 | |
| - | |
| 5,475.8 | |
| (13,687.0 | ) |
| 604.8 | | ||||||
Effect on cash of changes in exchange rates | - | |
| - | |
| - | |
| (33.1 | ) |
| - | |
| (33.1 | ) | ||||||
Net (decrease) increase in cash and cash equivalents | (0.1 | ) |
| 757.6 | |
| - | |
| 253.0 | |
| - | |
| 1,010.5 | | ||||||
Cash and cash equivalents - beginning of period | 0.1 | |
| 112.9 | |
| - | |
| 112.5 | |
| - | |
| 225.5 | | ||||||
Cash and cash equivalents - end of period | $ | - | |
| $ | 870.5 | |
| $ | - | |
| $ | 365.5 | |
| $ | - | |
| $ | 1,236.0 | |
Supplemental disclosures of cash flow information - |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Non-cash transactions: |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Contingent consideration | $ | - | |
| $ | - | |
| $ | - | |
| $ | 18.0 | |
| $ | - | |
| $ | 18.0 | |
Ordinary shares issued for acquisition | $ | 6,305.8 | |
| $ | - | |
| $ | - | |
| $ | - | |
| $ | - | |
| $ | 6,305.8 | |
164
Table of Contents
CONDENSED CONSOLIDATING STATEMENT OF CASH FLOWS
(In millions) | Mylan Inc. |
| Guarantor Subsidiaries |
| Non-Guarantor Subsidiaries |
| Eliminations |
| Consolidated | ||||||||||
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
| ||||||||||
Net cash (used in) provided by operating activities | $ | (705.1 | ) |
| $ | - | |
| $ | 1,719.9 | |
| $ | - | |
| $ | 1,014.8 | |
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
| ||||||||||
Capital expenditures | (72.1 | ) |
| - | |
| (253.2 | ) |
| - | |
| (325.3 | ) | |||||
Change in restricted cash | - | |
| - | |
| (5.1 | ) |
| - | |
| (5.1 | ) | |||||
Cash paid for acquisitions, net | - | |
| - | |
| (50.0 | ) |
| - | |
| (50.0 | ) | |||||
Purchase of marketable securities | (2.8 | ) |
| - | |
| (17.1 | ) |
| - | |
| (19.9 | ) | |||||
Proceeds from sale of marketable securities | 1.6 | |
| - | |
| 18.6 | |
| - | |
| 20.2 | | |||||
Investments in affiliates | (64.1 | ) |
| - | |
| - | |
| 64.1 | |
| - | | |||||
Loans to affiliates | (5,901.0 | ) |
| - | |
| (6,857.5 | ) |
| 12,758.5 | |
| - | | |||||
Repayments of loans from affiliates | 5.8 | |
| - | |
| 20.2 | |
| (26.0 | ) |
| - | | |||||
Payments for product rights and other, net | (1.9 | ) |
| - | |
| (418.3 | ) |
| - | |
| (420.2 | ) | |||||
Net cash (used in) provided by investing activities | (6,034.5 | ) |
| - | |
| (7,562.4 | ) |
| 12,796.6 | |
| (800.3 | ) | |||||
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
| ||||||||||
Payment of financing fees | (5.8 | ) |
| - | |
| - | |
| - | |
| (5.8 | ) | |||||
Change in short-term borrowings, net | - | |
| - | |
| (107.8 | ) |
| - | |
| (107.8 | ) | |||||
Proceeds from issuance of long-term debt | 2,235.0 | |
| - | |
| - | |
| - | |
| 2,235.0 | | |||||
Payment of long-term debt | (2,295.8 | ) |
| - | |
| - | |
| - | |
| (2,295.8 | ) | |||||
Proceeds from exercise of stock options | 53.8 | |
| - | |
| - | |
| - | |
| 53.8 | | |||||
Taxes paid related to net share settlement of equity awards | (17.3 | ) |
| - | |
| (10.4 | ) |
| - | |
| (27.7 | ) | |||||
Payments for contingent consideration | - | |
| - | |
| (150.0 | ) |
| - | |
| (150.0 | ) | |||||
Capital contribution from affiliates | - | |
| - | |
| 64.1 | |
| (64.1 | ) |
| - | | |||||
Proceeds from borrowings from affiliates | 6,857.5 | |
| - | |
| 5,901.0 | |
| (12,758.5 | ) |
| - | | |||||
Payments on borrowings from affiliates | (20.2 | ) |
| - | |
| (5.8 | ) |
| 26.0 | |
| - | | |||||
Other items, net | 30.9 | |
| - | |
| - | |
| - | |
| 30.9 | | |||||
Net cash provided by (used in) financing activities | 6,838.1 | |
| - | |
| 5,691.1 | |
| (12,796.6 | ) |
| (267.4 | ) | |||||
Effect on cash of changes in exchange rates | - | |
| - | |
| (12.9 | ) |
| - | |
| (12.9 | ) | |||||
Net increase (decrease) in cash and cash equivalents | 98.5 | |
| - | |
| (164.3 | ) |
| - | |
| (65.8 | ) | |||||
Cash and cash equivalents - beginning of period | 14.4 | |
| - | |
| 276.9 | |
| - | |
| 291.3 | | |||||
Cash and cash equivalents - end of period | $ | 112.9 | |
| $ | - | |
| $ | 112.6 | |
| $ | - | |
| $ | 225.5 | |
165
Table of Contents
16. | Restructuring |
On December 5, 2016, the Company announced restructuring programs in certain locations representing initial steps in a series of actions that are anticipated to further streamline its operations globally. Since 2015, the Company has made a number of significant acquisitions, and as part of the holistic, global integration of these acquisitions, the Company is focused on how to best optimize and maximize all of its assets across the organization and across all geographies. A pre-tax restructuring charge of $149.7 million was recorded for certain workforce reduction and cost savings initiatives during the year ended December 31, 2016 , the majority of which represents employee severance and other employee related costs. Of this amount, approximately $28.9 million is included in cost of sales, $7.7 million is included in R&D and $113.1 million is included in SG&A in the Consolidated Statements of Operations .
Charges for restructuring and ongoing cost reduction initiatives are recorded in the period the Company commits to a restructuring or cost reduction plan, or executes specific actions contemplated by the plan and all criteria for liability recognition have been met.
The Company continues to develop the details of the cost reduction initiatives, including workforce actions and other potential restructuring activities beyond the programs announced, including potential shutdown or consolidation of certain operations. The continued restructuring actions are expected to be implemented through fiscal year 2018. For the restructuring activities that have been approved to date, the Company estimates that it will incur aggregate pre-tax charges ranging between $175.0 million and $225.0 million , inclusive of the 2016 restructuring charge of $149.7 million , the majority of which are employee related costs. As additional restructuring activities are approved, the Company expects to incur additional costs including employee related costs, such as severance and continuation of healthcare and other benefits; asset impairments; accelerated deprecation; costs associated with contract terminations; and, other closure costs. At this time, the expenses related to the additional restructuring activities cannot be reasonably estimated.
The following table summarizes the restructuring charges and the reserve activity since inception and through the year ended December 31, 2016 :
(In millions) | Employee Related Costs |
| Other Exit Costs |
| Total | ||||||
Balance at December 31, 2015: | $ | 14.8 | |
| $ | - | |
| $ | 14.8 | |
Charges (1) | 148.1 | |
| 1.6 | |
| 149.7 | | |||
Cash payment | (24.3 | ) |
| - | |
| (24.3 | ) | |||
Balance at December 31, 2016: | $ | 138.6 | |
| $ | 1.6 | |
| $ | 140.2 | |
(1) | For the year ended December 31, 2016 , restructuring charges for employee related costs in North America, Europe and Rest of World were approximately $89.9 million , $53.7 million and $4.5 million , respectively. |
For the years ended December 31, 2015 and 2014 , the Company recorded approximately $18.7 million and $10.4 million of restructuring expenses, respectively. The expenses were primarily incurred in connection with various initiatives as part of cost saving efforts.
At December 31, 2016 and 2015 , accrued liabilities for restructuring and other costs reduction programs were included in other current liabilities on the Consolidated Balance Sheets .
17. | Collaboration and Licensing Agreements |
We periodically enter into collaboration and licensing agreements with other pharmaceutical companies for the development, manufacture, marketing and/or sale of pharmaceutical products. Our significant collaboration agreements are focused on the development, manufacturing, supply and commercialization of multiple, high-value generic biologic compounds, insulin analog products and respiratory products. Under these agreements, we have future potential milestone payments and co-development expenses payable to third parties as part of our licensing, development and co-development programs. Payments under these agreements generally become due and are payable upon the satisfaction or achievement of certain developmental, regulatory or commercial milestones or as development expenses are incurred on defined projects. Milestone payment obligations are uncertain, including the prediction of timing and the occurrence of events triggering a future obligation and are not reflected as liabilities in the Consolidated Balance Sheets, except for milestone and royalty obligations reflected as acquisition related contingent consideration. Refer to Note 7 Financial Instruments and Risk Management for
166
Table of Contents
further discussion of contingent consideration. Our potential maximum development milestones not accrued for at December 31, 2016 totaled approximately $596 million . We estimate that the amounts that may be paid in the next twelve months to be approximately $172 million . These agreements may also include potential sales-based milestones and call for us to pay a percentage of amounts earned from the sale of the product as a royalty or a profit share. The amounts disclosed do not include sales based milestones or royalty obligations on future sales of product as the timing and amount of future sales levels and costs to produce products subject to these obligations is not reasonably estimable. These sales-based milestones or royalty obligations may be significant depending upon the level of commercial sales for each product. A summary of our most significant collaboration and licensing agreements include the following:
On January 8, 2016, the Company entered into an agreement with Momenta Pharmaceuticals, Inc. (" Momenta ") to develop, manufacture and commercialize up to six of Momenta 's current biosimilar candidates, including Momenta 's biosimilar candidate, ORENCIA® (abatacept). Mylan paid an up-front cash payment of $45 million to Momenta. Under the terms of the agreement, the Company and Momenta are jointly responsible for product development and equally share in the costs and profits of the products with Mylan leading the worldwide commercialization efforts. Under the terms of the agreement, Momenta is eligible to receive additional contingent milestone payments of up to $200 million .
On November 2, 2016, the Company and Momenta announced that dosing had begun in a Phase 1 study to compare the pharmacokinetics, safety and immunogenicity of M834, a proposed biosimilar of ORENCIA® (abatacept), to U.S. and European Union sourced ORENCIA® in normal healthy volunteers. Under the agreement, Mylan paid $60 million related to certain milestones in 2016.
In accordance with ASC 730, Research and Development and based upon the cost sharing provisions of the agreement, the Company is accounting for the contingent milestone payments related to the Momenta collaboration as non-refundable advance payments for services to be used in future R&D activities, which are required to be capitalized until the related services have been performed. More specifically, as costs are incurred within the scope of the collaboration, the Company will record its share of the costs as R&D expense. In addition to the upfront cash payment, during the year ended December 31, 2016 the Company incurred approximately $29.2 million of R&D expense related to this collaboration. To the extent the contingent milestone payments made by the Company exceed the liability incurred, a prepaid asset will be reflected on the Company's Consolidated Balance Sheet. To the extent the contingent milestone payments made by the Company are less than the expense incurred, the difference between the payment and the expense will be recorded as a liability on the Company's Consolidated Balance Sheet. At December 31, 2016, approximately $30.8 million was recorded as a prepaid asset on the Consolidated Balance Sheet.
On January 30, 2015 , the Company entered into a development and commercialization collaboration with Theravance Biopharma, Inc. (" Theravance Biopharma ") for the development and, subject to FDA approval, commercialization of Revefenacin ("TD-4208"), a novel once-daily nebulized long-acting muscarinic antagonist for chronic obstructive pulmonary disease ("COPD") and other respiratory diseases. Under the terms of the agreement, Mylan and Theravance Biopharma are co-developing nebulized TD-4208 for COPD and other respiratory diseases. Theravance Biopharma is leading the U.S. registrational development program and Mylan is responsible for the reimbursement of Theravance Biopharma 's development costs for that program up until the approval of the first new drug application, after which costs will be shared. In addition, Mylan is responsible for commercial manufacturing. In the U.S., Mylan is leading commercialization and Theravance Biopharma retains the right to co-promote the product under a profit-sharing arrangement. On September 14, 2015, Mylan announced the initiation of the Phase 3 program that will support the registrational development program of TD-4208 in the U.S. In addition to funding the U.S. registrational development program, the Company made a $30 million investment in Theravance Biopharma 's common stock during the first quarter of 2015, which is being accounted for as an available-for-sale security. The Company also incurred $15 million in upfront development costs during the year ended December 31, 2015. Under the terms of the agreement, Theravance Biopharma is eligible to receive potential development and sales milestone payments totaling $220 million in the aggregate. As of December 31, 2016, Mylan has paid a total of $15 million in milestone payments to Theravance Biopharma .
In the fourth quarter of 2013, the Company entered into a licensing agreement with Pfizer Inc. for the exclusive worldwide rights to develop, manufacture and commercialize a novel long-acting muscarinic antagonist compound. Depending on the commercialization of this novel compound and the level of future sales and profits, the Company could also be obligated to make payments upon the occurrence of certain sales milestones, along with sales royalties and profit sharing payments. The Company has also entered into exclusive collaborations with Biocon Limited on the development, manufacturing, supply and commercialization of multiple, high value generic biologic compounds and three insulin analog products for the global marketplace. The Company plans to provide funding related to the collaborations over the next several years. As the timing of cash expenditures is dependent upon a number of factors, many of which are out of the Company's control, it is difficult to forecast the amount of payments to be made over the next few years, which could be significant.
