UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| [X] | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended June 30, 2014
OR
| [ ] | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to .
Commission File Number: 000-50484
MEI Pharma, Inc.
(Exact name of registrant as specified in its charter)
| DELAWARE | 51-0407811 | |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
11975 El Camino Real, Suite 101, San Diego, CA 92130
(Address of principal executive offices) (Zip Code)
(858) 792-6300
(Registrant's telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
Title of each class | Name of each exchange on which registered | |
| Common Stock, $0.00000002 par value | The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act:
None
(Title of Class)
Indicate by a check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by a check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (Section 229.405 of this chapter) is not contained herein, and will not be contained, to the best of the registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See definition of "large accelerated filer, "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer | ¨ | Accelerated filer | x | |||
| Non-accelerated filer | ¨ (Do not check if a smaller reporting company) | Smaller reporting company | ¨ | |||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The aggregate market value of the voting common equity held by non-affiliates of the registrant was approximately $132.3 million as of December 31, 2013, based on the closing price of the registrant's Common Stock as reported on the NASDAQ Capital Market on such date. For purposes of this calculation, shares of the registrant's common stock held by directors and executive officers have been excluded. This number is provided only for purposes of this Annual Report on Form 10-K and does not represent an admission that any particular person or entity is an affiliate of the registrant.
As of September 5, 2014, there were 21,607,296 shares of the registrant's common stock, par value $0.00000002 per share, outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Certain information required by Part III of this Annual Report on Form 10-K is incorporated by reference from the registrant's definitive proxy statement for the annual meeting of stockholders to be held in December 2014, which will be filed with the Securities and Exchange Commission within 120 days after the close of the registrant's fiscal year ended June 30, 2014.
MEI PHARMA, INC.
TABLE OF CONTENTS
| Page | ||||||
PART I | ||||||
| Item 1: | Business | 3 | ||||
| Item 1A: | Risk Factors | 15 | ||||
| Item 1B: | Unresolved Staff Comments | 24 | ||||
| Item 2: | Properties | 24 | ||||
| Item 3: | Legal Proceedings | 24 | ||||
| Item 4: | Mine Safety Disclosures | 24 | ||||
PART II | ||||||
| Item 5: | Market for the Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | 25 | ||||
| Item 6: | Selected Financial Data | 27 | ||||
| Item 7: | Management's Discussion and Analysis of Financial Condition and Results of Operations | 28 | ||||
| Item 7a: | Quantitative and Qualitative Disclosures about Market Risk | 36 | ||||
| Item 8: | Financial Statements and Supplementary Data | 37 | ||||
| Item 9: | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure | 57 | ||||
| Item 9A: | Controls and Procedures | 57 | ||||
| Item 9B: | Other Information | 59 | ||||
PART III | ||||||
| Item 10: | Directors, Executive Officers and Corporate Governance | 60 | ||||
| Item 11: | Executive Compensation | 60 | ||||
| Item 12: | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 60 | ||||
| Item 13: | Certain Relationships and Related Transactions, and Director Independence | 60 | ||||
| Item 14: | Principal Accountant Fees and Services | 60 | ||||
PART IV | ||||||
| Item 15: | Exhibits, Financial Statement Schedules | 61 | ||||
2
Forward-Looking Statements
This Annual Report on Form 10-K, or Annual Report, includes forward-looking statements, which involve a number of risks and uncertainties. These forward-looking statements can generally be identified as such because the context of the statement will include words such as "may," "will," "intend," "plan," "believe," "anticipate," "expect," "estimate," "predict," "potential," "continue," "likely," or "opportunity," the negative of these words or other similar words. Similarly, statements that describe our future plans, strategies, intentions, expectations, objectives, goals or prospects and other statements that are not historical facts are also forward-looking statements. Discussions containing these forward-looking statements may be found, among other places, in "Business" and "Management's Discussion and Analysis of Financial Condition and Results of Operations" in this Annual Report. For such statements, we claim the protection of the Private Securities Litigation Reform Act of 1995. Readers of this Annual Report are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the time this Annual Report was filed with the Securities and Exchange Commission, or SEC. These forward-looking statements are based largely on our expectations and projections about future events and future trends affecting our business, and are subject to risks and uncertainties that could cause actual results to differ materially from those anticipated in the forward-looking statements. These risks and uncertainties include, without limitation, those discussed in "Risk Factors" and in "Management's Discussion and Analysis of Financial Condition and Results of Operations" of this Annual Report. In addition, past financial or operating performance is not necessarily a reliable indicator of future performance, and you should not use our historical performance to anticipate results or future period trends. We can give no assurances that any of the events anticipated by the forward-looking statements will occur or, if any of them do, what impact they will have on our results of operations and financial condition. Except as required by law, we undertake no obligation to update publicly or revise our forward-looking statements to reflect events or circumstances that arise after the filing of this Annual Report or documents incorporated by reference herein that include forward-looking statements.
MEI Pharma, Inc. ® and our corporate logo are registered service marks of MEI Pharma. Any other brand names or trademarks appearing in this Annual Report are the property of their respective holders.
In this Annual Report, "MEI Pharma," "we," "us" and "our" refer to MEI Pharma, Inc. and our former wholly owned subsidiary Marshall Edwards Pty Ltd. ("MEPL"), which was dissolved in April 2012.
All financial data and share information in this Annual Report has been presented on an as-adjusted basis to give effect to our December 2012 1-for-6 reverse stock split.
PART I
| Item 1. | Business |
Overview
We are an oncology company focused on the clinical development of novel therapies for cancer. Our common stock is listed on the Nasdaq Capital Market under the symbol "MEIP".
Our business purpose is the development of drugs for the treatment of cancer. We are principally focused on the clinical development of our lead drug candidate, Pracinostat, which we are currently investigating in three Phase II clinical trials. Pracinostat is an orally available histone deacetylase (HDAC) inhibitor that is currently being developed for advanced hematologic diseases such as myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). In August 2012, we completed the acquisition of certain assets and intellectual property, including those related to Pracinostat, from S*Bio Pte Ltd ("S*Bio"). Our clinical development pipeline also includes our isoflavone-based mitochondrial inhibitor drug candidate, ME-344. Results from a Phase I clinical trial of ME-344 in patients with refractory solid tumors were presented in October 2013. A Phase Ib trial in patients with small cell lung cancer and ovarian cancer is ongoing. In September 2013, we acquired
3
PWT143, an oral inhibitor of phosphatidylinositide 3-kinase (PI3K) delta, a molecular target that has been shown to play a critical role in the proliferation and survival of hematologic cancer cells. We expect to complete the pre-clinical work required to support the filing of an Investigational New Drug (IND) application for PWT143 by the end of calendar year 2014. We own exclusive worldwide rights to all of our drug candidates, including Pracinostat, ME-344 and PWT143.
Clinical Development Programs
Lead Drug Candidate: Pracinostat
We are principally focused on the clinical development of our lead drug candidate, Pracinostat. Pracinostat is an orally available inhibitor of a group of enzymes called histone deacetylases, or HDACs. HDACs belong to a larger set of proteins collectively known as epigenetic regulators that can alter gene expression by chemically modifying DNA or its associated chromosomal proteins. Abnormal activity of these regulators is believed to play an important role in cancer and other diseases.
Pracinostat has been tested in multiple Phase I and Phase II clinical trials in advanced hematologic malignancies, such as MDS, AML and myelofibrosis, as well as in solid tumor indications in both adult and pediatric patients. Pracinostat has been generally well tolerated in more than 250 patients to date, with readily manageable side effects often associated with drugs of this class, the most frequent of which is fatigue. The results of these studies also suggest that Pracinostat has potential best-in-class pharmacokinetic properties when compared to other oral HDAC inhibitors.
Pracinostat has demonstrated clinical evidence of single-agent activity in patients with AML and myelofibrosis. In a Phase I dose-escalation trial in patients with advanced hematologic malignancies, 14% of evaluable AML patients (two of 14) achieved a complete remission (CR), with the responses enduring for more than 206 and 362 days, respectively. These results were presented at the American Society of Hematology (ASH) Annual Meeting in December 2010. In a Phase II clinical trial in intermediate or high-risk myelofibrosis, 36% of patients (eight of 22) demonstrated clinical response from Pracinostat treatment, with 9% of patients (two of 22) having a clinical improvement (anemia response) and 27% (six of 22) experiencing some reduction in splenomegaly. These results were published in the September 2012 issue of Leukemia Research .
Pracinostat has also shown evidence of synergistic activity when used in combination with the hypomethylating agent, azacitidine (or 5-azacitidine), in patients with advanced MDS. Results from a pilot Phase II study presented at the ASH Annual Meeting in December 2012 showed an overall response rate of 89% (eight of nine) among the nine patients treated at the MD Anderson Cancer Center, including seven patients who achieved either a CR or a complete remission with incomplete blood count recovery (CRi). An additional patient treated at the University of Wisconsin-Madison achieved a CR, increasing the overall response rate in the trial to 90% (nine of 10). The combination of Pracinostat and azacitidine was well tolerated in the study; the most frequent side effects were nausea and fatigue.