167
Table of Contents
18. | Litigation |
The Company is involved in various disputes, governmental and/or regulatory inquiries and proceedings, tax proceedings and litigation matters that arise from time to time, some of which are described below. The Company is also party to certain litigation matters including those for which Merck KGaA or Strides Arcolab has agreed to indemnify the Company, pursuant to the respective sale and purchase agreements.
While the Company believes that it has meritorious defenses with respect to the claims asserted against it and intends to vigorously defend its position, the process of resolving matters through litigation or other means is inherently uncertain, and it is not possible to predict the ultimate resolution of any such proceeding. It is possible that an unfavorable resolution of any of the matters described below, or the inability or denial of Merck KGaA, Strides Arcolab or another indemnitor or insurer to pay an indemnified claim, could have a material effect on the Company's business financial condition, results of operations, cash flows and/or ordinary share price. Unless otherwise disclosed below, the Company is unable to predict the outcome of the respective litigation or to provide an estimate of the range of reasonably possible losses. Legal costs are recorded as incurred and are classified in SG&A in the Company's Consolidated Statements of Operations .
Lorazepam and Clorazepate
On June 1, 2005, a jury verdict was rendered against Mylan , MPI , and co-defendants Cambrex Corporation and Gyma Laboratories in the U.S. District Court for the District of Columbia in the amount of approximately $12.0 million , which has been accrued for by the Company. The jury found that Mylan and its co-defendants willfully violated Massachusetts, Minnesota and Illinois state antitrust laws in connection with API supply agreements entered into between the Company and its API supplier (Cambrex) and broker (Gyma) for two drugs, Lorazepam and Clorazepate, in 1997, and subsequent price increases on these drugs in 1998. The case was brought by four health insurers who opted out of earlier class action settlements agreed to by the Company in 2001 and represents the last remaining antitrust claims relating to Mylan 's 1998 price increases for Lorazepam and Clorazepate. Following the verdict, the Company filed a motion for judgment as a matter of law, a motion for a new trial, a motion to dismiss two of the insurers and a motion to reduce the verdict. On December 20, 2006, the Company's motion for judgment as a matter of law and motion for a new trial were denied and the remaining motions were denied on January 24, 2008. In post-trial filings, the plaintiffs requested that the verdict be trebled and that request was granted on January 24, 2008. On February 6, 2008, a judgment was issued against Mylan and its co-defendants in the total amount of approximately $69.0 million , which, in the case of three of the plaintiffs, reflects trebling of the compensatory damages in the original verdict (approximately $11.0 million in total) and, in the case of the fourth plaintiff, reflects their amount of the compensatory damages in the original jury verdict plus doubling this compensatory damage award as punitive damages assessed against each of the defendants (approximately $58.0 million in total), some or all of which may be subject to indemnification obligations by Mylan . Plaintiffs are also seeking an award of attorneys' fees and litigation costs in unspecified amounts and prejudgment interest of approximately $8.0 million . The Company and its co-defendants appealed to the U.S. Court of Appeals for the D.C. Circuit and have challenged the verdict as legally erroneous on multiple grounds. The appeals were held in abeyance pending a ruling on the motion for prejudgment interest, which has been granted. Mylan has contested this ruling along with the liability finding and other damages awards as part of its appeal, which was filed in the Court of Appeals for the D.C. Circuit. On January 18, 2011, the Court of Appeals issued a judgment remanding the case to the District Court for further proceedings based on lack of diversity with respect to certain plaintiffs. On June 13, 2011, Mylan filed a certiorari petition with the U.S. Supreme Court requesting review of the judgment of the D.C. Circuit. On October 3, 2011, the certiorari petition was denied. The case is now proceeding before the District Court. On January 14, 2013, following limited court-ordered jurisdictional discovery, the plaintiffs filed a fourth amended complaint containing additional factual averments with respect to the diversity of citizenship of the parties, along with a motion to voluntarily dismiss 775 (of 1,387 ) self-funded customers whose presence would destroy the District Court's diversity jurisdiction. The plaintiffs also moved for a remittitur (reduction) of approximately $8.1 million from the full damages award. Mylan 's brief in response to the new factual averments in the complaint was filed on February 13, 2013. On July 29, 2014, the court granted both plaintiffs' motion to amend the complaint and their motion to dismiss 775 self-funded customers.
In connection with the Company's appeal of the judgment, the Company submitted a surety bond underwritten by a third-party insurance company in the amount of $74.5 million in February 2008. On May 30, 2012, the District Court ordered the amount of the surety bond reduced to $66.6 million .
Pricing and Medicaid Litigation
Dey L.P. (now known as Mylan Specialty L.P. and herein as " Mylan Specialty "), a wholly owned subsidiary of the Company, was named as a defendant in several class actions brought by consumers and third-party payors. Mylan Specialty reached a settlement of these class actions, which was approved by the court and all claims have been dismissed. Additionally, a
168
Table of Contents
complaint was filed under seal by a plaintiff on behalf of the United States of America against Mylan Specialty in August 1997. In August 2006, the Government filed its complaint-in-intervention and the case was unsealed in September 2006. The Government asserted that Mylan Specialty was jointly liable with a co-defendant and sought recovery of alleged overpayments, together with treble damages, civil penalties and equitable relief. Mylan Specialty completed a settlement of this action in December 2010. These cases all have generally alleged that Mylan Specialty falsely reported certain price information concerning certain drugs marketed by Mylan Specialty , that Mylan Specialty caused false claims to be made to Medicaid and to Medicare, and that Mylan Specialty caused Medicaid and Medicare to make overpayments on those claims.
Under the terms of the purchase agreement with Merck KGaA, Mylan is fully indemnified for the claims in the preceding paragraph and Merck KGaA is entitled to any income tax benefit the Company realizes for any deductions of amounts paid for such pricing litigation. Under the indemnity, Merck KGaA is responsible for all settlement and legal costs, and, as such, these settlements had no impact on the Company's Consolidated Statements of Operations . At December 31, 2016 , the Company has accrued approximately $63.3 million in other current liabilities, which represents its estimate of the remaining amount of anticipated income tax benefits due to Merck KGaA. We are not aware of any outstanding claims related to Merck KGaA.
Modafinil Antitrust Litigation and FTC Inquiry
Beginning in April 2006, Mylan and four other drug manufacturers have been named as defendants in civil lawsuits filed in or transferred to the U.S. District Court for the Eastern District of Pennsylvania by a variety of plaintiffs purportedly representing direct and indirect purchasers of the drug modafinil and in a lawsuit filed by Apotex, Inc., a manufacturer of generic drugs. These actions allege violations of federal antitrust and state laws in connection with the generic defendants' settlement of patent litigation with Cephalon relating to modafinil. Discovery has closed. On June 23, 2014, the court granted the defendants' motion for partial summary judgment dismissing plaintiffs' claims that the defendants had engaged in an overall conspiracy to restrain trade (and denied the corresponding plaintiffs' motion). On January 28, 2015, the District Court denied the defendants' summary judgment motions based on factors identified in the Supreme Court's Actavis decision. In an order on June 1, 2015, vacated and reissued on June 11, 2015, the District Court denied the indirect purchaser plaintiffs' motion for class certification. The indirect purchaser plaintiffs filed a petition for leave to appeal the certification decision, which was denied by the Court of Appeals for the Third Circuit on December 21, 2015. On July 27, 2015, the District Court granted the direct purchaser plaintiffs' motion for class certification. On October 9, 2015, the Third Circuit granted defendants' petition for leave to appeal the class certification decision. On October 16, 2015, defendants filed a motion to stay the liability trial, which had been set to begin on February 2, 2016, with the District Court pending the appeal of the decision to certify the direct purchaser class; this motion was denied on December 17, 2015. On December 17, 2015, the District Court approved the form and manner of notice to the certified class of direct purchasers; the notice was subsequently issued to the class. On December 21, 2015, the defendants filed a motion to stay with the Court of Appeals for the Third Circuit, which was granted on January 25, 2016; accordingly, the trial was stayed and the case was placed in suspense. The appeal was fully briefed on April 28, 2016. Oral arguments on the appeal took place on July 12, 2016. On September 13, 2016, the Third Circuit reversed the district court's certification order and remanded for further proceedings. On October 14, 2016 direct purchaser plaintiffs filed a petition seeking rehearing. On October 31, 2016 the petition seeking rehearing was denied. On December 12, 2016, the District Court removed the case from suspense and set the trial for June 5, 2017. On March 24, 2015, Mylan reached a settlement in principle with the putative indirect purchasers and on November 20, 2015, Mylan entered into a settlement agreement with the putative indirect purchasers for approximately $16 million . Plaintiffs have not yet moved for preliminary approval of that settlement. In December 2016, Mylan reached a settlement with the putative direct purchaser class and the retailer opt-out plaintiffs for $165 million , of which approximately $68.5 million was paid before December 31, 2016. The settlement with the retailer opt-out plaintiffs has been completed. The settlement with the putative direct purchaser class will undergo the court approval process. On February 3, 2017, the putative direct purchaser class moved for preliminary approval. At December 31, 2016 , the Company has accrued in total approximately $112.5 million related to this matter.
On June 29, 2015, the City of Providence, Rhode Island filed suit in the District of Rhode Island against the same parties named as defendants in litigation pending in the Eastern District of Pennsylvania, including Mylan, asserting state law claims based on the same underlying allegations. All defendants, including Mylan, moved to dismiss the suit on October 15, 2015, and the case was subsequently settled.
On July 10, 2015, the Louisiana Attorney General filed in the 19th Judicial District Court in Louisiana a petition against Mylan and three other drug manufacturers asserting state law claims based on the same underlying allegations as those made in litigation pending in the Eastern District of Pennsylvania. The petition was filed by the State of Louisiana purportedly in its capacity as an indirect purchaser. On May 16, 2016, the Judicial District Court deferred Mylan's declinatory exception of no personal jurisdiction and its peremptory exception of prescription, and granted in part and denied in part Mylan's peremptory exceptions of no cause of action and no right of action. On June 30, 2016, the plaintiff filed a supplemental and amended
169
Table of Contents
petition. The defendants filed a motion to strike and joint peremptory exceptions to the amended petition. On July 21, 2016, the plaintiff filed in the First Circuit Court of Appeal its application for a supervisory writ regarding the granting of defendant's exceptions, which the defendants opposed. The appeal was denied on October 31, 2016. On April 20, 2016, the State of Louisiana filed a motion to consolidate the pending action with four other actions against other pharmaceutical manufacturers concerning products not related to modafinil, which Mylan opposed. On June 27, 2016, the Judicial District Court declined to consolidate Mylan's case with the other four actions, with leave to renew the consolidation request after filing the above-referenced amended petition. On July 21, 2016, the plaintiff filed a motion to reurge consolidation. Subsequently, the action to which plaintiff seeks to join Mylan was stayed, resulting in a stay of the consolidation motion. On December 8, 2016, Mylan's peremptory exceptions of no cause of action with respect to the supplemental and amended petition were granted in their entirety and with prejudice and judgment was entered. On February 17, 2017, the plaintiff filed in the 19th Judicial District Court a motion for appeal, which the Judicial District Court granted on February 21, 2017.
On July 28, 2016, United Healthcare filed a complaint against Mylan and four other drug manufacturers in the United States District of Minnesota, asserting state law claims based on the same underlying allegations as those made in litigation pending in the Eastern District of Pennsylvania. Mylan's response deadline is March 31, 2017. On January 6, 2017, the case was transferred to the Eastern District of Pennsylvania.
In addition, by letter dated July 11, 2006, Mylan was notified by the U.S. Federal Trade Commission ("FTC") of an investigation relating to the settlement of the modafinil patent litigation. In its letter, the FTC requested certain information from Mylan , MPI and Mylan Technologies, Inc. pertaining to the patent litigation and the settlement thereof. On March 29, 2007, the FTC issued a subpoena, and on April 26, 2007, the FTC issued a civil investigative demand to Mylan , requesting additional information from the Company relating to the investigation. Mylan has cooperated fully with the government's investigation and completed all requests for information. On February 13, 2008, the FTC filed a lawsuit against Cephalon in the U.S. District Court for the District of Columbia and the case was subsequently transferred to the U.S. District Court for the Eastern District of Pennsylvania. On July 1, 2010, the FTC issued a third party subpoena to Mylan , requesting documents in connection with its lawsuit against Cephalon. Mylan has responded to the subpoena. The lawsuit against Cephalon settled and a Stipulated Order for Permanent Injunction and Equitable Monetary Relief was entered by the Court on June 17, 2015. The Company believes that it has strong defenses to the remaining cases. Although it is reasonably possible that the Company may incur additional losses from these matters, any amount cannot be reasonably estimated at this time.
Pioglitazone
Beginning in December 2013, Mylan , Takeda, and several other drug manufacturers have been named as defendants in civil lawsuits consolidated in the U.S. District Court for the Southern District of New York by plaintiffs which purport to represent indirect purchasers of branded or generic Actos® and Actoplus Met®. These actions allege violations of state and federal competition laws in connection with the defendants' settlements of patent litigation in 2010 relating to Actos and Actoplus Met®. Plaintiffs filed an amended complaint on August 22, 2014. Mylan and the other defendants filed motions to dismiss the amended complaint on October 10, 2014. Two additional complaints were subsequently filed by plaintiffs purporting to represent classes of direct purchasers of branded or generic Actos® and Actoplus Met®. On September 23, 2015, the District Court granted defendants' motions to dismiss the indirect purchasers amended complaints with prejudice. The indirect purchasers filed a notice of appeal on October 22, 2015; however they have since abandoned and dismissed their appeal of the District Court's dismissal of claims asserted against Mylan. The putative direct purchaser class filed an amended complaint on January 8, 2016. Defendants' motion to dismiss was filed on January 28, 2016 and the briefing has been completed. The case has been stayed pending the resolution of the indirect purchasers' appeal against the defendants remaining in that case. A decision was issued by the Second Circuit on February 8, 2017, reversing in part and affirming in part, the District Court's decision as to the remaining defendants.