In August 2014, we completed enrollment in a randomized, double-blind, placebo-controlled Phase II clinical trial of Pracinostat in combination with azacitidine in intermediate-2 or high-risk patients with previously untreated MDS. The multicenter trial enrolled 108 patients with a one-to-one randomization. We expect to unblind this study after a six month follow-up period and report topline data by the first quarter of 2015. The primary endpoint of the study is CR. Secondary endpoints include overall response rate, hematologic improvement, duration of response, progression-free survival, rate of leukemic transformation, overall survival and safety.
In addition, two open-label Phase II trials of Pracinostat are ongoing: one in combination with azacitidine or decitabine in patients with MDS who either failed to respond or maintain a response to their hypomethylating agent alone and the other in combination with azacitidine in elderly patients with newly diagnosed AML who are not suited for intensive chemotherapy. In June 2014, we announced preliminary data
4
from the front line AML trial. Of the first nine patients enrolled in the study, three achieved a CR or CRi, each following one or two treatment cycles. In addition, three patients achieved a partial response (PR) or a partial response with incomplete blood count recovery (PRi) after their initial or second treatment evaluation, for an overall response rate of 67%. We expect to report additional data from this open-label study at a scientific conference in December 2014.
In February 2014, the U.S. Food & Drug Administration (FDA) granted orphan drug designation to Pracinostat for the treatment of AML. The designation provides orphan status to drugs defined by the FDA as those intended for the safe and effective treatment, diagnosis or prevention of rare diseases that affect fewer than 200,000 people in the U.S. Orphan designation qualifies us for certain development incentives, including tax credits for qualified clinical testing, prescription drug user fee exemptions and seven-year marketing exclusivity upon FDA approval.
Mitochondrial Inhibitor Drug Candidate: ME-344
ME-344 is our isoflavone-derived mitochondrial inhibitor drug candidate. In preclinical studies, ME-344 has been shown to cause cell death in multiple human tumor cell lines, including ovarian cancer stem cells, by interfering with mitochondrial energy generation. In April 2013, Dr. Ayesha Alvero, Yale University School of Medicine, presented data at the American Association for Cancer Research (AACR) Annual Meeting showing the ability of ME-344 to decrease tumor burden and delay recurrence in a pre-clinical in vivo model of recurrent epithelial ovarian cancer, the most lethal of all gynecological malignancies.
In October 2013, results from our first-in-human, single-agent Phase I clinical trial of ME-344 in patients with refractory solid tumors were presented at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics. The results indicated that eight of 21 evaluable patients (38%) treated with ME-344 achieved stable disease or better, including five who experienced progression-free survival that was at least twice the duration of their last prior treatment before entry into the study. In addition, one of these patients, a heavily pre-treated patient with small cell lung cancer, achieved a confirmed partial response. This patient remains on study and has continued weekly dosing for more than two years (as of September 2014). ME-344 was generally well tolerated at doses equal to or less than 10 mg/kg delivered on a weekly schedule for extended durations. Dose limiting toxicities were observed at both the 15 mg/kg and 20 mg/kg dose levels, consisting primarily of Grade 3 peripheral neuropathy. Other medically significant adverse events observed in single patients included angina and QTc prolongation at the 10 mg/kg dose.
In May 2014, we initiated a Phase Ib clinical trial of ME-344 in combination with topotecan (trade name Hycamtin ® ) in patients with solid tumors, including small cell lung cancer and ovarian cancers. The Phase Ib study is evaluating the safety and tolerability of intravenous ME-344 in combination with topotecan, a chemotherapy approved by the FDA for the treatment of small cell lung, ovarian and cervical cancers. The initial stage of the study will establish the maximum tolerated dose (MTD) of ME-344 in combination with topotecan in up to 24 patients. Once the MTD has been determined, the study will enroll an additional 40 patients into two cohorts: relapsed/refractory small cell lung cancer and platinum-refractory ovarian cancer.
PI3-Kinase Delta Drug Candidate: PWT143
In September 2013, we acquired exclusive worldwide rights to PWT143 from Pathway Therapeutics, Inc. for an undisclosed upfront cash payment with no future milestone or royalty obligations. In pre-clinical studies, PWT143 has been found to be a potent and selective oral inhibitor of PI3-kinase delta, a molecular target that has been shown to play a critical role in the proliferation and survival of certain hematologic cancer cells. We expect to complete the required pre-clinical studies necessary for an IND filing by the end of calendar year 2014.
5
Scientific Overview
Epigenetics Program
HDACs play a key role in epigenetic regulation of gene expression by regulating chromatin structure. Acetylation of positively charged lysine residues present in histone proteins by the histone acetyltransferase (HATs) reduces the affinity between histones and negatively charged DNA, resulting in the opening of the chromatin structure. This makes it easier for the transcriptional machinery to access the DNA, enhancing RNA transcription. Conversely, deacetylation by the HDACs closes the chromatin structure leading to a repression of gene transcription. In normal cells, HDACs and HATs together control histone acetylation levels to maintain a balance. In diseases such as cancer, this regulation can be disturbed. HDAC inhibitors cause accumulation of acetylated histones, enhance transcription and result in changes of a variety of cellular responses including differentiation, proliferation, migration, survival and response to metabolic and hypoxic stress. In general, tumor cells are more susceptible than normal cells to the anti-proliferative and pro-apoptotic effects of HDAC inhibitors.
There are currently three HDAC inhibitors, one oral and two injectable, approved by the FDA for the treatment of T-cell lymphoma. Other HDAC inhibitors are being evaluated in clinical trials as single agents and in combination with chemotherapy for various hematologic diseases, including acute myeloid leukemia, MDS and myelofibrosis, as well as for solid tumors.
Pracinostat
Our lead drug candidate, Pracinostat, is an orally available, potent HDAC inhibitor with potentially improved physicochemical, pharmaceutical and pharmacokinetic properties when compared to other compounds of this class, including increased bioavailability and increased half-life.
Pracinostat has been tested in more than 250 patients in multiple Phase I and Phase II clinical trials and found to be generally well tolerated with readily manageable side effects often associated with drugs of this class, including fatigue, myelosuppresion and gastrointestinal toxicity. Results from a Phase I dose-escalation study, presented at the ASH Annual Meeting in December 2010, demonstrated clinical evidence of single-agent activity in patients with AML, including two CRs. In addition, data from a Phase II clinical trial of Pracinostat showed single-agent activity in patients with intermediate or high-risk myelofibrosis. These results were published in the September 2012 issue of Leukemia Research .
Pracinostat has also shown evidence of activity when used in combination with a wide range of therapies in clinical and pre-clinical studies. Pre-clinical data published in the May 2012 issue of Blood Cancer Journal demonstrated synergistic activity when Pracinostat was combined with Pacritinib, an experimental JAK2 inhibitor. In addition, encouraging preliminary results were observed in a pilot Phase II clinical trial of Pracinostat in combination with azacitidine in patients with advanced MDS, including eight responses out of nine patients treated at the MD Anderson Cancer Center. These results were presented at the ASH Annual Meeting in December 2012.
We are currently investigating Pracinostat in three Phase II studies, each in combination with an approved hypomethylating agent (HMA): 1) A randomized, double-blind, placebo-controlled study in combination with azacitidine in intermediate-2 or high-risk patients with previously untreated MDS; 2) an open-label study in combination with azacitidine in elderly patients with newly diagnosed AML who are not suited for intensive chemotherapy; and 3) an open-label study in combination with azacitidine or decitabine in patients with MDS who either failed to respond or maintain a response to their HMA alone. We anticipate data from each of these studies by the first quarter of 2015.
6
Cancer Metabolism Programs
Our Company was originally formed to develop novel cancer therapeutics based on a group of compounds known as isoflavones. More than 400 new chemical structures were created based on the central design of these naturally occurring plant isoflavones. We believe that some of these synthetic compounds, including our drug candidate ME-344, interact with specific enzyme targets, resulting in the inhibition of tumor cell metabolism, a function critical for the survival of cancer cells.
ME-344
ME-344 is the active metabolite of a first generation compound, named NV-128. The proposed target for NV-128 and ME-344 is found in the tumor cell mitochondria, the specialized area in the cell that produces energy in the form of adenosine triphosphate (ATP). When these compounds interact with their target, a rapid reduction in ATP occurs, which leads to a cascade of biochemical events within the cell and ultimately to cell death. One outcome that is believed to be critical for cell death induction by ME-344 is the disruption of both mammalian target of rapamycin (mTOR1 and mTOR2) pathways. In cancer cells, the mTOR protein is involved in enhancing tumor growth and may be associated with resistance to chemotherapeutic drugs. Inhibition of both mTOR pathways appears to shut down many of the cellular survival pathways of cancer cells.