SEC Investigation
On September 10, 2015, Mylan N.V. received a subpoena from the SEC seeking documents with regard to certain related party matters. Mylan is fully cooperating with the SEC in its investigation, and we are unable to predict the outcome of this matter at this time.
MDRP Classification of EpiPen® Auto-Injector and EpiPen Jr® Auto-Injector
In November 2014, the Company received a subpoena from the U.S. Department of Justice ("DOJ") related to the classification of the EpiPen® Auto-Injector for purposes of the Medicaid Drug Rebate Program. The Company complied with various information requests received from the DOJ pursuant to the subpoena. The question in the underlying matter was whether EpiPen® Auto-Injector was properly classified with the Centers for Medicare and Medicaid Services ("CMS") as a
170
Table of Contents
non-innovator drug under the applicable definition in the Medicaid Rebate statute and subject to the formula that is used to calculate rebates to Medicaid for such drugs. EpiPen® Auto-Injector has been classified with CMS as a non-innovator drug since before Mylan acquired the product in 2007 based on longstanding written guidance from the federal government. Beginning in August 2016, questions regarding the pricing of the EpiPen® Auto-Injector significantly increased and the Company received additional inquiries, including with respect to the classification of EpiPen® Auto-Injector for purposes of the Medicaid Drug Rebate Program, from committees and members of Congress and from other federal and state governmental agencies.
Subsequent to these developments, on October 7, 2016, Mylan agreed to the terms of a $465 million settlement with the DOJ and other government agencies related to the classification of the EpiPen® Auto-Injector for purposes of the Medicaid Drug Rebate Program. The terms of the settlement do not provide for any finding of wrongdoing on the part of Mylan Inc. or any of its affiliated entities or personnel. The settlement terms provide for resolution of all potential Medicaid rebate liability claims by federal and state governments as to whether the product should have been classified as an innovator drug for Medicaid Drug Rebate Program purposes, and subject to a higher rebate formula. In connection with the settlement, EpiPen® Auto-Injector will begin being classified as an innovator drug on April 1, 2017. Also in connection with the settlement, Mylan expects to enter into a corporate integrity agreement with the Office of Inspector General of the Department of Health and Human Services. Mylan continues to work with the government to finalize the settlement. During the year ended December 31, 2016, the Company recorded an accrual of $465 million related to the DOJ settlement which is included in other current liabilities in the Consolidated Balance Sheets.
SEC Document Request/Subpoena
On October 7, 2016, Mylan received a document request from the Division of Enforcement at the SEC seeking communications with CMS and documents concerning Mylan products sold and related to the Medicaid Drug Rebate Program, and any related complaints. On November 15, 2016, Mylan received a follow-up letter, modifying the initial document request, seeking information on and public disclosures regarding the settlement with the DOJ announced on October 7, 2016 and the classification of the EpiPen® Auto-Injector under the Medicaid Drug Rebate Program. On February 6, 2017, Mylan received a subpoena from the SEC in this matter, seeking additional documents. Mylan is fully cooperating with the SEC's inquiry.
FTC Request for Information
On November 18, 2016, Mylan received a request from the FTC Bureau of Competition seeking documents and information relating to its preliminary investigation into potential anticompetitive practices in the market for epinephrine auto-injectors. Mylan is fully cooperating with the FTC's inquiry.
EpiPen® Auto-Injector Federal Securities Litigation
Purported class action complaints were filed in October 2016 against Mylan N.V., Mylan Inc. and certain of their directors and officers (collectively, for purposes of this paragraph, the "defendants") in the United States District Court for the Southern District of New York on behalf of certain purchasers of securities of Mylan N.V. and/or Mylan Inc. The complaints allege that defendants made false or misleading statements and omissions of purportedly material fact, in violation of federal securities laws, in connection with disclosures relating to Mylan N.V. and Mylan Inc.'s classification of their EpiPen® Auto-Injector as a non-innovator drug for purposes of the Medicaid Drug Rebate Program. The complaints seek damages, as well as the plaintiffs' fees and costs. On January 9, 2017, the Court consolidated the cases for pre-trial purposes and appointed co-lead plaintiffs. On January 20, 2017, the Court entered a scheduling order, with co-lead plaintiffs' consolidated amended complaint due by March 20, 2017, and defendants' response to that complaint due by May 30, 2017. We believe that the claims in these lawsuits are without merit and intend to defend against them vigorously.
EpiPen® Auto-Injector Israeli Securities Litigation
On October 13, 2016, a purported shareholder of Mylan N.V. filed a lawsuit, together with a motion to certify the lawsuit as a class action on behalf of certain Mylan N.V. shareholders, against Mylan N.V. and four of its directors and officers (collectively, for purposes of this paragraph, the "defendants") in the Tel Aviv District Court (Economic Division). The plaintiff alleges that the defendants made false or misleading statements and omissions of purportedly material fact in Mylan N.V.'s reports to the Tel Aviv Stock Exchange regarding Mylan N.V.'s classification of its EpiPen® Auto-Injector for purposes of the Medicaid Drug Rebate Program, in violation of both U.S. and Israeli securities laws, the Israeli Companies Law and the Israeli Torts Ordinance. The plaintiff seeks damages, among other remedies. On January 19. 2017, the Court stayed this case until a final judgment is issued in the securities litigation currently pending in the United States District Court for the Southern District of New York. We believe that the claims in this lawsuit are without merit and intend to defend against them vigorously.
171
Table of Contents
EpiPen® Auto-Injector Civil Litigation
Beginning in August 2016, Mylan Specialty L.P. and other Mylan-affiliated entities (as well as a Mylan officer and other non-Mylan affiliated companies) have been named as defendants in nine putative class action lawsuits relating to the pricing and/or marketing of the EpiPen® Auto-Injector, which were filed in the U.S. District Courts for the Northern District of California, Northern District of Illinois, District of Kansas, Eastern District of Michigan, and the Western District of Pennsylvania, as well as the Hamilton County, Ohio Court of Common Pleas (later removed to the Southern District of Ohio). These putative class action lawsuits have either been dismissed or consolidated into a single putative class action pending in the U.S. District Court for the District of Kansas. The plaintiffs assert violations of various state antitrust and consumer protection laws, the Racketeer Influenced and Corrupt Organizations Act ("RICO"), as well as common law claims. Plaintiffs' claims include purported challenges to the prices charged for the EpiPen® Auto-Injector and/or the marketing of the product in packages containing two auto-injectors. We believe that the claims in this lawsuit are without merit and intend to defend against them vigorously.
EpiPen® Auto-Injector State AG Investigations
Beginning in August 2016, the Company and certain of its affiliated entities received subpoenas and informal requests from various state attorneys general seeking information and documents relating to the pricing and/or marketing of the EpiPen® Auto-Injector. The Company is fully cooperating with these inquiries.
EpiPen® Auto-Injector U.S. Congress/State Requests for Information and Documents
Beginning in August 2016, Mylan has received several requests for information and documents from various Committees of the U.S. Congress and federal and state lawmakers concerning the marketing, distribution and sales of Mylan products. Mylan has cooperated and intends to continue cooperating with federal and state lawmakers as appropriate in response to their requests.
The Company believes that it has strong defenses to current and future potential civil litigation, as well as governmental investigations and enforcement proceedings. Although it is reasonably possible that the Company may incur additional losses from these matters, any amount cannot be reasonably estimated at this time. In addition, the Company expects to incur additional legal and other professional service expenses associated with the EpiPen® Auto-Injector pricing matters in future periods and will recognize these expenses as services are received. The Company believes that the ultimate amount paid for these services and claims could have a material effect on the Company's business, consolidated financial condition, results of operations, cash flows and/or ordinary share price in future periods.
Drug Pricing Matters
Department of Justice/Connecticut Subpoenas
On December 3, 2015, a subsidiary of Mylan N.V. received a subpoena from the Antitrust Division of the U.S. DOJ seeking information relating to the marketing, pricing, and sale of our generic Doxycycline products and any communications with competitors about such products. On September 8, 2016, a subsidiary of Mylan N.V., as well as certain employees and a member of senior management, received subpoenas from the DOJ seeking additional information relating to the marketing, pricing and sale of our generic Cidofovir, Glipizide-metformin, Propranolol and Verapamil products and any communications with competitors about such products. Related search warrants also were executed. The Company is fully cooperating with the DOJ's inquiry.
On December 21, 2015, the Company received a subpoena and interrogatories from the Connecticut Office of the Attorney General seeking information relating to the marketing, pricing and sale of certain of the Company's generic products (including Doxycycline) and communications with competitors about such products. The Company is fully cooperating with Connecticut's inquiry.
Civil Litigation
Twenty putative class action complaints are pending against Mylan Inc., Mylan Pharmaceuticals Inc. and other pharmaceutical manufacturers in a Multi-district Litigation ("MDL") in the United States District Court for the Eastern District of Pennsylvania; plaintiff indirect and direct purchasers generally allege anticompetitive conduct with respect to certain Doxycycline and Digoxin products. Mylan and its subsidiary believe that the claims in these lawsuits are without merit and intend to deny liability and to defend against them vigorously.
172
Table of Contents
Ten putative class action complaints are pending against Mylan Inc., Mylan Pharmaceuticals Inc. and other pharmaceutical manufacturers in the United States District Court for the Eastern District of Pennsylvania; plaintiff indirect and direct purchasers generally allege anticompetitive conduct with respect to certain Pravastatin products and to allocate markets and customers for those products. Mylan and its subsidiary believes that the claims in these lawsuits are without merit and intend to deny liability and to defend against them vigorously.
Seven putative class action complaints are pending against Mylan Inc., Mylan Pharmaceuticals Inc. and other pharmaceutical manufacturers in the United States District Court for the Eastern District of Pennsylvania; plaintiff indirect and direct purchasers generally allege anticompetitive conduct with respect to certain Divalproex products and to allocate markets and customers for those products. Mylan and its subsidiary believe that the claims in these lawsuits are without merit and intend to deny liability and to defend against them vigorously.
Nine putative class action complaints are pending against Mylan Pharmaceuticals Inc. and other pharmaceutical manufacturers in the United States District Court for the Southern District of New York and the Eastern District of Pennsylvania; plaintiff indirect and direct purchasers generally allege anticompetitive conduct with respect to certain Levothyroxine products. Mylan and its subsidiaries believe that the claims in these lawsuits are without merit and intend to deny liability and to defend against them vigorously.
Seven putative class action complaints are pending against Mylan Inc., Mylan Pharmaceuticals Inc., UDL Laboratories, Inc. and other pharmaceutical manufacturers in the United States District Court for the Southern District of New York and the Eastern District of Pennsylvania; plaintiff indirect and direct purchasers generally allege anticompetitive conduct with respect to certain Propranolol products. Mylan and its subsidiaries believe that the claims in these lawsuits are without merit and intend to deny liability and to defend against them vigorously.
Four putative class action complaints are pending against Mylan Inc. and other pharmaceutical manufacturers in the United States District Court for the District of Puerto Rico, the District of New Jersey and the Eastern District of Pennsylvania; plaintiff indirect and direct purchasers generally allege anticompetitive conduct with respect to certain Clomipramine products. Mylan and its subsidiaries believe that the claims in these lawsuits are without merit and intend to deny liability and to defend against them vigorously.
A complaint was filed on January 31, 2017 by putative classes of direct and indirect purchasers against Mylan Pharmaceuticals Inc. and other pharmaceutical manufacturers in the United States District Court for the District of Connecticut. Plaintiffs generally allege anticompetitive conduct and RICO violations with respect to, among other things, certain Doxycycline products. Mylan Pharmaceuticals Inc. believes that the claims in this lawsuit are without merit and intends to deny liability and to defend against them vigorously.
Attorney General Litigation
On December 14, 2016, attorneys general of twenty states filed a complaint in the United States District Court for the District of Connecticut against several generic pharmaceutical drug manufacturers, including Mylan, alleging anticompetitive conduct with respect to, among other things, Doxycycline Hyclate Delayed Release in violation of federal antitrust laws. We believe that the claims in this lawsuit against Mylan are without merit and intend to defend against them vigorously.
European Commission Proceedings
Perindopril
On or around July 8, 2009, the European Commission ( the "Commission") stated that it had initiated antitrust proceedings pursuant to Article 11(6) of Regulation No. 1/2003 and Article 2(1) of Regulation No. 773/2004 to explore possible infringement of Articles 81 and 82 EC and Articles 53 and 54 of the European Economic Area Agreement by Les Laboratoires Servier ("Servier") as well as possible infringement of Article 81 EC by the Company's Indian subsidiary, Mylan Laboratories Limited , and four other companies, each of which entered into agreements with Servier relating to the product Perindopril. On July 30, 2012, the Commission issued a Statement of Objections to Servier SAS, Servier Laboratories Limited, Les Laboratories Servier, Adir, Biogaran, Krka, d.d. Novo mesto, Lupin Limited, Mylan Laboratories Limited , Mylan , Niche Generics Limited, Teva UK Limited, Teva Pharmaceutical Industries Ltd., Teva Pharmaceuticals Europe B.V. and Unichem Laboratories Limited. Mylan Inc. and Mylan Laboratories Limited filed responses to the Statement of Objections. On July 9, 2014, the Commission issued a decision finding that Mylan Laboratories Limited and Mylan , as well as the companies noted above (with the exception of Adir, a subsidiary of Servier), had violated European Union competition rules and fined Mylan
173
Table of Contents
Laboratories Limited approximately €17.2 million , including approximately €8.0 million jointly and severally with Mylan Inc. The Company paid approximately $21.7 million related to this matter during the fourth quarter of 2014. In September 2014, the Company filed an appeal of the Commission's decision to the General Court of the European Union. The briefing on appeal is complete and we are awaiting the scheduling of the hearing date.