ME-344 has demonstrated broad activity against a panel of human cancer cell lines both as a single agent and as a chemosensitizing agent. Results from ongoing laboratory research studies conducted in collaboration with the Department of Obstetrics, Gynecology, and Reproductive Sciences at the Yale School of Medicine demonstrate that NV-128 and ME-344 are active against chemotherapy-resistant ovarian tumor stem cells. In April 2011, at the AACR Annual Meeting, Dr. Alvero from the Yale School of Medicine presented pre-clinical data demonstrating the ability of NV-128 to induce mitochondrial instability, ultimately leading to cell death in chemotherapy-resistant ovarian cancer stem cells. This cell death was associated with the activation of the MEK/ERK pathway leading to mitochondrial depolarization and DNA fragmentation. The study further characterized the mechanism of action of NV-128 and demonstrated that NV-128 also promotes a state of cellular starvation, resulting in the activation of the AMP kinase pathway, leading to inhibition of both mTOR pathways and the induction of destructive autophagy. In April 2013, Dr. Alvero presented new data at the AACR Annual Meeting showing the ability of ME-344 to decrease tumor burden and delay recurrence in a pre-clinical in vivo model of recurrent epithelial ovarian cancer, the most lethal of all gynecologic malignancies. In October 2013, results from our first-in-human, single-agent Phase I clinical trial of ME-344 in patients with refractory solid tumors were presented at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics indicating that eight of 21 evaluable patients (38%) treated with ME-344 achieved stable disease or better.
ME-143
ME-143, an NADH oxidase inhibitor, is also from our isoflavone-based library. Results from a Phase I dose-escalation study of intravenous ME-143 in patients with solid refractory tumors were presented at the American Society of Clinical Oncology Annual Meeting in June 2012. The data showed that ME-143 was generally well tolerated with the exception of a serious infusion reaction in one patient at the highest dose level. No additional clinical studies of ME-143 are planned at this time.
Signaling Program
PWT143
The PI3K/AKT/mTOR pathway is an important signaling pathway for many cellular functions such as cell survival, cell cycle progression and cellular growth. PI3Ks are a family of enzymes within this pathway that have been shown to play a critical role in the proliferation and survival of certain cancer cells. There are several isoforms of PI3K that are expressed in different types of cells. The PI3K delta isoform is believed to be important for survival of certain B-cell leukemias and lymphomas.
7
In July 2014, the FDA approved the first PI3K delta selective inhibitor, idelalisib (trade name Zydelig ® ), for the treatment of three B-cell blood cancers: chronic lymphocytic leukemia, follicular lymphoma and small lymphocytic lymphoma.
In pre-clinical studies, PWT143 has been found to be a potent and selective oral inhibitor of PI3K delta, with a distinct chemical structure and evidence of improved pre-clinical activity compared to other PI3K delta inhibitors. We expect to complete the required pre-clinical studies necessary for an IND filing by the end of calendar year 2014.
Competition
The marketplace for our drug candidates is highly competitive. A number of other companies have products or drug candidates in various stages of pre-clinical or clinical development that are intended for the same therapeutic indications for which our drug candidates are being developed. Some of these potential competing drugs are further advanced in development than our drug candidates and may be commercialized sooner. Even if we are successful in developing effective drugs, our drug candidates may not compete successfully with products produced by our competitors.
Our competitors include pharmaceutical companies and biotechnology companies, as well as universities and public and private research institutions. In addition, companies active in different but related fields represent substantial competition for us. Many of our competitors developing oncology drugs have significantly greater capital resources, larger research and development staffs and facilities, and greater experience in drug development, regulation, manufacturing, and marketing than we do. They compete with us in recruiting eligible patients to participate in clinical studies and in attracting development and/or commercialization partners. They also license technologies that are competitive with our technologies. As a result, our competitors may be able to more easily develop technologies and products that would render our technologies or our drug candidates obsolete or non-competitive.
Intellectual Property
We own, by assignment, worldwide rights to each of our current drug candidates. Our intellectual property portfolio includes approximately 22 issued U.S. patents, 161 issued foreign patents, 10 pending U.S. patent applications and 57 pending foreign applications.
We have acquired, by assignment, patents and patent applications from S*Bio relating to a family of heterocyclic compounds, which include Pracinostat, that inhibit histone deacetylases. The U.S. Patent and Trademark Office (USPTO) has issued three patents covering a number of these heterocyclic-based compounds, including Pracinostat, and their composition of matter, pharmaceutical compositions, and methods of use to treat proliferative diseases. The composition of matter claims covering Pracinostat are projected to expire in May 2028, not including patent term extension.
We have acquired, by assignment, patents and patent applications from Novogen, our former majority shareholder, which relate to a large family of isoflavonoid compounds, including ME-344.
The USPTO has issued two patents covering ME-344, including its composition of matter, pharmaceutical compositions and methods of use to treat cancer. The composition of matter claims covering ME-344 are expected to expire in September 2025, not including patent term extension.
We have acquired by assignment worldwide rights to PWT143 and other related compounds from Pathway Therapeutics, Inc. The USPTO has issued one patent covering the composition of matter and pharmaceutical compositions of PWT143 which is projected to expire in January 2031, not including any patent term extension. There are currently three U.S. and 29 foreign applications for PWT143 and related compounds pending.
8
Our success depends in large part on our ability to protect our proprietary technologies, compounds and information, and to operate without infringing the proprietary rights of third parties. We rely on a combination of patent, trade secret, copyright, and trademark laws, as well as confidentiality, licensing and other agreements, to establish and protect our proprietary rights. We seek patent protection for our key inventions, including drug candidates we identify, routes for chemical synthesis and pharmaceutical formulations. There is no assurance that any of our pending patent applications will issue, or that any of our patents will be enforceable or will cover a drug or other commercially significant product or method. In addition, we regularly review our patent portfolio to identify patents and patent applications that we deem to have relatively low value to our ongoing business operations for potential abandonment. There is also no assurance that we will correctly identify which of our patents and patent applications should be maintained and which should be abandoned. The term of most of our other current patents commenced, and most of our future patents, if any, will commence, on the date of issuance and terminate 20 years from the earliest effective filing date of the patent application. Because any marketing and regulatory approval for a drug often occurs several years after the related patent application is filed, the resulting market exclusivity afforded by any patent on our drug candidates and technologies will likely be substantially less than 20 years.
As most patent applications in the U.S. are maintained as confidential until published by the U.S. Patent and Trademark Office at 18 months from filing for all cases filed after November 29, 2000, or at issue, for cases filed prior to November 29, 2000, we cannot be certain that we, S*Bio, Novogen or Pathway Therapeutics, Inc. were the first to make the inventions covered by the patents and applications referred to above. Additionally, publication of discoveries in the scientific or patent literature often lags behind the actual discoveries. Moreover, pursuant to the terms of the Uruguay Round Agreements Act, patents filed on or after June 8, 1995 have a term of twenty years from the date of such filing except for provisional applications, irrespective of the period of time it may take for such patent to ultimately issue. This may shorten the period of patent protection afforded to therapeutic uses of Pracinostat, ME-344 or PWT143, as patent applications in the biopharmaceutical sector often take considerable time to issue. However, in some countries the patent term may be extended.
In order to protect the confidentiality of our technology, including trade secrets and know-how and other proprietary technical and business information, we require all of our consultants, advisors and collaborators to enter into agreements that prohibit the use or disclosure of information that is deemed confidential. These agreements also oblige our consultants, advisors and collaborators to assign to us, or negotiate a license to developments, discoveries and inventions made by such persons in connection with their work relating to our products. We cannot be sure that confidentiality will be maintained by those from whom we have acquired technology or disclosure prevented by these agreements. We also cannot be sure that our proprietary information or intellectual property will be protected by these agreements or that others will not independently develop substantially equivalent proprietary information or intellectual property.
The pharmaceutical industry is highly competitive and patents may have been applied for by, and issued to, other parties relating to products competitive with Pracinostat, ME-344 or PWT143. Use of these compounds and any other drug candidates may give rise to claims that they infringe the patents or proprietary rights of other parties, existing now and in the future. An adverse claim could subject us to significant liabilities to such other parties and/or require disputed rights to be licensed from such other parties. We cannot be sure that any license required under any such patents or proprietary rights would be made available on terms acceptable to us, if at all. If we do not obtain such licenses, we may encounter delays in product market introductions, or may find that the development, manufacture or sale of products requiring such licenses may be precluded.