Citalopram
On March 19, 2010, Mylan and Generics [U.K.] Limited , a wholly owned subsidiary of the Company, received notice that the Commission had opened proceedings against Lundbeck with respect to alleged unilateral practices and/or agreements related to Citalopram in the European Economic Area. On July 25, 2012, a Statement of Objections was issued to Lundbeck, Merck KGaA, Generics [U.K.] Limited , Arrow, Resolution Chemicals, Xelia Pharmaceuticals, Alpharma, A.L. Industrier and Ranbaxy. Generics [U.K.] Limited filed a response to the Statement of Objections and vigorously defended itself against allegations contained therein. On June 19, 2013, the Commission issued a decision finding that Generics [U.K.] Limited , as well as the companies noted above, had violated European Union competition rules and fined Generics [U.K.] Limited approximately €7.8 million , jointly and severally with Merck KGaA. Generics [U.K.] Limited has appealed the Commission's decision to the General Court of the EU. Briefing on the appeal has been completed and a hearing took place on October 8, 2015. On September 8, 2016, the General Court dismissed all appeals against the European Commission's decision. Mylan filed an appeal of the decision on November 18, 2016 to the European Court of Justice. The Company has accrued approximately $8.2 million and $9.8 million as of December 31, 2016 and December 31, 2015 , respectively, related to this matter. It is reasonably possible that we will incur additional losses above the amount accrued but we cannot estimate a range of such reasonably possible losses at this time. There are no assurances, however, that settlements reached and/or adverse judgments received, if any, will not exceed amounts accrued. Generics [U.K.] Limited has also sought indemnification from Merck KGaA with respect to the €7.8 million portion of the fine for which Merck KGaA and Generics [U.K.] Limited were held jointly and severally liable. Merck KGaA has counterclaimed against Generics [U.K.] Limited seeking the same indemnification.
U.K. Competition and Markets Authority
Paroxetine
On August 12, 2011, Generics [U.K.] Limited received notice that the Office of Fair Trading (subsequently changed to the Competition and Markets Authority (the "CMA")) was opening an investigation to explore the possible infringement of the Competition Act 1998 and Articles 101 and 102 of the Treaty on the Functioning of the European Union, with respect to alleged agreements related to Paroxetine. On April 19, 2013, a Statement of Objections was issued to Beecham Group plc, GlaxoSmithKline UK Limited, GlaxoSmithKline plc and SmithKline Beecham Limited (formerly, SmithKline Beecham plc) (together, "GlaxoSmithKline"), Generics [U.K.] Limited , Merck KGaA, Actavis UK Limited (formerly, Alpharma Limited), Xellia Pharmaceuticals ApS (formerly, Alpharma ApS) and Alpharma LLC (formerly, Zoetis Products LLC, Alpharma LLC, and Alpharma Inc.) (together, "Alpharma"), and Ivax LLC (formerly, Ivax Corporation) and Norton Healthcare Limited (which previously traded as Ivax Pharmaceuticals UK) (together, "Ivax"). Generics [U.K.] Limited filed a response to the Statement of Objections, defending itself against the allegations contained therein. The CMA issued a Supplementary Statement of Objections ("SSO") to the above-referenced parties on October 21, 2014 and a hearing with regard to the SSO took place on December 19, 2014. The CMA issued a decision on February 12, 2016, finding that GlaxoSmithKline, Generics [U.K.] Limited , Merck KGaA and Alpharma, were liable for infringing EU and U.K. competition rules. With respect to Merck KGaA and Generics [U.K.] Limited , the CMA issued a penalty of approximately £5.8 million , for which Merck KGaA is liable for the entire amount; and of that amount Generics [U.K.] Limited is jointly and severally liable for approximately £2.7 million , which was accrued for at December 31, 2016 . Generics [U.K.] Limited has appealed the decision. The hearing commenced on February 27, 2017 before the Competition Appeals Tribunal.
Product Liability
The Company is involved in a number of product liability lawsuits and claims related to alleged personal injuries arising out of certain products manufactured and/or distributed by the Company, including but not limited to Phenytoin, Alendronate and Reglan. The Company believes that it has meritorious defenses to these lawsuits and claims and is vigorously defending itself with respect to those matters. From time to time, the Company has agreed to settle or otherwise resolve certain lawsuits and claims on terms and conditions that are in the best interests of the Company. The Company has accrued approximately $31.5 million and $9.5 million at December 31, 2016 and 2015, respectively. It is reasonably possible that we will incur additional losses and fees above the amount accrued but we cannot estimate a range of such reasonably possible losses or legal fees related to these claims at this time. There are no assurances, however, that settlements reached and/or adverse judgments received, if any, will not exceed amounts accrued.
174
Table of Contents
Intellectual Property
In certain situations, the Company has used its business judgment to decide to market and sell products, notwithstanding the fact that allegations of patent infringement(s) or other potential third party rights have not been finally resolved by the courts. The risk involved in doing so can be substantial because the remedies available to the owner of a patent for infringement may include, a reasonable royalty on sales or damages measured by the profits lost by the patent owner. If there is a finding of willful infringement, damages may be increased up to three times. Moreover, because of the discount pricing typically involved with bioequivalent products, patented branded products generally realize a substantially higher profit margin than bioequivalent products. An adverse decision could have an adverse effect that is material to our business, financial condition, results of operations, cash flows and/or ordinary share price.
Other Litigation
The Company is involved in various other legal proceedings that are considered normal to its business, including but not limited to certain proceedings assumed as a result of the acquisition of the former Merck Generics business, Agila and the acquired EPD Business . The Company has approximately $20 million accrued related to these various other legal proceedings at December 31, 2016. While it is not possible to predict the ultimate outcome of such other proceedings, the ultimate outcome of any such proceeding is not currently expected to be material to the Company's business, financial condition, results of operations, cash flows and/or ordinary share price.
175
Table of Contents
Mylan N.V.
Supplementary Financial Information
Quarterly Financial Data
(Unaudited, in millions, except per share data)
Year Ended December 31, 2016
| Three-Month Period Ended | ||||||||||||||
| March 31, |
| June 30, |
| September 30, |
| December 31, | ||||||||
Total revenues | $ | 2,191.3 | |
| $ | 2,560.7 | |
| $ | 3,057.1 | |
| $ | 3,267.8 | |
Gross profit | 907.0 | |
| 1,171.7 | |
| 1,283.3 | |
| 1,335.0 | | ||||
Net earnings (loss) | 13.9 | |
| 168.4 | |
| (119.8 | ) |
| 417.5 | | ||||
Net earnings (loss) attributable to Mylan N.V. ordinary shareholders | 13.9 | |
| 168.4 | |
| (119.8 | ) |
| 417.5 | | ||||
Earnings (loss) per share (2) : |
|
|
|
|
|
|
| ||||||||
Basic | $ | 0.03 | |
| $ | 0.33 | |
| $ | (0.23 | ) |
| $ | 0.78 | |
Diluted | $ | 0.03 | |
| $ | 0.33 | |
| $ | (0.23 | ) |
| $ | 0.78 | |
Share prices (3) : |
|
|
|
|
|
|
| ||||||||
High | $ | 54.09 | |
| $ | 48.80 | |
| $ | 49.92 | |
| $ | 38.92 | |
Low | $ | 41.42 | |
| $ | 38.62 | |
| $ | 38.12 | |
| $ | 34.14 | |
Year Ended December 31, 2015
| Three-Month Period Ended | ||||||||||||||
| March 31, 2015 (1) |
| June 30, |
| September 30, |
| December 31, | ||||||||
Total revenues | $ | 1,871.7 | |
| $ | 2,371.7 | |
| $ | 2,695.2 | |
| $ | 2,490.7 | |
Gross profit | 830.1 | |
| 1,008.1 | |
| 1,315.3 | |
| 1,062.6 | | ||||
Net earnings | 56.6 | |
| 167.9 | |
| 428.6 | |
| 194.6 | | ||||
Net earnings attributable to Mylan N.V. ordinary shareholders | 56.6 | |
| 167.8 | |
| 428.6 | |
| 194.6 | | ||||
Earnings per share (2) : |
|
|
|
|
|
|
| ||||||||
Basic | $ | 0.14 | |
| $ | 0.34 | |
| $ | 0.87 | |
| $ | 0.40 | |
Diluted | $ | 0.13 | |
| $ | 0.32 | |
| $ | 0.83 | |
| $ | 0.38 | |
Share prices (3) : |
|
|
|
|
|
|
| ||||||||
High | $ | 64.96 | |
| $ | 76.06 | |
| $ | 71.49 | |
| $ | 55.28 | |
Low | $ | 52.74 | |
| $ | 57.94 | |
| $ | 39.80 | |
| $ | 39.16 | |
____________
(1) | On February 27, 2015 , Mylan Inc. became an indirect wholly owned subsidiary of Mylan N.V. |
(2) | The sum of earnings per share for the quarters may not equal earnings per share for the total year due to changes in the average number of ordinary shares outstanding. |
(3) | Closing prices are as reported on NASDAQ. |
176
Table of Contents
ITEM 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosures |
None.
ITEM 9A. | Controls and Procedures |
The Company's management performed an evaluation under the supervision and with the participation of the Company's Principal Executive Officer and the Principal Financial Officer of the effectiveness of the design and operation of the Company's disclosure controls and procedures as of December 31, 2016 . Based upon that evaluation, the Principal Executive Officer and the Principal Financial Officer concluded that the Company's disclosure controls and procedures were effective.
On August 5, 2016, the Company completed its acquisition of Meda AB (publ.) ("Meda"). Meda represented 8% of the Company's consolidated total revenues for the year ended December 31, 2016 , and assets (including intangible assets and goodwill) represented 34% of the Company's consolidated total assets, as of December 31, 2016 . Management did not include Meda when conducting its assessment of the effectiveness of the Company's internal control over financial reporting as of December 31, 2016 .
In addition, during the quarter ended December 31, 2016, the Company continued to implement and use a new Enterprise Resource Planning ("ERP") system in certain countries, which, when completed, will handle the business, financial and administrative processes for the Company. The Company has modified and will continue to modify its internal controls relating to its business and financial processes through the entire ERP system implementation, which is expected to progress through the end of calendar 2017. While the Company believes that this new system and the related changes to internal controls will ultimately strengthen its internal control over financial reporting, there are inherent risks in implementing any new ERP system and the Company has evaluated and tested control changes in order to provide certification for the year ended December 31, 2016 on the effectiveness of internal control over financial reporting.
Management has not identified any other changes in the Company's internal control over financial reporting that occurred during the fourth quarter of 2016 that have materially affected, or are reasonably likely to materially affect, the Company's internal control over financial reporting.
Management's Report on Internal Control over Financial Reporting is on page 88 , which is incorporated herein by reference. The effectiveness of the Company's internal control over financial reporting as of December 31, 2016 has been audited by Deloitte & Touche LLP, an independent registered public accounting firm, as stated in their report on page 90 , which is incorporated herein by reference.
ITEM 9B. | Other Information |
None.
177
Table of Contents
PART III
ITEM 10. | Directors, Executive Officers and Corporate Governance |
Certain information required by this Item will be provided in an amendment to this Form 10-K in accordance with General Instruction G(3) to Form 10-K.
Code of Ethics
The Company has adopted a Code of Ethics for our Chief Executive Officer, Chief Financial Officer and Controller. This Code of Ethics for our Chief Executive Officer, Chief Financial Officer and Controller is posted on Mylan 's website at http://www.mylan.com/company/corporate-governance, and Mylan intends to post any amendments to and waivers from the Code of Ethics for our Chief Executive Officer, Chief Financial Officer and Controller on that website.
ITEM 11. | Executive Compensation |
The information required by this Item will be provided in an amendment to this Form 10-K in accordance with General Instruction G(3) to Form 10-K.
ITEM 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The additional information required by this Item will be provided in an amendment to this Form 10-K in accordance with General Instruction G(3) to Form 10-K.
Equity Compensation Plan Information
| Number of Securities to be |
| Weighted-Average Exercise |
| Number of Securities | ||||
Plan Category |
|
| |||||||
Equity compensation plans approved by security holders | 13,367,615 | |
| $ | 37.23 | |
| 10,776,317 | |
Equity compensation plans not approved by security holders | - | |
| - | |
| - | | |
Total | 13,367,615 | |
| $ | 37.23 | |
| 10,776,317 | |
ITEM 13. | Certain Relationships and Related Transactions, and Director Independence |
The information required by this Item will be provided in an amendment to this Form 10-K in accordance with General Instruction G(3) to Form 10-K.
ITEM 14. | Principal Accounting Fees and Services |
The information required by this Item will be provided in an amendment to this Form 10-K in accordance with General Instruction G(3) to Form 10-K.
178
Table of Contents
PART IV
ITEM 15. | Exhibits, Consolidated Financial Statement Schedules |
1. | Consolidated Financial Statements |
The Consolidated Financial Statements listed in the Index to Consolidated Financial Statements are filed as part of this Form.
2. | Consolidated Financial Statement Schedules |
MYLAN N.V. AND SUBSIDIARIES
SCHEDULE II - VALUATION AND QUALIFYING ACCOUNTS
(In millions)
Mylan N.V. is the successor to Mylan Inc., the information set forth below refers to Mylan Inc. for periods prior to February 27, 2015, and to Mylan N.V. on and after February 27, 2015.