Research and Development
The objective of our research and development program is the generation of data sufficient to achieve regulatory approval of our drug candidates in one or more dosage forms in major markets such as the U.S., to develop commercially attractive attributes, and/or to allow us to enter into a development and/or commercial relationship with another party. The data are generated by our pre-clinical studies and clinical trial programs.
9
The key aspects of our research and development program are to provide more complete characterization of the following:
| • | the relevant molecular targets of action of our drug candidates; |
| • | the relative therapeutic benefits and indications for use of our drug candidates as a monotherapy or as part of combinational therapy with other agents; |
| • | the most appropriate therapeutic indications and dosage forms for Pracinostat, ME-344 and PWT143. |
Our research and development expenses include clinical and preclinical study fees, personnel costs, and manufacturing costs. Research and development expenses were $19.3 million, $6.1 million, and $4.9 million for the years ended June 30, 2014, 2013 and 2012, respectively. We expect research and development expenses to increase during the fiscal year ending June 30, 2015, related primarily to our clinical trials.
Government Regulation
U.S. Regulatory Requirements
The FDA, and comparable regulatory agencies in other countries, regulate and impose substantial requirements upon the research, development, pre-clinical and clinical testing, labeling, manufacture, quality control, storage, approval, advertising, promotion, marketing, distribution and export of pharmaceutical products including biologics, as well as significant reporting and record-keeping obligations. State governments may also impose obligations in these areas.
In the U.S., pharmaceutical products are regulated by the FDA under the Federal Food, Drug, and Cosmetic Act or FDCA and other laws, including in the case of biologics, the Public Health Service Act. We believe, but cannot be certain, that our products will be regulated as drugs by the FDA. The process required by the FDA before drugs may be marketed in the U.S. generally involves the following:
| • | pre-clinical laboratory evaluations, including formulation and stability testing, and animal tests performed under the FDA's Good Laboratory Practices regulations to assess pharmacological activity and toxicity potential; |
| • | submission and approval of an Investigational New Drug Application, or IND, including results of pre-clinical tests, manufacturing information, and protocols for clinical tests, which must become effective before clinical trials may begin in the U.S.; |
| • | obtaining approval of Institutional Review Boards, or IRBs, to administer the products to human subjects in clinical trials; |
| • | adequate and well-controlled human clinical trials to establish the safety and efficacy of the product for the product's intended use; |
| • | development of manufacturing processes which conform to FDA current Good Manufacturing Practices, or cGMPs, as confirmed by FDA inspection; |
| • | submission of results for pre-clinical and clinical studies, and chemistry, manufacture and control information on the product to the FDA in a New Drug Approval Application, or NDA; and |
| • | FDA review and approval of a NDA, prior to any commercial sale or shipment of a product. |
The testing and approval process requires substantial time, effort, and financial resources, and we cannot be certain that any approval will be granted on a timely basis, if at all.
The results of the pre-clinical studies, together with initial specified manufacturing information, the proposed clinical trial protocol, and information about the participating investigators are submitted to the FDA as part of an IND, which must become effective before we may begin human clinical trials in the U.S. Additionally,
10
an independent IRB must review and approve each study protocol and oversee conduct of the trial. An IND becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day period, raises concerns or questions about the conduct of the trials as outlined in the IND and imposes a clinical hold. If the FDA imposes a clinical hold, the IND sponsor must resolve the FDA's concerns before clinical trials can begin. Pre-clinical tests and studies can take several years to complete, and there is no guarantee that an IND we submit based on such tests and studies will become effective within any specific time period, if at all.
Human clinical trials are typically conducted in three sequential phases that may overlap.
| • | Phase I: The drug is initially introduced into healthy human subjects or patients and tested for safety and dosage tolerance. Absorption, metabolism, distribution, and excretion testing is generally performed at this stage. |
| • | Phase II: The drug is studied in controlled, exploratory therapeutic trials in a limited number of subjects with the disease or medical condition for which the new drug is intended to be used in order to identify possible adverse effects and safety risks, to determine the preliminary or potential efficacy of the product for specific targeted diseases or medical conditions, and to determine dosage tolerance and the optimal effective dose. |
| • | Phase III: When Phase II studies demonstrate that a specific dosage range of the drug is likely to be effective and the drug has an acceptable safety profile, controlled, large-scale therapeutic Phase III trials are undertaken at multiple study sites to demonstrate clinical efficacy and to further test for safety in an expanded patient population. |
We cannot be certain that we will successfully complete clinical testing of our products within any specific time period, if at all. Furthermore, the FDA, the IRB or we may suspend or terminate clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
Results of pre-clinical studies and clinical trials, as well as detailed information about the manufacturing process, quality control methods, and product composition, among other things, are submitted to the FDA as part of a NDA seeking approval to market and commercially distribute the product on the basis of a determination that the product is safe and effective for its intended use. Before approving a NDA, the FDA will inspect the facilities at which the product is manufactured and will not approve the product unless cGMP compliance is satisfactory. If applicable regulatory criteria are not satisfied, the FDA may deny the NDA or require additional testing or information. As a condition of approval, the FDA also may require post-marketing testing or surveillance to monitor the product's safety or efficacy. Even after a NDA is approved, the FDA may impose additional obligations or restrictions (such as labeling changes, or clinical post-marketing requirements), or even suspend or withdraw a product approval on the basis of data that arise after the product reaches the market, or if compliance with regulatory standards is not maintained. We cannot be certain that any NDA we submit will be approved by the FDA on a timely basis, if at all. Also, any such approval may limit the indicated uses for which the product may be marketed. Any refusal to approve, delay in approval, suspension or withdrawal of approval, or restrictions on indicated uses could have a material adverse impact on our business prospects.
Each NDA must be accompanied by a user fee, pursuant to the requirements of the Prescription Drug User Fee Act, or PDUFA, and its amendments. According to the FDA's fee schedule, effective on October 1, 2014 for the fiscal year 2015, the user fee for an application requiring clinical data, such as a NDA, is $2,335,200. The FDA adjusts the PDUFA user fees on an annual basis. PDUFA also imposes an annual product fee for prescription drugs and biologics ($110,370), and an annual establishment fee ($569,200) on facilities used to manufacture prescription drugs and biologics. A written request can be submitted for a waiver for the application fee for the first human drug application that is filed by a small business, but there are no waivers for product or establishment fees. We are not at the stage of development with our products where we are subject to these fees, but they are significant expenditures that may be incurred in the future and must be paid at the time of application submissions to the FDA.
11
Satisfaction of FDA requirements typically takes several years. The actual time required varies substantially, based upon the type, complexity, and novelty of the pharmaceutical product, among other things. Government regulation imposes costly and time-consuming requirements and restrictions throughout the product life cycle and may delay product marketing for a considerable period of time, limit product marketing, or prevent marketing altogether. Success in pre-clinical or early stage clinical trials does not ensure success in later stage clinical trials. Data obtained from pre-clinical and clinical activities are not always conclusive and may be susceptible to varying interpretations that could delay, limit, or prevent marketing approval. Even if a product receives marketing approval, the approval is limited to specific clinical indications. Further, even after marketing approval is obtained, the discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market.
After product approval, there are continuing significant regulatory requirements imposed by the FDA, including record-keeping requirements, obligations to report adverse side effects in patients using the products, and restrictions on advertising and promotional activities. Quality control and manufacturing procedures must continue to conform to cGMPs, and the FDA periodically inspects facilities to assess cGMP compliance. Additionally, post-approval changes in ingredient composition, manufacturing processes or facilities, product labeling, or other areas may require submission of a NDA Supplement to the FDA for review and approval. New indications will require additional clinical studies and submission of a NDA Supplement. Failure to comply with FDA regulatory requirements may result in an enforcement action by the FDA, including Warning Letters, product recalls, suspension or revocation of product approval, seizure of product to prevent distribution, impositions of injunctions prohibiting product manufacture or distribution, and civil and criminal penalties. Maintaining compliance is costly and time-consuming. We cannot be certain that we, or our present or future suppliers or third-party manufacturers, will be able to comply with all FDA regulatory requirements, and potential consequences of noncompliance could have a material adverse impact on our business prospects.
The FDA's policies may change, and additional governmental regulations may be enacted that could delay, limit, or prevent regulatory approval of our products or affect our ability to manufacture, market, or distribute our products after approval. Moreover, increased attention to the containment of healthcare costs in the U.S. and in foreign markets could result in new government regulations that could have a material adverse effect on our business. Our failure to obtain coverage, an adequate level of reimbursement, or acceptable prices for our future products could diminish any revenues we may be able to generate. Our ability to commercialize future products will depend in part on the extent to which coverage and reimbursement for the products will be available from government and health administration authorities, private health insurers, and other third-party payers. European Union member states and U.S. government and other third-party payers increasingly are attempting to contain healthcare costs by consideration of new laws and regulations limiting both coverage and the level of reimbursement for new drugs. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the U.S. or abroad.
Our activities also may be subject to state laws and regulations that affect our ability to develop and sell our products. We are also subject to numerous federal, state, and local laws relating to such matters as safe working conditions, clinical, laboratory, and manufacturing practices, environmental protection, fire hazard control, and disposal of hazardous or potentially hazardous substances. We may incur significant costs to comply with such laws and regulations now or in the future, and the failure to comply may have a material adverse impact on our business prospects.
The FDCA includes provisions designed to facilitate the development and expedite the review of drugs and biological products intended for treatment of serious or life-threatening conditions that demonstrate the potential to address unmet medical needs for such conditions. These provisions set forth a procedure for designation of a drug as a "fast track product". The fast track designation applies to the combination of the product and specific indication for which it is being studied. A product designated as fast track is ordinarily eligible for additional programs for expediting development and review, but products that are not in fast track drug development programs may also be able to take advantage of these programs. These programs include
12
priority review of NDAs and accelerated approval. Drug approval under the accelerated approval regulations may be based on evidence of clinical effect on a surrogate endpoint that is reasonably likely to predict clinical benefit. A post-marketing clinical study will be required to verify clinical benefit, and other restrictions to assure safe use may be imposed. We do not currently have fast track designation for any of our clinical programs. If we should seek such designation for any of our programs, however, we cannot be assured that it will be granted by the FDA.
Under the Drug Price Competition and Patent Term Restoration Act of 1984, a sponsor may obtain marketing exclusivity for a period of time following FDA approval of certain drug applications, regardless of patent status, if the drug is a new chemical entity or if new clinical studies were required to support the marketing application for the drug. This marketing exclusivity prevents a third party from obtaining FDA approval for an identical or nearly identical drug under an Abbreviated New Drug Application or a "505(b)(2) New Drug Application". The statute also allows a patent owner to obtain an extension of applicable patent terms for a period equal to one-half the period of time elapsed between the filing of an IND and the filing of the corresponding NDA plus the period of time between the filing of the NDA and FDA approval, with a five year maximum patent extension. We cannot be certain that we will be able to take advantage of either the patent term extension or marketing exclusivity provisions of these laws.
The Best Pharmaceuticals for Children Act, or BPCA, signed into law on January 4, 2002, was reauthorized and amended by the FDA Amendments Act of 2007 or FDAAA. The reauthorization of BPCA provides an additional six months of patent protection to NDA applicants that conduct acceptable pediatric studies of new and currently-marketed drug products for which pediatric information would be beneficial, as identified by the FDA in a Pediatric Written Request. The Pediatric Research Equity Act, or PREA, signed into law on December 3, 2003, also was reauthorized and amended by the FDAAA. The reauthorization of PREA requires that most applications for drugs and biologics include a pediatric assessment (unless waived or deferred) to ensure the drugs' and biologics' safety and effectiveness in children. Such pediatric assessment must contain data, gathered using appropriate formulations for each age group for which the assessment is required, that are adequate to assess the safety and effectiveness of the drug or the biological product for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the drug or the biological product is safe and effective. The pediatric assessments can only be deferred provided there is a timeline for the completion of such studies. The FDA may waive (partially or fully) the pediatric assessment requirement for several reasons, including if the applicant can demonstrate that reasonable attempts to produce a pediatric formulation necessary for that age group have failed. The Food and Drug Administration Safety and Innovation Act (FDASIA) signed into law on July 9, 2012, permanently renewed and strengthened BPCA and PREA.
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a "rare disease or condition," which generally is a disease or condition that affects fewer than 200,000 individuals in the U.S. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. If a product which has an orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, i.e., the FDA may not approve any other applications to market the same drug for the same indication for a period of seven years, except in limited circumstances. Pracinostat has been granted orphan drug designation by the FDA for the treatment of AML, but it may not receive orphan designation for other indications. Our other products may not be eligible for orphan drug status or be designated as orphan drugs. Even if designated as orphan drugs, our products may not be approved before other applications or granted orphan drug exclusivity if approved.
Foreign Regulatory Requirements
Outside the U.S., our ability to market our products will also be contingent upon receiving marketing authorizations from the appropriate regulatory authorities and compliance with applicable post-approval regulatory requirements. Although the specific requirements and restrictions vary from country to country, as a general matter, foreign regulatory systems include risks similar to those associated with FDA regulation, described above.
13
Under European Union regulatory systems, marketing authorizations may be submitted either under a centralized or a national procedure. Under the centralized procedure, a single application to the European Medicines Agency (EMA) leads to an approval granted by the European Commission which permits the marketing of the product throughout the EU. The centralized procedure is mandatory for certain classes of medicinal products, but optional for others. For example, all medicinal products developed by certain biotechnological means, and those developed for cancer and other specified diseases and disorders, must be authorized via the centralized procedure. We assume that the centralized procedure will apply to our products that are developed by means of a biotechnology process or are intended for treatment of cancer. The national procedure is used for products that are not required to be authorized by the centralized procedure. Under the national procedure, an application for a marketing authorization is submitted to the competent authority of one member state of the EU. The holders of a national marketing authorization may submit further applications to the competent authorities of the remaining member states via either the decentralized or mutual recognition procedure. The decentralized procedure enables applicants to submit an identical application to the competent authorities of all member states where approval is sought at the same time as the first application, while under the mutual recognition procedure, products are authorized initially in one member state, and other member states where approval is sought are then requested to recognize the original authorization based upon an assessment report prepared by the original authorizing competent authority. Both the decentralized and mutual recognition procedures should take no longer than 90 days, but if one member state makes an objection, which under the legislation can only be based on a possible risk to human health, the application will be automatically referred to the Committee for Medicinal Products for Human Use (CHMP) of the EMA. If a referral for arbitration is made, the procedure is suspended. However, member states that have already approved the application may, at the request of the applicant, authorize the product in question without waiting for the result of the arbitration. Such authorizations will be without prejudice to the outcome of the arbitration. For all other concerned member states, the opinion of the CHMP, which is binding, could support or reject the objection or alternatively could reach a compromise position acceptable to all EU countries concerned. The arbitration procedure may take an additional year before a final decision is reached and may require the delivery of additional data.
As with FDA approval, we may not be able to secure regulatory approvals in Europe in a timely manner, if at all. Additionally, as in the U.S., post-approval regulatory requirements, such as those regarding product manufacture, marketing, or distribution, would apply to any product that is approved in Europe, and failure to comply with such obligations could have a material adverse effect on our ability to successfully commercialize any product.
The conduct of clinical trials in the European Union is governed by the European Clinical Trials Directive (2001/20/EC), which was implemented in May 2004. This Directive governs how regulatory bodies in member states control clinical trials. No clinical trial may be started without a clinical trial authorization granted by the national competent authority and favorable ethics approval. New legislation to revise and replace the European Clinical Trials Directive is currently proposed by the European Commission and is under consideration by European Union institutions.
Accordingly, there is a marked degree of change and uncertainty both in the regulation of clinical trials and in respect of marketing authorizations which we face for our products in Europe.
Manufacturing
We do not have the facilities or capabilities to commercially manufacture any of our products and product candidates. We are and expect to continue to be dependent on contract manufacturers for supplying our existing and future product candidates for clinical trials and commercial scale manufacturing of our product candidates in accordance with regulatory requirements, including current Good Manufacturing Practices. Contract manufacturers may utilize their own technology, technology developed by us, or technology acquired or licensed from third parties. FDA approval of the manufacturing procedures and the site will be required prior to commercial distribution.
14
Employees
As of June 30, 2014, we had nineteen employees, seven of whom hold a Ph.D. or M.D. degree. Other personnel resources are used from time to time as consultants or third party service organizations on an as-needed basis. All members of our senior management team have prior experience with pharmaceutical, biotechnology or medical product companies. We believe that we have been successful in attracting skilled and experienced personnel, but there can be no assurance that we will be able to attract and retain the individuals needed. None of our employees are represented by a collective bargaining agreement, nor have we experienced work stoppages. We believe that our relations with our employees are good.
Available Information
Our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed with or furnished to the Securities and Exchange Commission pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended, are available free of charge through our website at www.meipharma.com as soon as reasonably practicable after they are electronically filed with, or furnished to, the Securities and Exchange Commission.
Item 1A. Risk Factors
Investment in our securities involves a high degree of risk. You should consider carefully the risks described below, together with other information in this Annual Report and other public filings, before making investment decisions regarding our securities. If any of the following events actually occur, our business, operating results, prospects or financial condition could be materially and adversely affected. This could cause the trading price of our common stock to decline and you may lose all or part of your investment. Moreover, the risks described below are not the only ones that we face. Additional risks not presently known to us or that we currently deem immaterial may also affect our business, operating results, prospects or financial condition.