Description | Beginning |
| Additions Charged to Costs and Expenses |
| Additions |
| Deductions |
| Ending | |||||||
Allowance for doubtful accounts: |
|
|
|
|
|
|
|
|
| |||||||
Year ended December 31, 2016 | $ | 33.6 | |
| 15.6 | |
| 13.0 | |
| (3.2 | ) |
| $ | 59.0 | |
Year ended December 31, 2015 | $ | 25.7 | |
| 10.5 | |
| 0.3 | |
| (2.9 | ) |
| $ | 33.6 | |
Year ended December 31, 2014 | $ | 24.6 | |
| 6.0 | |
| 1.2 | |
| (6.1 | ) |
| $ | 25.7 | |
Valuation allowance for deferred tax assets: |
|
|
|
|
|
|
|
|
| |||||||
Year ended December 31, 2016 | $ | 355.7 | |
| 108.8 | |
| 3.4 | |
| (7.2 | ) |
| $ | 460.7 | |
Year ended December 31, 2015 | $ | 304.5 | |
| 75.6 | |
| 6.1 | |
| (30.5 | ) |
| $ | 355.7 | |
Year ended December 31, 2014 | $ | 279.5 | |
| 49.8 | |
| 17.7 | |
| (42.5 | ) |
| $ | 304.5 | |
____________
(1) | In 2016, this amount includes opening balances of businesses acquired in the period. |
3. | Exhibits |
2.1 |
| Amended and Restated Business Transfer Agreement and Plan of Merger, dated November 4, 2014, between and among Abbott Laboratories, Mylan Inc., New Moon B.V. and Moon of PA Inc., filed as Annex A to the Registration Statement on Form S-4 filed with the SEC on November 5, 2014, as amended on December 9 and December 23, 2014, and incorporated herein by reference.^ |
|
|
|
2.2 |
| Shareholder Agreement, dated as of February 27, 2015, between and among Mylan N.V., Abbott Laboratories, Laboratoires Fournier S.A.S., Abbott Established Products Holdings (Gibraltar) Limited, and Abbott Investments Luxembourg S.à r.l., filed as Exhibit 2.2 to the Report on Form 8-K filed with the SEC on February 27, 2015, and incorporated herein by reference.^ |
2.3(a) |
| Irrevocable Undertaking, dated February 10, 2016, between Mylan N.V. and Stena Sessan Rederi AB, filed as Exhibit 2.1 to the Report on Form 8-K filed with the SEC on February 17, 2016, and incorporated herein by reference. |
2.3(b) |
| Irrevocable Undertaking, dated February 10, 2016, between Mylan N.V. and Fidim S.r.l., filed as Exhibit 2.2 to the Report on Form 8-K filed with the SEC on February 17, 2016, and incorporated herein by reference. |
|
|
|
2.3(c) |
| Shareholder Agreement, dated February 10, 2016, between Mylan N.V. and Stena Sessan Rederi AB, filed as Exhibit 2.3 to the Report on Form 8-K filed with the SEC on February 17, 2016, and incorporated herein by reference.^ |
|
|
|
2.3(d) |
| Shareholder Agreement, dated February 10, 2016, between Mylan N.V. and Fidim S.r.l., filed as Exhibit 2.4 to the Report on Form 8-K filed with the SEC on February 17, 2016, and incorporated herein by reference.^ |
|
|
|
3.1 |
| Amended and Restated Articles of Association of Mylan N.V., filed as Exhibit 3.1 to the Report on Form 8-K filed with the SEC on February 27, 2015, and incorporated herein by reference. |
|
|
|
179
Table of Contents
4.1(a) |
| Indenture, dated December 21, 2012, between and among Mylan Inc., the guarantors named therein, and The Bank of New York Mellon, as trustee, filed by Mylan Inc. as Exhibit 4.1 to the Report on Form 8-K filed with the SEC on December 24, 2012, and incorporated herein by reference. |
|
|
|
4.1(b) |
| First Supplemental Indenture, dated February 27, 2015, between and among Mylan Inc., as Issuer, Mylan N.V., as Guarantor, and The Bank of New York Mellon, as Trustee, to the Indenture, dated December 21, 2012, filed as Exhibit 4.4 to the Report on Form 8-K filed with the SEC on February 27, 2015, and incorporated herein by reference. |
|
|
|
4.1(c) |
| Second Supplemental Indenture, dated March 12, 2015, between and among Mylan Inc., as Issuer, Mylan N.V., as Parent, and The Bank of New York Mellon, as Trustee, to the Indenture, dated December 21, 2012, filed as Exhibit 4.3(b) to Form 10-Q for the quarter ended March 31, 2015, and incorporated herein by reference. |
|
|
|
4.2(a) |
| Indenture, dated June 25, 2013, among Mylan Inc., the guarantors thereto and The Bank of New York Mellon, as trustee, filed by Mylan Inc. as Exhibit 4.1 to the Report on Form 8-K filed with the SEC on June 27, 2013, and incorporated herein by reference. |
|
|
|
4.2(b) |
| First Supplemental Indenture, dated February 27, 2015, between and among Mylan Inc., as Issuer, Mylan N.V., as Guarantor, and The Bank of New York Mellon, as Trustee, to the Indenture, dated June 25, 2013, filed as Exhibit 4.5 to the Report on Form 8-K filed with the SEC on February 27, 2015, and incorporated herein by reference. |
|
|
|
4.2(c) |
| Second Supplemental Indenture, dated March 12, 2015, between and among Mylan Inc., as Issuer, Mylan N.V., as Parent, and The Bank of New York Mellon, as Trustee, to the Indenture, dated June 25, 2013, filed as Exhibit 4.4(b) to Form 10-Q for the quarter ended March 31, 2015, and incorporated herein by reference. |
|
|
|
4.3 |
| Registration Rights Agreement, dated June 25, 2013, among Mylan Inc., the guarantors thereto, and the representatives of the initial purchasers of Mylan Inc.'s $500 million aggregate principal amount of Mylan Inc.'s 1.800% Senior Notes due 2016 and $650 million aggregate principal amount of Mylan Inc.'s 2.600% senior notes due 2018, filed by Mylan Inc. as Exhibit 10.1 to the Report on the Form 8-K filed with the SEC on June 27, 2013, and incorporated herein by reference. |
|
|
|
4.4(a) |
| Indenture, dated November 29, 2013, by and between Mylan Inc. and The Bank of New York Mellon, as trustee, filed by Mylan Inc. as Exhibit 4.1 to the Report on Form 8-K filed with the SEC on November 29, 2013, and incorporated herein by reference. |
|
|
|
4.4(b) |
| First Supplemental Indenture, dated November 29, 2013, by and between Mylan Inc. and The Bank of New York Mellon, as trustee, filed by Mylan Inc. as Exhibit 4.2 to the Report on Form 8-K filed with the SEC on November 29, 2013, and incorporated herein by reference. |
|
|
|
4.4(c) |
| Second Supplemental Indenture, dated February 27, 2015, between and among Mylan Inc., as Issuer, Mylan N.V., as Guarantor, and The Bank of New York Mellon, as Trustee, to the Indenture, dated November 29, 2013, filed as Exhibit 4.6 to the Report on Form 8-K filed with the SEC on February 27, 2015, and incorporated herein by reference. |
|
|
|
4.4(d) |
| Third Supplemental Indenture, dated March 12, 2015, between and among Mylan Inc., as Issuer, Mylan N.V., as Parent, and The Bank of New York Mellon, as Trustee, to the Indenture, dated November 29, 2013, filed as Exhibit 4.5(b) to Form 10-Q for the quarter ended March 31, 2015, and incorporated herein by reference. |
|
|
|
4.5 |
| Indenture, dated as of December 9, 2015, among Mylan N.V., Mylan Inc., as guarantor, and The Bank of New York Mellon, as trustee, filed as Exhibit 4.1 to the Report on Form 8-K filed with the SEC on December 15, 2015, and incorporated herein by reference. |
|
|
|
4.6 |
| Registration Rights Agreement, dated December 9, 2015, among Mylan N.V., Mylan Inc., as guarantor, and Goldman, Sachs & Co., Deutsche Bank Securities Inc. and Mitsubishi UFJ Securities (USA), Inc., as representatives of the initial purchasers of $500 million aggregate principal amount of Mylan N.V.'s 3.000% senior notes due 2018 and $500 million aggregate principal amount of Mylan N.V.'s 3.750% senior notes due 2020, filed as Exhibit 10.1 to the Report on Form 8-K filed with the SEC on December 15, 2015, and incorporated herein by reference. |
|
|
|
4.7 |
| Indenture, dated as of June 9, 2016, among Mylan N.V., as issuer, Mylan Inc., as guarantor, and The Bank of New York Mellon, as trustee, filed as Exhibit 4.1 to the Report on Form 8-K filed with the SEC on June 15, 2016, and incorporated herein by reference. |
|
|
|
180
Table of Contents
4.8 |
| Registration Rights Agreement, dated as of June 9, 2016, among Mylan N.V., as issuer, Mylan Inc., as guarantor, and Deutsche Bank Securities Inc., Goldman, Sachs & Co. and Merrill Lynch, Pierce, Fenner & Smith Incorporated, as representatives of the initial purchasers of the $1 billion aggregate principal amount of Mylan N.V.'s 2.500% Senior Notes due 2019, $2.25 billion aggregate principal amount of Mylan N.V.'s 3.150% Senior Notes due 2021, $2.25 billion aggregate principal amount of Mylan N.V.'s 3.950% Senior Notes due 2026, and $1 billion aggregate principal amount of Mylan N.V.'s 5.250% Senior Notes due 2046, filed as Exhibit 10.1 to the Report on Form 8-K filed with the SEC on June 15, 2016, and incorporated herein by reference. |
|
|
|
4.9 |
| Indenture, dated November 22, 2016, among Mylan N.V., as issuer, Mylan Inc., as guarantor and Citibank, N.A., London Branch, as trustee. |
|
|
|
10.1(a) |
| Amended and Restated 2003 Long-Term Incentive Plan, filed as Appendix B to the Definitive Proxy Statement on Schedule 14A filed on May 25, 2016, and incorporated herein by reference.* |
|
|
|
10.1(b) |
| Amendment to Amended and Restated 2003 Long-Term Incentive Plan, filed as Appendix B to the Definitive Proxy Statement on Schedule 14A filed on May 25, 2016, and incorporated herein by reference.* |
|
|
|
10.1(c) |
| Amended and Restated Form of Stock Option Agreement under the 2003 Long-Term Incentive Plan for Robert J. Coury, Heather Bresch, and Rajiv Malik, filed by Mylan Inc. as Exhibit 10.2 to Form 10-Q for the quarter ended September 30, 2013, and incorporated herein by reference.* |
|
|
|
10.1(d) |
| Amended and Restated Form of Stock Option Agreement under the 2003 Long-Term Incentive Plan for awards granted following fiscal year 2012, filed by Mylan Inc. as Exhibit 10.4(i) to Form 10-K for the fiscal year ended December 31, 2013, and incorporated herein by reference.* |
|
|
|
10.1(e) |
| Amended and Restated Form of Restricted Stock Unit Award Agreement under the 2003 Long-Term Incentive Plan for awards granted following fiscal year 2012, filed by Mylan Inc. as Exhibit 10.4(j) to Form 10-K for the fiscal year ended December 31, 2013, and incorporated herein by reference.* |
|
|
|
10.1(f) |
| Amended and Restated Form of Performance-Based Restricted Stock Unit Award Agreement under the 2003 Long-Term Incentive Plan for awards granted following fiscal year 2012, filed by Mylan Inc. as Exhibit 10.4(k) to Form 10-K for the fiscal year ended December 31, 2013, and incorporated herein by reference.* |
|
|
|
10.1(g) |
| Form of Performance-Based Stock Appreciation Rights Award Agreement under the Mylan Inc. One-Time Special Five-Year Performance-Based Realizable Value Incentive Program, filed by Mylan Inc. as Exhibit 10.5 to the Report on Form 8-K filed with the SEC on February 28, 2014, and incorporated herein by reference.* |
|
|
|
10.1(h) |
| Form of Performance-Based Restricted Stock Unit Award Agreement under the Mylan Inc. One-Time Special Five-Year Performance-Based Realizable Value Incentive Program, filed by Mylan Inc. as Exhibit 10.6 to the Report on Form 8-K filed with the SEC on February 28, 2014, and incorporated herein by reference.* |
|
|
|
10.1(i) |
| Form of Stock Option Agreement under the 2003 Long-Term Incentive Plan for Robert J. Coury, Heather Bresch, and Rajiv Malik for awards granted after February 27, 2015, filed as Exhibit 10.1(i) to Form 10-K for the fiscal year ended December 31, 2015, and incorporated herein by reference.* |
|
|
|
10.1(j) |
| Form of Restricted Stock Unit Award Agreement under the 2003 Long-Term Incentive Plan for Robert J. Coury, Heather Bresch, and Rajiv Malik for awards granted after February 27, 2015, filed as Exhibit 10.1(j) to Form 10-K for the fiscal year ended December 31, 2015, and incorporated herein by reference.* |
|
|
|
10.1(k) |
| Form of Performance-Based Restricted Stock Unit Award Agreement under the 2003 Long-Term Incentive Plan for Robert J. Coury, Heather Bresch, and Rajiv Malik for awards granted after February 27, 2015, filed as Exhibit 10.1(k) to Form 10-K for the fiscal year ended December 31, 2015, and incorporated herein by reference.* |
|
|
|
10.1(l) |
| Form of Stock Option Agreement under the 2003 Long-Term Incentive Plan for awards granted after February 27, 2015, filed as Exhibit 10.1(l) to Form 10-K for the fiscal year ended December 31, 2015, and incorporated herein by reference.* |
|
|
|
10.1(m) |
| Form of Restricted Stock Unit Award Agreement under the 2003 Long-Term Incentive Plan for awards granted after February 27, 2015, filed as Exhibit 10.1(m) to Form 10-K for the fiscal year ended December 31, 2015, and incorporated herein by reference.* |