Risks Related to Our Business
We will need substantial additional funds to progress the clinical trial program for our drug candidates Pracinostat, ME-344 and PWT143, and to develop new compounds. The actual amount of funds we will need will be determined by a number of factors, some of which are beyond our control.
We will need substantial additional funds to progress the clinical trial program for our drug candidates Pracinostat, ME-344 and PWT143 and to develop any additional compounds. The factors which will determine the actual amount of funds that we will need to progress the clinical trial programs for Pracinostat, ME-344 and PWT143 may include, but are not limited to, the following:
| • | the therapeutic indications for use being developed; |
| • | the clinical trial endpoint required to achieve regulatory approval; |
| • | the number of clinical trials required to achieve regulatory approval; |
| • | the number of sites included in the trials; |
| • | the length of time required to enroll suitable patients; |
| • | the number of patients who participate in the trials and the rate that they are recruited; |
| • | the number of treatment cycles patients complete while they are enrolled in the trials; and |
| • | the efficacy and safety profile of the product. |
We have been opportunistic in our efforts to obtain cash, and we expect to continue to evaluate various funding alternatives from time to time. If we obtain additional funding, it may adversely affect the market price of our common stock. If we are unable to obtain additional funds on favorable terms or at all, we may be required
15
to cease or reduce our operations. We may sell additional shares of common stock, and securities exercisable for or convertible into shares of our common stock, to satisfy our capital and operating needs; however, such transactions will be subject to market conditions and there can be no assurance any such transactions will be completed.
The number of shares of our common stock outstanding has increased substantially as a result of our December 2012 private placement, our April 2013 underwritten registered offering, and our October 2013 underwritten registered offering, and some of the purchasers in the private placement beneficially own significant amounts of our common stock.
In December 2012, we completed the private placement of an aggregate of (i) 9,166,665 shares of our common stock and (ii) warrants to purchase an aggregate of 6,416,665 shares of our common stock. In April 2013, we completed an underwritten registered offering of 2,030,000 shares of our common stock. In October 2013, we completed an underwritten registered offering of 4,375,000 shares of our common stock. Some of the purchasers in the private placement beneficially own significant amounts of our common stock and will have a corresponding influence over the outcome of any stockholder vote, including the election of directors and the approval of mergers or other business combination transactions.
Negative U.S. and global economic conditions may pose challenges to our business strategy, which relies on funding from the financial markets or collaborators.
Negative conditions in the U.S. or global economy, including financial markets, may adversely affect our business and the business of current and prospective vendors, licensees and collaborators, and others with whom we do or may conduct business. The duration and severity of these conditions is uncertain. If negative economic conditions occur, we may be unable to secure funding to sustain our operations or to find suitable collaborators to advance our internal programs, even if we achieve positive results from our drug development programs.
We have a limited operating history and are likely to incur operating losses for the foreseeable future.
You should consider our prospects in light of the risks and difficulties frequently encountered by early stage and developmental companies. We were incorporated in December 2000, and have been in operation since May 2002. We have incurred net losses of $123.4 million from our inception through June 30, 2014, including net losses of $27.1 million, $11.2 million and $7.5 million for the years ended June 30, 2014, 2013, and 2012, respectively. We anticipate that we will incur operating losses and negative operating cash flow for the foreseeable future. We have not yet commercialized any drug candidates and cannot be sure that we will ever be able to do so, or that we may ever become profitable.
Our stockholders may not realize a benefit from the purchase of intellectual property commensurate with the associated ownership dilution experienced.
In August 2012, we completed the acquisition of certain assets and intellectual property, including those related to Pracinostat, from S*Bio. Additionally, in May 2011, we completed the acquisition of certain assets (the "Isoflavone Transaction") used in or generated under or in connection with the discovery, development, manufacture and marketing of intellectual property and products based on the field of isoflavonoid technology and on compounds known as isoflavones, including those related to the drug candidate ME-344 (the "Isoflavone-related Assets"), from Novogen.
If we are unable to realize the expected strategic and financial benefits from the purchase of intellectual property, our stockholders will have experienced dilution of their ownership interest as a result of the issuance of shares of common stock to Novogen to acquire the Isoflavone-related Assets, and as a result of the issuance of 195,756 shares of common stock to S*Bio to acquire certain assets and intellectual property, including those related to Pracinostat, without receiving any commensurate benefit. In the asset purchase agreement relating to
16
the acquisition of certain assets and intellectual property from S*Bio, S*Bio made certain representations and warranties regarding its intellectual property rights to such assets; however, its indemnification obligations with respect to such representations and warranties are limited. Similarly, upon consummation of the Isoflavone Transaction, we issued to Novogen 1,000 shares of our Series A Convertible Preferred Stock which were subsequently converted into an aggregate of 804,500 shares of our common stock in November 2012.
Accordingly, we do not expect to be adequately compensated, if at all, for the loss of any such intellectual property rights acquired in the acquisition from S*Bio or in the Isoflavone Transaction.
The results of pre-clinical studies and completed clinical trials are not necessarily predictive of future results, and our current drug candidates may not have favorable results in later studies or trials.
Pre-clinical studies and Phase I and Phase II clinical trials are an expensive and uncertain process that may take years to complete. Pre-clinical studies and Phase I and Phase II clinical trials are not primarily designed to test the efficacy of a drug candidate, but rather to test safety, to study pharmacokinetics and pharmacodynamics, and to understand the drug candidate's side effects at various doses and schedules. Favorable results in early studies or trials may not be repeated in later studies or trials, including ongoing pre-clinical studies and large-scale Phase III clinical trials, and our drug candidates in later-stage trials may fail to show desired safety and efficacy despite having progressed through earlier-stage trials. Unfavorable results from ongoing pre-clinical studies or clinical trials could result in delays, modifications or abandonment of ongoing or future clinical trials, or abandonment of a clinical program. Pre-clinical and clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals or commercialization. Negative or inconclusive results or adverse medical events during a clinical trial could cause a clinical trial to be delayed, repeated or terminated, or a clinical program to be abandoned.
Final approval by regulatory authorities of our drug candidates for commercial use may be delayed, limited or prevented, any of which would adversely affect our ability to generate operating revenues.
We will not generate any operating revenue until we successfully license or commercialize one of our drug candidates. Currently, we have drug candidates at different stages of development, and each will need to successfully complete a number of studies and obtain regulatory approval before potential commercialization.
The preclinical and clinical development, manufacturing, labeling, packaging, storage, recordkeeping, export, marketing and distribution, and other possible activities relating to our drug candidates are subject to extensive regulation by the FDA and other regulatory agencies. Failure to comply with applicable regulatory requirements may, either before or after product approval, subject us to administrative or judicially imposed sanctions that may negatively impact the approval of one or more of our drug candidates or otherwise negatively impact our business.
Neither collaborators nor we are permitted to market a drug candidate in the United States until the particular drug candidate is approved for marketing by the FDA. Specific preclinical data, chemistry, manufacturing and controls data, a proposed clinical trial protocol and other information must be submitted to the FDA as part of an IND, application, and clinical trials may commence only after the IND application becomes effective. To market a new drug in the United States, we must submit to the FDA and obtain FDA approval of an NDA. An NDA must be supported by extensive clinical and preclinical data, as well as extensive information regarding chemistry, manufacturing and controls to demonstrate the safety and effectiveness of the drug candidate.
Obtaining approval of an NDA can be a lengthy, expensive and uncertain process. Regulatory approval of an NDA is not guaranteed. The number and types of preclinical studies and clinical trials that will be required for FDA approval varies depending on the drug candidate, the disease or condition that the drug candidate is designed to target and the regulations applicable to any particular drug candidate. Despite the time and expense
17
exerted in preclinical and clinical studies, failure can occur at any stage, and we could encounter problems that cause us to abandon clinical trials or to repeat or perform additional preclinical studies and clinical trials. The FDA can delay, limit or deny approval of a drug candidate for many reasons, including but not limited to, the following:
| • | a drug candidate may not be deemed adequately safe or effective; |
| • | FDA officials may not find the data from preclinical studies and clinical trials sufficient; |
| • | the FDA's interpretation and our interpretation of data from preclinical studies and clinical trials may differ significantly; |
| • | our or our contractors' or collaborators' failure to comply with applicable FDA and other regulatory requirements, including those identified in other risk factors; |
| • | the FDA may not approve the manufacturing processes or facilities; |
| • | the FDA may change its approval policies or adopt new regulations; or |
| • | the FDA may not accept an NDA or other submission due to, among other reasons, the content or formatting of the submission. |
Our preclinical and clinical data, other information and procedures relating to a drug candidate may not be sufficient to support approval by the FDA or any other U.S. or foreign regulatory authority, or regulatory interpretation of these data and procedures may be unfavorable. Our business and reputation may be harmed by any failure or significant delay in receiving regulatory approval for the sale of any drugs resulting from our drug candidates. As a result, we cannot predict when or whether regulatory approval will be obtained for any drug we develop.