|
|
|
10.1(n) |
| Form of Performance-Based Restricted Stock Unit Award Agreement under the 2003 Long-Term Incentive Plan for awards granted after February 27, 2015, filed as Exhibit 10.1(n) to Form 10-K for the fiscal year ended December 31, 2015, and incorporated herein by reference.* |
|
|
|
10.2(a) |
| Mylan Inc. Severance Plan, amended as of August, 2009, filed by Mylan Inc. as Exhibit 10.6 to Form 10-Q for the quarter ended September 30, 2009, and incorporated herein by reference.* |
|
|
|
10.2(b) |
| Amendment to Mylan Inc. Severance Plan, dated July 13, 2014, filed by Mylan Inc. as Exhibit 10.1 to Form 10-Q for the quarter ended September 30, 2014, and incorporated herein by reference.* |
181
Table of Contents
|
|
|
10.3(a) |
| Confirmation of OTC Convertible Note Hedge Transaction, dated September 9, 2008, among Mylan Inc., Merrill Lynch International and Merrill Lynch, Pierce, Fenner & Smith Incorporated, filed by Mylan Inc. as Exhibit 10.1 to the Report on Form 8-K filed with the SEC on September 15, 2008, and incorporated herein by reference. |
|
|
|
10.3(b) |
| Confirmation of OTC Convertible Note Hedge Transaction, amended as of November 25, 2008, among Mylan Inc., Merrill Lynch International and Merrill Lynch, Pierce, Fenner & Smith Incorporated, filed by Mylan Inc. as Exhibit 10.7(b) to Form 10-K for the fiscal year ended December 31, 2008, and incorporated herein by reference. |
|
|
|
10.4 |
| Confirmation of OTC Convertible Note Hedge Transaction, dated September 9, 2008, between Mylan Inc. and Wells Fargo Bank, National Association, filed by Mylan Inc. as Exhibit 10.2 to the Report on Form 8-K filed with the SEC on September 15, 2008, and incorporated herein by reference. |
|
|
|
10.5 |
| Confirmation of OTC Warrant Transaction, dated September 9, 2008, among Mylan Inc., Merrill Lynch International and Merrill Lynch, Pierce, Fenner & Smith Incorporated, filed by Mylan Inc. as Exhibit 10.3 to the Report on Form 8-K filed with the SEC on September 15, 2008, and incorporated herein by reference. |
|
|
|
10.6 |
| Confirmation of OTC Warrant Transaction, dated September 9, 2008, between Mylan Inc. and Wells Fargo Bank, National Association, filed by Mylan Inc. as Exhibit 10.4 to the Report on Form 8-K filed with the SEC on September 15, 2008, and incorporated herein by reference. |
|
|
|
10.7 |
| Amendment to Confirmation of OTC Warrant Transaction, dated September 15, 2008 among Mylan Inc., Merrill Lynch International and Merrill Lynch, Pierce, Fenner & Smith Incorporated, filed by Mylan Inc. as Exhibit 10.5 to the Report on Form 8-K filed with the SEC on September 15, 2008, and incorporated herein by reference. |
|
|
|
10.8 |
| Amendment to Confirmation of OTC Warrant Transaction, dated September 15, 2008, between Mylan Inc. and Wells Fargo Bank, National Association, filed by Mylan Inc. as Exhibit 10.6 to the Report on Form 8-K filed with the SEC on September 15, 2008, and incorporated herein by reference. |
|
|
|
10.9 |
| Amendment to Confirmation of OTC Warrant Transaction, dated September 9, 2008 among Mylan Inc., Merrill Lynch International and Merrill Lynch, Pierce, Fenner & Smith Incorporated, filed by Mylan Inc. as Exhibit 10.7 to the Report on Form 8-K filed with the SEC on September 15, 2008, and incorporated herein by reference. |
|
|
|
10.10 |
| Amendment to Confirmation of OTC Warrant Transaction, dated September 9, 2008 among by Mylan Inc., Merrill Lynch International and Merrill Lynch, Pierce, Fenner & Smith Incorporated, filed by Mylan Inc. as Exhibit 10.8 to the Report on Form 8-K filed with the SEC on September 15, 2008, and incorporated herein by reference. |
|
|
|
10.11 |
| Amendment to the Confirmation of OTC Warrant Transaction, dated September 9, 2008, among Mylan Inc., Merrill Lynch International and Merrill Lynch Pierce, Fenner & Smith Incorporated, dated September 9, 2011, and filed by Mylan Inc. as Exhibit 10.1 to Form 10-Q for the quarter ended September 30, 2011, and incorporated herein by reference. |
|
|
|
10.12 |
| Amendment to the Confirmation of OTC Warrant Transaction, dated September 9, 2008, between Mylan Inc. and Goldman, Sachs & Co., as successor to Wells Fargo Bank, National Association, dated September 13, 2011, and filed by Mylan Inc. as Exhibit 10.2 to Form 10-Q for the quarter ended September 30, 2011, and incorporated herein by reference. |
|
|
|
10.13 |
| Amendment to the Confirmation of OTC Warrant Transaction, dated September 9, 2008, between Mylan Inc. and Goldman, Sachs & Co., as successor to Wells Fargo Bank, National Association, dated September 14, 2011, and filed by Mylan Inc. as Exhibit 10.3 to Form 10-Q for the quarter ended September 30, 2011, and incorporated herein by reference. |
|
|
|
10.14 |
| Executive Employment Agreement, dated July 31, 2013, between Mylan Inc. and John Sheehan, filed by Mylan Inc. as Exhibit 10.1 to Form 10-Q for the quarter ended September 30, 2013, and incorporated herein by reference.* |
|
|
|
10.15 |
| Amended and Restated Executive Employment Agreement, dated January 8, 2016 and effective January 1, 2016, by and between Mylan Inc. and Anthony Mauro, filed as Exhibit 10.16 to Form 10-K for the fiscal year ended December 31, 2015, and incorporated herein by reference.* |
|
|
|
10.16(a) |
| Retirement Benefit Agreement, dated December 31, 2004, between Mylan Inc. and Robert J. Coury, filed by Mylan Inc. as Exhibit 10.7 to Form 10-Q for the quarter ended December 31, 2004, and incorporated herein by reference.* |
|
|
|
10.16(b) |
| Amendment to Retirement Benefit Agreement, dated April 3, 2006, between Mylan Inc. and Robert J. Coury, filed by Mylan Inc. as Exhibit 10.11(b) to Form 10-K for the fiscal year ended March 31, 2006, and incorporated herein by reference.* |
|
|
|
182
Table of Contents
10.16(c) |
| Amendment to Retirement Benefit Agreement, dated December 22, 2008, between Mylan Inc. and Robert J. Coury, filed by Mylan Inc. as Exhibit 10.20(c) to Form 10-K for the fiscal year ended December 31, 2008, and incorporated herein by reference.* |
|
|
|
10.16(d) |
| Amendment to Retirement Benefit Agreement, dated March 3, 2010, by and between Mylan Inc. and Robert J. Coury, filed by Mylan Inc. as Exhibit 10.1 to Form 8-K filed with the SEC on March 5, 2010, and incorporated herein by reference.* |
|
|
|
10.16(e) |
| Amendment to Retirement Benefit Agreement, effective as of January 1, 2012, by and between Mylan Inc. and Robert J. Coury, filed by Mylan Inc. as Exhibit 10.6 to Form 8-K filed with the SEC on October 28, 2011, and incorporated herein by reference.* |
|
|
|
10.16(f) |
| Amendment to Retirement Benefit Agreement, effective as of January 1, 2014, by and between Mylan Inc. and Robert J. Coury, filed by Mylan Inc. as Exhibit 10.2 to the Report on Form 8-K filed with the SEC on February 28, 2014, and incorporated herein by reference.* |
|
|
|
10.17 |
| Retirement Benefit Agreement, dated August 31, 2009, by and between Mylan Inc. and Heather Bresch filed by Mylan Inc. as Exhibit 10.3 to Form 10-Q for the quarter ended September 30, 2009, and incorporated herein by reference.* |
|
|
|
10.18(a) |
| Retirement Benefit Agreement, dated August 28, 2009, by and between Mylan Inc. and Rajiv Malik, filed by Mylan Inc. as Exhibit 10.4 to Form 10-Q for the quarter ended September 30, 2009, and incorporated herein by reference.* |
|
|
|
10.18(b) |
| The Executive Nonqualified Excess Plan Adoption Agreement, effective as of December 28, 2007, between Mylan International Holdings, Inc. and Rajiv Malik, filed by Mylan Inc. as Exhibit 10.27(b) to Form 10-K for the fiscal year ended December 31, 2013, and incorporated herein by reference.* |
|
|
|
10.19 |
| Retirement Benefit Agreement, dated February 22, 2011, by and between Mylan Inc. and John D. Sheehan, filed by Mylan Inc. as Exhibit 10.23 to Form 10-K for the fiscal year ended December 31, 2010, and incorporated herein by reference.* |
|
|
|
10.20(a) |
| Transition and Succession Agreement, dated December 15, 2003, between Mylan Inc. and Robert J. Coury, filed by Mylan Inc. as Exhibit 10.19 to Form 10-Q for the quarter ended December 31, 2003, and incorporated herein by reference.* |
|
|
|
10.20(b) |
| Amendment No. 1 to Transition and Succession Agreement, dated December 2, 2004, between Mylan Inc. and Robert J. Coury, filed by Mylan Inc. as Exhibit 10.1 to Form 10-Q for the quarter ended December 31, 2004, and incorporated herein by reference.* |
|
|
|
10.20(c) |
| Amendment No. 2 to Transition and Succession Agreement, dated April 3, 2006, between Mylan Inc. and Robert J. Coury filed by Mylan Inc. as Exhibit 10.19(c) to Form 10-K for the fiscal year ended March 31, 2006, and incorporated herein by reference.* |
|
|
|
10.20(d) |
| Amendment No. 3 to Transition and Succession Agreement, dated December 22, 2008, between Mylan Inc. and Robert J. Coury, filed by Mylan Inc. as Exhibit 10.25(d) to Form 10-K for the fiscal year ended December 31, 2008, and incorporated herein by reference.* |
|
|
|
10.21(a) |
| Amended and Restated Transition and Succession Agreement, dated December 31, 2007, between Mylan Inc. and Heather Bresch, filed by Mylan Inc. as Exhibit 10.2 to Form 10-Q for the quarter ended March 31, 2008, and incorporated herein by reference.* |
|
|
|
10.21(b) |
| Amendment No. 1 to Transition and Succession Agreement, dated December 22, 2008, between Mylan Inc. and Heather Bresch, filed by Mylan Inc. as Exhibit 10.27(b) to Form 10-K for the fiscal year ended December 31, 2008, and incorporated herein by reference.* |
|
|
|
10.22(a) |
| Transition and Succession Agreement, dated January 31, 2007, between Mylan Inc. and Rajiv Malik, filed by Mylan Inc. as Exhibit 10.5 to Form 10-Q for the quarter ended March 31, 2008, and incorporated herein by reference.* |
|
|
|
10.22(b) |
| Amendment No. 1 to Transition and Succession Agreement, dated December 22, 2008, between Mylan Inc. and Rajiv Malik, filed by Mylan Inc. as Exhibit 10.28(b) to Form 10-K for the fiscal year ended December 31, 2008, and incorporated herein by reference.* |
|
|
|
10.23 |
| Transition and Succession Agreement, dated April 1, 2010, by and between Mylan Inc. and John Sheehan, filed by Mylan Inc. as Exhibit 10.3 to Form 10-Q for the quarter ended March 31, 2010, and incorporated herein by reference.* |
|
|
|
10.24(a) |
| Transition and Succession Agreement, dated February 25, 2008, by and between Mylan Inc. and Anthony Mauro, filed by Mylan Inc. as Exhibit 10.5(a) to Form 10-Q for the quarter ended March 31, 2012, and incorporated herein by reference.* |
|
|
|
183
Table of Contents
10.24(b) |
| Amendment No. 1 to Transition and Succession Agreement, dated December 15, 2008, by and between Mylan Inc. and Anthony Mauro, filed by Mylan Inc. as Exhibit 10.5(b) to Form 10-Q for the quarter ended March 31, 2012, and incorporated herein by reference.* |
|
|
|
10.24(c) |
| Amendment No. 2 to Transition and Succession Agreement, dated October 15, 2009, by and between Mylan Inc. and Anthony Mauro, filed by Mylan Inc. as Exhibit 10.5(c) to Form 10-Q for the quarter ended March 31, 2012, and incorporated herein by reference.* |
|
|
|
10.25 |
| Supplemental Health Insurance Program For Certain Officers of Mylan Inc., effective December 15, 2001, filed by Mylan Inc. as Exhibit 10.1 to Form 10-Q for the quarter ended December 31, 2001, and incorporated herein by reference.* |
|
|
|
10.26(a) |
| Amended and Restated Form of Indemnification Agreement between Mylan Inc. and each Director, filed by Mylan Inc. as Exhibit 10.38 to Form 10-K for the fiscal year ended December 31, 2013, and incorporated herein by reference.* |
|
|
|
10.26(b) |
| Form of indemnification agreement between Mylan N.V. and each director, filed as Exhibit 10.1 to the Report on Form 8-K filed with the SEC on February 27, 2015, and incorporated herein by reference.* |
|
|
|
10.27 |
| Agreement Regarding Consulting Services and Shareholders Agreement dated December 31, 2007 by and among Mylan Inc., MP Laboratories (Mauritius) Ltd, Prasad Nimmagadda, Globex and G2 Corporate Services Limited, filed by Mylan Inc. as Exhibit 10.26 to Form 10-KT/A for the period ended December 31, 2007, and incorporated herein by reference. |
|
|
|
10.28(a) |
| Share Purchase Agreement, dated May 12, 2007, by and among Merck Generics Holding GmbH, Merck S.A., Merck Internationale Beteiligung GmbH, Merck KGaA and Mylan Inc., filed by Mylan Inc. as Exhibit 10.1 to the Report on Form 8-K filed with the SEC on May 17, 2007, and incorporated herein by reference. |
|
|
|
10.28(b) |
| Amendment No. 1 to Share Purchase Agreement, dated October 1, 2007, by and among Mylan Inc. and Merck Generics Holding GmbH, Merck S.A., Merck Internationale Beteiligung GmbH and Merck KGaA, filed by Mylan Inc. as Exhibit 10.1 to the Report on Form 8-K filed with the SEC on October 5, 2007, and incorporated herein by reference. |
|
|
|
10.29 |
| Purchase Agreement, dated May 12, 2010, among Mylan Inc., the guarantors named therein and Goldman, Sachs & Co., as representative of the several purchasers named therein, filed by Mylan Inc. as Exhibit 10.1 to Form 10-Q for the quarter ended June 30, 2010, and incorporated herein by reference. |
|
|
|
10.30 |
| Share Purchase Agreement, dated July 14, 2010, by and among Mylan Inc., Mylan Luxembourg L3 S.C.S., Bioniche Pharma Holdings Limited, the shareholders party thereto and the optionholders party thereto, filed by Mylan Inc. as Exhibit 2.1 to the Report on Form 8-K filed with the SEC on July 16, 2010, and incorporated herein by reference. |
|
|
|
10.31 |
| Purchase Agreement, dated July 30, 2010, among Mylan Inc., the guarantors named therein and Goldman, Sachs & Co., filed by Mylan Inc. as Exhibit 10.1 to Form 10-Q for the quarter ended September 30, 2010, and incorporated herein by reference. |
|
|
|
10.32(a) |
| Mylan 401(k) Restoration Plan, filed by Mylan Inc. as Exhibit 10.1 to the Report on Form 8-K filed by Mylan Inc. with the SEC on December 14, 2009, and incorporated herein by reference.* |
|
|
|
10.32(b) |
| Amendment to Mylan 401(k) Restoration Plan, dated November 4, 2014, filed by Mylan Inc. as Exhibit 10.41(b) to Form 10-K for the fiscal year ended December 31, 2014, and incorporated herein by reference.* |
|
|
|
10.33(a) |
| Mylan Executive Income Deferral Plan, filed by Mylan Inc. as Exhibit 10.2 to the Report on Form 8-K filed with the SEC on December 14, 2009, and incorporated herein by reference.* |
|
|
|
10.33(b) |
| Amendment to Mylan Executive Income Deferral Plan, dated November 4, 2014, filed by Mylan Inc. as Exhibit 10.42(b) to Form 10-K for the fiscal year ended December 31, 2014, and incorporated herein by reference.* |
|
|
|
10.34 |
| Performance Guaranty, dated February 21, 2012, by Mylan Inc. in favor of The Bank of Tokyo-Mitsubishi UFJ, Ltd., New York Branch, as Agent, filed by Mylan Inc. as Exhibit 10.3 to Form 10-Q for the quarter ended March 31, 2012, and incorporated herein by reference. |
|
|
|
10.35 |
| Amended and Restated Sale and Purchase Agreement, dated December 4, 2013, by and among Mylan Inc., Mylan Institutional Inc., Strides Pharma Asia Pte Ltd (Agila Specialties Asia Pte Ltd), and the promoters named therein, filed by Mylan Inc. as Exhibit 10.50 to Form 10-K for the fiscal year ended December 31, 2013, and incorporated herein by reference.† |
|
|
|
10.36 |
| Amended and Restated Sale and Purchase Agreement, dated December 4, 2013, by and among Mylan Inc., Mylan Laboratories Limited, Strides Arcolab Limited, and the promoters named therein, filed by Mylan Inc. as Exhibit 10.51 to Form 10-K for the fiscal year ended December 31, 2013, and incorporated herein by reference.† |
|
|
|
184
Table of Contents
10.37 |
| Restrictive Covenant Agreement, effective February 27, 2013, by and among Mylan Inc., Strides Arcolab Limited, and the promoters named therein, filed by Mylan Inc. as Exhibit 10.3 to Form 10-Q for the quarter ended March 31, 2013, and incorporated herein by reference.† |
|
|
|
10.38(a) |
| Completion Deed, effective February 27, 2013, by and among Mylan Inc., Strides Arcolab Limited, Agila Specialties Asia Pte Ltd, and the promoters named therein, filed by Mylan Inc. as Exhibit 10.4 to Form 10-Q for the quarter ended March 31, 2013, and incorporated herein by reference.† |
|
|
|
10.38(b) |
| Amendment to Completion Deed, effective December 4, 2013, by and among Mylan Institutional Inc., Mylan Laboratories Limited, Strides Arcolab Limited, Strides Pharma Asia Pte Ltd (f/k/a Agila Specialties Asia Pte Ltd), and the promoters named therein, filed by Mylan Inc. as Exhibit 10.53(b) to Form 10-K for the fiscal year ended December 31, 2013, and incorporated herein by reference.† |
|
|
|
10.39 |
| Agila Global Guarantee Deed, effective February 27, 2013, by and between Mylan Inc. and Strides Arcolab Ltd., filed by Mylan Inc. as Exhibit 10.5 to Form 10-Q for the quarter ended March 31, 2013, and incorporated herein by reference.† |
|
|
|
10.40 |
| The Executive Nonqualified Excess Plan, filed by Mylan Inc. as Exhibit 10.57 to Form 10-K for the fiscal year ended December 31, 2013, and incorporated herein by reference.* |
|
|
|
10.41 |
| Third Amended and Restated Executive Employment Agreement, entered into on February 25, 2014, by and between Mylan Inc. and Robert J. Coury, filed by Mylan Inc. as Exhibit 10.1 to the Report on Form 8-K filed with the SEC on February 28, 2014, and incorporated herein by reference.* |
|
|
|
10.42 |
| Second Amended and Restated Executive Employment Agreement, entered into on February 25, 2014, by and between Mylan Inc. and Heather Bresch, filed by Mylan Inc. as Exhibit 10.3 to the Report on Form 8-K filed with the SEC on February 28, 2014, and incorporated herein by reference.* |
|
|
|
10.43 |
| Second Amended and Restated Executive Employment Agreement, entered into on February 25, 2014, by and between Mylan Inc. and Rajiv Malik, filed by Mylan Inc. as Exhibit 10.4 to the Report on Form 8-K filed with the SEC on February 28, 2014, and incorporated herein by reference.* |
|
|
|
10.44(a) |
| Form of One-Time Special Five-Year Performance-Based Realizable Value Incentive Program Waiver Letter by and between Mylan Inc. and certain executive officers of Mylan Inc., filed by Mylan Inc. as Exhibit 10.56(a) to Form 10-K for the fiscal year ended December 31, 2014, and incorporated herein by reference.* |
|
|
|
10.44(b) |
| Form of One-Time Special Five-Year Performance-Based Realizable Value Incentive Program Waiver Letter by and between Mylan Inc. and certain employees of Mylan Inc., filed by Mylan Inc. as Exhibit 10.56(b) to Form 10-K for the fiscal year ended December 31, 2014, and incorporated herein by reference.* |
|
|
|
10.45 |
| Form of Transition and Succession Agreement Waiver Letter by and between Mylan Inc. and certain executive officers of Mylan Inc., filed by Mylan Inc. as Exhibit 10.57 to Form 10-K for the fiscal year ended December 31, 2014, and incorporated herein by reference.* |
|
|
|
10.46 |
| Form of Retirement Benefit Agreement Waiver Letter by and between Mylan Inc. and certain executive officers of Mylan Inc., filed by Mylan Inc. as Exhibit 10.58 to Form 10-K for the fiscal year ended December 31, 2014, and incorporated herein by reference.* |
|
|
|
10.47 |
| Letter Agreement, entered into on November 4, 2014, by and between Mylan Inc. and Robert J. Coury, filed by Mylan Inc. as Exhibit 10.59 to Form 10-K for the fiscal year ended December 31, 2014, and incorporated herein by reference.* |
|
|
|
10.48 |
| Form of Transition and Succession Agreement Waiver Letter by and between Mylan Inc. and certain executive officers of Mylan Inc., filed as Exhibit 10.2 to Form 10-Q for the quarter ended September 30, 2015, and incorporated herein by reference.* |
|
|
|
10.49 |
| Retirement and Consulting Agreement, dated April 13, 2016, between Mylan Inc. and John D. Sheehan, filed as Exhibit 10.1 to Form 10-Q for the quarter ended June 30, 2016, and incorporated herein by reference.* |
|
|
|
10.50(a) |
| Executive Employment Agreement, dated April 27, 2016 and effective June 6, 2016, between Mylan Inc. and Kenneth S. Parks, filed as Exhibit 10.2 to Form 10-Q for the quarter ended June 30, 2016, and incorporated herein by reference.* |
|
|
|
10.50(b) |
| Transition and Succession Agreement, dated April 27, 2016 and effective June 6, 2016, between Mylan Inc. and Kenneth S. Parks, filed as Exhibit 10.3 to Form 10-Q for the quarter ended June 30, 2016, and incorporated herein by reference.* |
|
|
|
10.51 |
| Letter Agreement, dated June 3, 2016, among Mylan N.V., Mylan Inc., and Robert J. Coury, filed as Exhibit 10.5 to Form 10-Q for the quarter ended June 30, 2016, and incorporated herein by reference.* |
|
|
|
185
Table of Contents
10.52(a) |
| Revolving Credit Agreement, dated December 19, 2014, among Mylan Inc., Mylan N.V., the lenders and issuing banks party thereto and Bank of America, N.A., as Administrative Agent, filed by Mylan Inc. as Exhibit 10.60 to Form 10-K for the fiscal year ended December 31, 2014, and incorporated herein by reference. |
|
|
|
10.52(b) |
| Amendment No. 1, dated May 1, 2015, to the Revolving Credit Agreement, dated December 19, 2014, between and among Mylan Inc., Mylan N.V., the lenders and issuing banks party thereto and Bank of America, N.A., as Administrative Agent, filed as Exhibit 10.1 to the Report on Form 8-K filed with the SEC on May 7, 2015, and incorporated herein by reference. |
|
|
|
10.52(c) |
| Additional Credit Extension Amendment, dated June 19, 2015, between and among Mylan Inc., Mylan N.V., ING Bank N.V., Dublin Branch, as Augmenting Lender, certain issuing banks and Bank of America, N.A., as Administrative Agent, to the Revolving Credit Agreement, dated December 19, 2014, between and among Mylan Inc., Mylan N.V., the lenders and issuing banks party thereto, and Bank of America, N.A., as Administrative Agent, filed as Exhibit 10.1 to Form 10-Q for the quarter ended June 30, 2015, and incorporated herein by reference. |
|
|
|
10.52(d) |
| Amendment No. 2, dated October 28, 2015, to the Revolving Credit Agreement, dated December 19, 2014, between and among Mylan Inc., Mylan N.V., the lenders and issuing banks party thereto and Bank of America, N.A., as Administrative Agent, filed as Exhibit 10.5 to Form 10-Q for the quarter ended September 30, 2015, and incorporated herein by reference. |
|
|
|
10.52(e) |
| Amendment No. 3, dated as of February 22, 2016, to the Revolving Credit Agreement, dated as of December 19, 2014, among Mylan Inc., Mylan N.V., the lenders and issuing banks party thereto and Bank of America, N.A., as Administrative Agent, filed as Exhibit 10.1 to the Report on Form 8-K filed with the SEC on February 26, 2016, and incorporated herein by reference. |
|
|
|
10.53(a) |
| Term Credit Agreement, dated December 19, 2014, among Mylan Inc., Mylan N.V., the lenders party thereto and Bank of America, N.A., as Administrative Agent, filed by Mylan Inc. as Exhibit 10.61 to Form 10-K for the fiscal year ended December 31, 2014, and incorporated herein by reference. |
|
|
|
10.53(b) |
| Amendment No. 1, dated May 1, 2015, to the Term Credit Agreement, dated December 19, 2014, among Mylan Inc., Mylan N.V., the lenders party thereto and Bank of America, N.A., as Administrative Agent, filed as Exhibit 10.2 to the Report on Form 8-K filed with the SEC on May 7, 2015, and incorporated herein by reference. |
|
|
|
10.53(c) |
| Amendment No. 2, dated October 28, 2015, to the Term Credit Agreement, dated December 19, 2014, among Mylan Inc., Mylan N.V., the lenders party thereto and Bank of America, N.A., as Administrative Agent, filed as Exhibit 10.4 to Form 10-Q for the quarter ended September 30, 2015, and incorporated herein by reference. |
|
|
|
10.53(d) |
| Amendment No. 3, dated as of February 22, 2016, to the Term Credit Agreement, dated as of December 19, 2014, among Mylan Inc., Mylan N.V., the lenders party thereto and Bank of America, N.A., as Administrative Agent, filed as Exhibit 10.2 to the Report on Form 8-K filed with the SEC on February 26, 2016, and incorporated herein by reference. |
|
|
|
10.54(a) |
| Amended and Restated Receivables Purchase Agreement, dated January 27, 2015, among Mylan Pharmaceuticals Inc., individually and as Servicer, Mylan Securitization LLC, as Seller, the Conduit Purchasers from time to time party thereto, the Committed Purchasers from time to time party thereto, the Purchaser Agents from time to time party thereto, the LOC Issuers from time to time party thereto, and The Bank of Tokyo-Mitsubishi UFJ, Ltd., New York Branch, as Agent, filed by Mylan Inc. as Exhibit 10.62 to Form 10-K for the fiscal year ended December 31, 2014, and incorporated herein by reference. |