Additionally, any of the following factors may serve to delay, limit or prevent the final approval by regulatory authorities of our drug candidates for commercial use, including, but not limited to:
| • | Pracinostat, ME-344 and PWT143 are in the early-to-intermediate stages of development, and we will need to conduct significant clinical testing to demonstrate safety and efficacy of these drug candidates before applications for marketing can be filed with the FDA, or with the regulatory authorities of other countries; |
| • | development and testing of product formulation, including identification of suitable excipients, or chemical additives intended to facilitate delivery of our drug candidates; |
| • | it may take us many years to complete the testing of our drug candidates, and failure can occur at any stage of this process; and |
| • | negative or inconclusive results or adverse medical events during a clinical trial could cause us to delay or terminate our development efforts. |
The successful development of any of these drug candidates is uncertain and, accordingly, we may never commercialize any of these drug candidates or generate revenue.
Even if we receive regulatory approval to commercialize our drug candidates, our ability to generate revenues from any resulting products will be subject to a variety of risks, many of which are out of our control.
Even if our drug candidates obtain regulatory approval, resulting products may not gain market acceptance among physicians, patients, healthcare payers or the medical community. We believe that the degree of market acceptance and our ability to generate revenues from such products will depend on a number of factors, including, but not limited to, the following:
| • | timing of market introduction of our drugs and competitive drugs; |
| • | actual and perceived efficacy and safety of our drug candidates; |
18
| • | prevalence and severity of any side effects; |
| • | potential or perceived advantages or disadvantages over alternative treatments; |
| • | strength of sales, marketing and distribution support; |
| • | price of our future products, both in absolute terms and relative to alternative treatments; |
| • | the effect of current and future healthcare laws on our drug candidates; and |
| • | availability of coverage and reimbursement from government and other third-party payers. |
If any of our drugs are approved and fail to achieve market acceptance, we may not be able to generate significant revenue to achieve or sustain profitability.
If we receive regulatory approval to market Pracinostat, we anticipate that its label may be limited to use in combination with a hypomethylating agent, such as azacitidine, the cost and availability of which are not in our control.
Our current clinical strategy is to develop Pracinostat for use in combination with an approved hypomethylating agent such as azacitidine. If we receive regulatory approval to market Pracinostat, we anticipate that its label may be limited to use in combination with a hypomethylating agent such as azacitidine. If approved hypomethylating agents are not available at an acceptable cost, or at all, this could prevent us from marketing Pracinostat, which could have a material adverse impact on our business and operations.
We may not be able to establish the contractual arrangements necessary to develop, market and distribute our product candidates.
A key part of our business plan is to establish contractual relationships with third parties to package, market and distribute our product candidates. There is no assurance that we will be able to negotiate commercially acceptable licensing or other agreements for the future exploitation of our drug product candidates, including continued clinical development, manufacture or marketing. If we are unable to successfully contract for these services, or if arrangements for these services are terminated, we may have to delay our commercialization program which will adversely affect our ability to generate operating revenues.
Our commercial opportunity will be reduced or eliminated if competitors develop and market products that are more effective, have fewer side effects or are less expensive than our drug candidates.
The development of drug candidates is highly competitive. A number of other companies have products or drug candidates that have either been approved or are in various stages of pre-clinical or clinical development that are intended for the same therapeutic indications for which our drug candidates are being developed. Some of these potential competing drugs are further advanced in development than our drug candidates and may be commercialized sooner. Even if we are successful in developing effective drugs, our compounds may not compete successfully with products produced by our competitors.
Our competitors include pharmaceutical companies and biotechnology companies, as well as universities and public and private research institutions. In addition, companies active in different but related fields represent substantial competition for us. Many of our competitors developing oncology drugs have significantly greater capital resources, larger research and development staffs and facilities and greater experience in drug development, regulation, manufacturing and marketing than we do. These organizations also compete with us and our service providers, to recruit qualified personnel, and with us to attract partners for joint ventures and to license technologies that are competitive with us. As a result, our competitors may be able to more easily develop technologies and products that would render our technologies or our drug candidates obsolete or non-competitive.
19
We rely on third parties to conduct our clinical trials and many of our pre-clinical studies. If those parties do not successfully carry out their contractual duties or meet expected deadlines, our drug candidates may not advance in a timely manner or at all.
In the course of our pre-clinical testing and clinical trials, we rely on third parties, including laboratories, investigators, clinical contract research organizations, or CROs, and manufacturers, to perform critical services for us. For example, we rely on third parties to conduct our clinical trials and many of our pre-clinical studies. CROs are responsible for many aspects of the trials, including finding and enrolling subjects for testing and administering the trials. Although we rely on these third parties to conduct our clinical trials, we are responsible for ensuring that each of our clinical trials is conducted in accordance with its investigational plan and protocol. Moreover, the FDA and foreign regulatory authorities require us to comply with regulations and standards, commonly referred to as good clinical practices, or GCPs, for conducting, monitoring, recording, and reporting the results of clinical trials to ensure that the data and results are scientifically credible and accurate, and that the trial subjects are adequately informed of the potential risks of participating in clinical trials. Our reliance on third parties does not relieve us of these responsibilities and requirements. These third parties may not be available when we need them or, if they are available, may not comply with all regulatory and contractual requirements or may not otherwise perform their services in a timely or acceptable manner, and we may need to enter into new arrangements with alternative third parties and our clinical trials may be extended, delayed or terminated. These independent third parties may also have relationships with other commercial entities, some of which may compete with us. In addition, if such third parties fail to perform their obligations in compliance with our clinical trial protocols or GCPs, our clinical trials may not meet regulatory requirements or may need to be repeated. As a result of our dependence on third parties, we may face delays or failures outside of our direct control. These risks also apply to the development activities of collaborators, and we do not control their research and development, clinical trial or regulatory activities.
We have no direct control over the cost of manufacturing our drug candidates. Increases in the cost of manufacturing our drug candidates would increase our costs of conducting clinical trials and could adversely affect our future profitability.
We do not intend to manufacture our drug product candidates ourselves, and we will rely on third parties for our drug supplies both for clinical trials and for commercial quantities in the future. We have taken the strategic decision not to manufacture active pharmaceutical ingredients ("API") for our drug candidates, as these can be more economically supplied by third parties with particular expertise in this area. We have identified contract facilities that are registered with the FDA, have a track record of large scale API manufacture, and have already invested in capital and equipment. We have no direct control over the cost of manufacturing our product candidates. If the cost of manufacturing increases, or if the cost of the materials used increases, these costs will be passed on to us, making the cost of conducting clinical trials more expensive. Increases in manufacturing costs could adversely affect our future profitability if we are unable to pass all of the increased costs along to our customers. We also rely on the contract manufacturers to comply with FDA regulatory requirements for good manufacturing practices.
We rely on acquisitions or licenses from third parties to expand our pipeline of drug candidates.
We are not presently engaged in drug discovery activities. In order to expand our pipeline of drug candidates for future development, we may need to purchase or in-license any such drug candidates. However, we may not be able to purchase or in-license future drug candidates from third parties on favorable terms, or at all.
We face a risk of product liability claims and may not be able to obtain adequate insurance.
Our business exposes us to the risk of product liability claims. This risk is inherent in the manufacturing, testing and marketing of human therapeutic products. Our product liability insurance coverage is subject to deductibles and coverage limitations. We may not be able to obtain or maintain adequate protection
20
against potential liabilities, or claims may exceed our insurance limits. If we cannot or do not sufficiently insure against potential product liability claims, we may be exposed to significant liabilities, which may materially and adversely affect our business development and commercialization efforts.
Our efforts will be seriously jeopardized if we are unable to retain and attract key employees.
Our success depends on the continued contributions of our principal management, development and scientific personnel. We face competition for such personnel, and we believe that risks and uncertainties related to our business, including the timing and risk associated with research and development, our available and anticipated cash resources, and the volatility of our stock price, may impact our ability to hire and retain key and other personnel. The loss of services of our Chief Executive Officer or other key employees could adversely impact our operations and ability to generate or raise additional capital.
Laws, rules and regulations relating to public companies may be costly and impact our ability to attract and retain directors and executive officers.
Laws and regulations affecting public companies, including rules adopted by the SEC and by Nasdaq, may result in increased costs to us. These laws, rules and regulations could make it more difficult or costly for us to obtain certain types of insurance, including director and officer liability insurance, and we may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. The impact of these events could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, on our board committees or as executive officers. We cannot estimate accurately the amount or timing of additional costs we may incur to respond to these laws, rules and regulations.