|
|
|
10.54(b) |
| Amendment No. 1, dated May 20, 2016, to the Amended and Restated Receivables Purchase Agreement, dated January 27, 2015, among Mylan Pharmaceuticals Inc., individually and as Servicer, Mylan Securitization LLC, as Seller, the Conduit Purchasers from time to time party thereto, the Committed Purchasers from time to time party thereto, the Purchaser Agents from time to time party thereto, the LOC Issuers from time to time party thereto, and The Bank of Tokyo-Mitsubishi UFJ, Ltd., New York Branch, as Agent, filed as Exhibit 10.4 to Form 10-Q for the quarter ended June 30, 2016, and incorporated herein by reference. |
|
|
|
10.55 |
| Amended and Restated Purchase and Contribution Agreement, dated January 27, 2015, between Mylan Pharmaceuticals Inc., as Originator and as Servicer, and Mylan Securitization LLC, as Buyer, filed by Mylan Inc. as Exhibit 10.63 to Form 10-K for the fiscal year ended December 31, 2014, and incorporated herein by reference. |
|
|
|
10.56 |
| Call Option Agreement between Mylan N.V. and Stichting Preferred Shares Mylan, dated April 3, 2015, filed as Exhibit 10.1 to the Report on Form 8-K filed with the SEC on April 3, 2015, and incorporated herein by reference. |
|
|
|
186
Table of Contents
10.57(a) |
| Bridge Credit Agreement, dated April 24, 2015, among Mylan N.V., Mylan Inc., the lenders party thereto and Goldman Sachs Bank USA, as Administrative Agent, filed as Exhibit 10.1 to the Report on Form 8-K filed with the SEC on April 24, 2015, and incorporated herein by reference. |
|
|
|
10.57(b) |
| Amendment No. 1, dated April 29, 2015, to the Bridge Credit Agreement, dated April 24, 2015, among Mylan N.V., Mylan Inc., the lenders party thereto and Goldman Sachs Bank USA, as Administrative Agent, filed as Exhibit 10.1 to the Report on Form 8-K filed with the SEC on May 1, 2015, and incorporated herein by reference. |
|
|
|
10.57(c) |
| Amendment No. 2, dated August 6, 2015, to the Bridge Credit Agreement, dated April 24, 2015, among Mylan N.V., Mylan Inc., the lenders party thereto and Goldman Sachs Bank USA, as Administrative Agent, filed as Exhibit 10.1 to the Report on Form 8-K filed with the SEC on August 7, 2015, and incorporated herein by reference. |
|
|
|
10.58(a) |
| Term Credit Agreement, dated July 15, 2015, among Mylan Inc., Mylan N.V., the lenders party thereto and PNC Bank, National Association, as Administrative Agent, filed as Exhibit 10.1 to Form 10-Q for the quarter ended September 30, 2015, and incorporated herein by reference. |
|
|
|
10.58(b) |
| Amendment No. 1, dated October 28, 2015, to the Term Credit Agreement, dated July 15, 2015, between and among Mylan Inc., Mylan N.V., the lenders party thereto and PNC Bank, National Association, as Administrative Agent, filed as Exhibit 10.6 to Form 10-Q for the quarter ended September 30, 2015, and incorporated herein by reference. |
|
|
|
10.58(c) |
| Amendment No. 2, dated as of February 22, 2016, to the Term Credit Agreement, dated July 15, 2015, among Mylan Inc., Mylan N.V., the lenders party thereto and PNC Bank, National Association, as Administrative Agent, filed as Exhibit 10.3 to the Report on Form 8-K filed with the SEC on February 26, 2016, and incorporated herein by reference. |
|
|
|
10.59 |
| Bridge Credit Agreement, dated as of February 10, 2016, among Mylan N.V., as borrower, Mylan Inc., as a guarantor, Deutsche Bank AG Cayman Islands Branch, as administrative agent and a lender, Goldman Sachs Bank USA, as a lender, Goldman Sachs Lending Partners LLC, as a lender, and other lenders party thereto from time to time, filed as Exhibit 10.1 to the Report on Form 8-K filed with the SEC on February 17, 2016, and incorporated herein by reference. |
|
|
|
10.60(a) |
| Facilities Agreement, dated December 17, 2014, among Meda AB (publ), as borrower, the lenders party thereto and Danske Bank A/S, as agent, filed as Exhibit 10.1 to Form 10-Q for the quarter ended September 30, 2016, and incorporated herein by reference. |
|
|
|
10.60(b) |
| Amendment Letter, dated October 29, 2015, to the Facilities Agreement, dated December 17, 2014, among Meda AB (publ), as borrower, the lenders party thereto and Danske Bank A/S, as agent, filed as Exhibit 10.2 to Form 10-Q for the quarter ended September 30, 2016, and incorporated herein by reference. |
|
|
|
10.60(c) |
| Amendment and Waiver Letter, dated August 30, 2016, to the Facilities Agreement, dated December 17, 2014, among Meda AB (publ), as borrower, the lenders party thereto and Danske Bank A/S, as agent, filed as Exhibit 10.3 to Form 10-Q for the quarter ended September 30, 2016, and incorporated herein by reference. |
|
|
|
10.60(d) |
| Amendment Letter, dated as of November 14, 2016, to the Facilities Agreement dated as of December 17, 2014, among Meda AB (publ), as borrower, the lenders party thereto and Danske Bank A/S, as agent. |
|
|
|
10.61(a) |
| Loan Agreement, dated September 17, 2014, between Meda AB (publ), as borrower, and AB Svensk Exportkredit (publ), as lender, filed as Exhibit 10.4 to Form 10-Q for the quarter ended September 30, 2016, and incorporated herein by reference. |
|
|
|
10.61(b) |
| Amendment and Waiver Agreement, dated as of December 22, 2016, to the Loan Agreement, dated as of September 17, 2014, between Meda AB (publ), as borrower, and AB Svensk Exportkredit (publ), as lender. |
|
|
|
10.62 |
| Revolving Credit Agreement, dated November 22, 2016, among Mylan N.V., Mylan Inc., as a guarantor, the lenders and issuing banks party thereto and Bank of America, N.A., as the administrative agent. |
|
|
|
10.63 |
| Term Credit Agreement, dated November 22, 2016, among Mylan N.V., Mylan Inc., as a guarantor, the lenders party thereto and Goldman Sachs Bank USA, as administrative agent. |
|
|
|
10.64 |
| Guarantee Agreement, dated as of December 22, 2016, among Meda AB (publ), Mylan N.V. and AB Svensk Exportkredit (publ). |
|
|
|
10.65 |
| Guarantee, dated December 20, 2016, by Mylan N.V. of Meda AB (publ)'s obligations under the 2013/2018 SEK 600,000,000 floating rate notes and 2014/2019 SEK 750,000,000 floating rate notes issued by Meda AB (publ). |
|
|
|
10.66 |
| Form of Performance-Based Restricted Stock Unit Award Agreement under the One-Time Special Five-Year Performance-Based Realizable Value Incentive Program for Kenneth S. Parks.* |
|
|
|
187
Table of Contents
12.1 |
| Statement of Computation of Ratios of Earnings to Fixed Charges and Preferred Stock Dividends. |
|
|
|
21.1 |
| Subsidiaries of the registrant. |
|
|
|
23 |
| Consent of Independent Registered Public Accounting Firm. |
|
|
|
31.1 |
| Certification of Principal Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
|
|
31.2 |
| Certification of Principal Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
|
|
32 |
| Certification of Principal Executive Officer and Principal Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
|
|
|
101.INS |
| XBRL Instance Document |
|
|
|
101.SCH |
| XBRL Taxonomy Extension Schema |
|
|
|
101.CAL |
| XBRL Taxonomy Extension Calculation Linkbase |
|
|
|
101.LAB |
| XBRL Taxonomy Extension Label Linkbase |
|
|
|
101.PRE |
| XBRL Taxonomy Extension Presentation Linkbase |
|
|
|
101.DEF |
| XBRL Taxonomy Extension Definition Linkbase |
* | Denotes management contract or compensatory plan or arrangement. |
† | The Company's request for confidential treatment with respect to certain portions of this exhibit has been accepted. |
^ | Exhibits and schedules have been omitted pursuant to Item 601(b)(2) of Regulation S-K. The Company will furnish a copy of any omitted exhibits and schedules to the Securities and Exchange Commission upon request but may request confidential treatment for any exhibit or schedule so furnished. |
188
Table of Contents
SIGNATURES
Pursuant to the requirements of section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this Form to be signed on its behalf by the undersigned, thereunto duly authorized on March 1, 2017 .
|
|
| Mylan N.V. |
|
|
|
|
| by |
| /s/ HEATHER BRESCH |
|
|
| Heather Bresch |
|
|
| Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this Form has been signed below by the following persons on behalf of the registrant and in the capacities indicated as of March 1, 2017 .
Signature |
| Title |
|
|
|
/s/ HEATHER BRESCH |
| Chief Executive Officer and Director |
Heather Bresch |
| (Principal Executive Officer) |
|
|
|
/s/ KENNETH S. PARKS |
| Chief Financial Officer |
Kenneth S. Parks |
| (Principal Financial Officer) |
|
|
|
/s/ PAUL B. CAMPBELL |
| Senior Vice President and Chief Accounting Officer |
Paul B. Campbell |
| (Principal Accounting Officer) |
|
|
|
/s/ ROBERT J. COURY |
| Chairman and Director |
Robert J. Coury |
|
|
|
|
|
/s/ RODNEY L. PIATT |
| Vice Chairman and Director |
Rodney L. Piatt |
|
|
|
|
|
/s/ WENDY CAMERON |
| Director |
Wendy Cameron |
|
|
|
|
|
/s/ ROBERT J. CINDRICH |
| Director |
Robert J. Cindrich |
|
|
|
|
|
/s/ JOELLEN LYONS DILLON |
| Director |
JoEllen Lyons Dillon |
|
|
|
|
|
/s/ NEIL DIMICK |
| Director |
Neil Dimick |
|
|
|
|
|
/s/ MELINA HIGGINS |
| Director |
Melina Higgins |
|
|
|
|
|
/s/ DOUGLAS J. LEECH |
| Director |
Douglas J. Leech |
|
|
|
|
|
/s/ RAJIV MALIK |
| President and Director |
Rajiv Malik |
|
|
|
|
|
/s/ JOSEPH C. MAROON, M.D. |
| Director |
Joseph C. Maroon, M.D. |
|
|
|
|
|
/s/ MARK W. PARRISH |
| Director |
Mark W. Parrish |
|
|
|
|
|
/s/ RANDALL L. VANDERVEEN, PH.D. |
| Director |
Randall L. Vanderveen, Ph.D. |
|
|
189
Table of Contents
EXHIBIT INDEX
4.9 |
| Indenture, dated November 22, 2016, among Mylan N.V., as issuer, Mylan Inc., as guarantor and Citibank, N.A., London Branch, as trustee. |
|
|
|
10.60(d) |
| Amendment Letter, dated as of November 14, 2016, to the Facilities Agreement dated as of December 17, 2014, among Meda AB (publ), as borrower, the lenders party thereto and Danske Bank A/S, as agent. |
|
|
|
10.61(b) |
| Amendment and Waiver Agreement, dated as of December 22, 2016, to the Loan Agreement, dated as of September 17, 2014, between Meda AB (publ), as borrower, and AB Svensk Exportkredit (publ), as lender. |
|
|
|
10.62 |
| Revolving Credit Agreement, dated November 22, 2016, among Mylan N.V., Mylan Inc., as a guarantor, the lenders and issuing banks party thereto and Bank of America, N.A., as the administrative agent. |
|
|
|
10.63 |
| Term Credit Agreement, dated November 22, 2016, among Mylan N.V., Mylan Inc., as a guarantor, the lenders party thereto and Goldman Sachs Bank USA, as administrative agent. |
|
|
|
10.64 |
| Guarantee Agreement, dated as of December 22, 2016, among Meda AB (publ), Mylan N.V. and AB Svensk Exportkredit (publ). |
|
|
|
10.65 |
| Guarantee, dated December 20, 2016, by Mylan N.V. of Meda AB (publ)'s obligations under the 2013/2018 SEK 600,000,000 floating rate notes and 2014/2019 SEK 750,000,000 floating rate notes issued by Meda AB (publ). |
|
|
|
10.66 |
| Form of Performance-Based Restricted Stock Unit Award Agreement under the One-Time Special Five-Year Performance-Based Realizable Value Incentive Program for Kenneth S. Parks.* |
|
|
|
12.1 |
| Statement of Computation of Ratios of Earnings to Fixed Charges and Preferred Stock Dividends. |
|
|
|
21.1 |
| Subsidiaries of the registrant. |
|
|
|
23 |
| Consent of Independent Registered Public Accounting Firm. |
|
|
|
31.1 |
| Certification of Principal Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
|
|
31.2 |
| Certification of Principal Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
|
|
32 |
| Certification of Principal Executive Officer and Principal Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
|
|
|
101.INS |
| XBRL Instance Document |
|
|
|
101.SCH |
| XBRL Taxonomy Extension Schema |
|
|
|
101.CAL |
| XBRL Taxonomy Extension Calculation Linkbase |
|
|
|
101.LAB |
| XBRL Taxonomy Extension Label Linkbase |
|
|
|
101.PRE |
| XBRL Taxonomy Extension Presentation Linkbase |
|
|
|
101.DEF |
| XBRL Taxonomy Extension Definition Linkbase |
* | Denotes management contract or compensatory plan or arrangement. |
190