Risks Relating to Our Intellectual Property
Our commercial success is dependent, in part, on obtaining and maintaining patent protection and preserving trade secrets, which cannot be guaranteed.
Patent protection and trade secret protection are important to our business and our future will depend, in part on our ability to maintain trade secret protection, obtain patents and operate without infringing the proprietary rights of others both in the United States and abroad. Litigation or other legal proceedings may be necessary to defend against claims of infringement, to enforce our patents or to protect our trade secrets. Such litigation could result in substantial costs and diversion of our management's attention.
The patent positions of pharmaceutical and biotechnology companies can be highly uncertain and involve complex legal and factual questions. In August 2012, we acquired patents and patent applications related to Pracinostat from S*Bio. In September 2013, we acquired patents and patent applications related to PWT143 from Pathway Therapeutics, Inc. Additionally, prior to the Isoflavone Transaction, Novogen had applied for patents in a number of countries with respect to the use of their isoflavone compounds, including ME-344. We acquired both issued patents and pending patent applications from Novogen in relation to our Isoflavone-based compounds, which we previously licensed from Novogen. The patent applications may not proceed to grant or may be amended to reduce the scope of protection of any patent granted. The applications and patents may also be opposed or challenged by third parties. Our commercial success will depend, in part, on our ability to obtain and maintain effective patent protection for our compounds and their use in treating, preventing, or curing cancer, and to successfully defend patent rights in those technologies against third-party challenges. As patent applications in the United States are maintained in secrecy until published or issued and as publication of discoveries in the scientific or patent literature often lag behind the actual discoveries, we cannot be certain that we, S*Bio, Novogen, or Pathway Therapeutics, Inc. were the first to make the inventions covered by the pending patent applications or issued patents referred to above or that we or they were the first to file patent applications for such inventions. Additionally, the breadth of claims allowed in biotechnology and pharmaceutical patents or their enforceability cannot be predicted. We cannot be sure that, should any patents issue, we will be provided
21
with adequate protection against potentially competitive products. Furthermore, we cannot be sure that should patents issue, they will be of commercial value to us, or that private parties, including competitors, will not successfully challenge our patents or circumvent our patent position in the United States or abroad.
Claims by other companies that we infringe on their proprietary technology may result in liability for damages or stop our development and commercialization efforts.
The pharmaceutical industry is highly competitive and patents have been applied for by, and issued to, other parties relating to products competitive with the compounds that we have acquired. Therefore, Pracinostat, ME-344 and PWT143 and any other drug candidates may give rise to claims that they infringe the patents or proprietary rights of other parties existing now and in the future.
Furthermore, to the extent that we or our consultants or research collaborators use intellectual property owned by others in work performed for us, disputes may also arise as to the rights in such intellectual property or in resulting know-how and inventions. An adverse claim could subject us to significant liabilities to such other parties and/or require disputed rights to be licensed from such other parties.
We have contracted formulation development and manufacturing process development work for our product candidates. This process has identified a number of excipients, or additives to improve drug delivery, which may be used in the formulations. Excipients, among other things, perform the function of a carrier of the active drug ingredient. Some of these identified excipients or carriers may be included in third party patents in some countries. We intend to seek a license if we decide to use a patented excipient in the marketed product or we may choose one of those excipients that does not have a license requirement.
We cannot be sure that any license required under any such patents or proprietary rights would be made available on terms acceptable to us, if at all. If we do not obtain such licenses, we may encounter delays in product market introductions, or may find that the development, manufacture or sale of products requiring such licenses may be precluded.
We may be subject to substantial costs stemming from our defense against third-party intellectual property infringement claims.
Third parties may assert that we are using their proprietary information without authorization. Third parties may also have or obtain patents and may claim that technologies licensed to or used by us infringe their patents. If we are required to defend patent infringement actions brought by third parties, or if we sue to protect our own patent rights, we may be required to pay substantial litigation costs and managerial attention may be diverted from business operations even if the outcome is not adverse to us. In addition, any legal action that seeks damages or an injunction to stop us from carrying on our commercial activities relating to the affected technologies could subject us to monetary liability and require us or any third party licensors to obtain a license to continue to use the affected technologies. We cannot predict whether we would prevail in any of these types of actions or that any required license would be made available on commercially acceptable terms or at all.
Risks Related to Securities Markets and Investment in Our Stock
The trading price of the shares of our common stock has been and may continue to be highly volatile and could decline in value and we may incur significant costs from class action litigation.
The trading price of our common stock could be highly volatile in response to various factors, many of which are beyond our control, including, but not limited to, the following:
| • | failure to successfully develop our lead drug candidate, Pracinostat; |
| • | design, results and timing of clinical trials and preclinical studies; |
| • | announcements of technological innovations by us or our competitors; |
22
| • | new products introduced or announced by us or our competitors; |
| • | changes in financial estimates by securities analysts; |
| • | actual or anticipated variations in operating results; |
| • | expiration or termination of licenses, research contracts or other collaboration agreements; |
| • | conditions or trends in the regulatory climate and the biotechnology, pharmaceutical and genomics industries; |
| • | instability in the stock market as a result of current or future domestic and global events; |
| • | changes in the market valuations of similar companies; |
| • | the liquidity of any market for our securities; and |
| • | threatened or actual delisting of our common stock from a national stock exchange. |
Equity markets in general, and the market for biotechnology and life sciences companies in particular, have experienced substantial price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of companies traded in those markets. In addition, changes in economic conditions in the U.S., Europe or globally, particularly in the context of current global events, could impact upon our ability to grow profitably. Adverse economic changes are outside our control and may result in material adverse impacts on our business or our results of operations. These broad market and industry factors may materially affect the market price of shares of our common stock, regardless of our development and operating performance. In the past, following periods of volatility in the market price of a company's securities, securities class-action litigation has often been instituted against that company. Such litigation, if instituted against us, could cause us to incur substantial costs and divert management's attention and resources.
Future sales of our common stock, including common stock issued upon exercise of outstanding warrants or options, may depress the market price of our common stock and cause stockholders to experience dilution.
The market price of our common stock could decline as a result of sales of substantial amounts of our common stock in the public market, including upon exercise of outstanding warrants or stock options, or upon issuance of shares related to restricted stock units, and any subsequent sales of such shares. As of June 30, 2014, we had outstanding (i) warrants issued in our December 2012 private placement exercisable to purchase 4,369,794 shares of Common Stock at an exercise price of $3.12 per share, which expire in December 2017; (ii) warrants issued in our May 2012 rights offering exercisable to purchase 315,484 shares of Common Stock at an exercise price of $7.14, which expire in May 2017; and (iii) Series A warrants issued in our May 2011 private placement exercisable to purchase 215,721 shares of Common Stock at an exercise price of $6.00, which expire in November 2016. We also had outstanding options to purchase 1,194,854 shares of common stock and restricted stock units representing the right to receive 400,000 shares of common stock. We may seek additional capital through one or more additional equity transactions in the future; however, such transactions will be subject to market conditions and there can be no assurance any such transactions will be completed. If we sell shares in the future, the prices at which we sell these future shares will vary, and these variations may be significant. Stockholders will experience significant dilution if we sell these future shares at prices significantly below the price at which previous shareholders invested.
Because we do not intend to pay, and have not paid, any cash dividends on our shares of common stock, our stockholders will not be able to receive a return on their shares unless the value of our common stock appreciates and they sell their shares.
We have never paid or declared any cash dividends on our common stock, and we intend to retain any future earnings to finance the development and expansion of our business. We do not anticipate paying any cash dividends on our common stock in the foreseeable future. Therefore, our stockholders will not be able to receive a return on their investment unless the value of our common stock appreciates and they sell their shares.
23
We will have broad discretion over the use of the net proceeds from any exercise of outstanding warrants and options.
We will have broad discretion to use the net proceeds to us upon any exercise of outstanding warrants and options, and investors in our stock will be relying on the judgment of our board of directors and management regarding the application of these proceeds. Although we expect to use a substantial portion of the net proceeds from any exercise of the warrants and options for general corporate purposes and progression of our clinical trial programs, we have not allocated these net proceeds for specific purposes.
We are authorized to issue blank check preferred stock, which could adversely affect the holders of our common stock.
Our restated certificate of incorporation allows us to issue blank check preferred stock with rights potentially senior to those of our common stock without any further vote or action by the holders of our common stock. The issuance of a class of preferred stock could decrease the amount of earnings and assets available for distribution to the holders of our common stock or could adversely affect the rights and powers, including voting rights, of such holders. In certain circumstances, such issuance could have the effect of decreasing the market price of our shares, or making a change in control of the Company more difficult.
Our executive officers and directors may sell shares of their stock, and these sales could adversely affect our stock price.
Sales of our stock by our executive officers and directors, or the perception that such sales may occur, could adversely affect the market price of our stock. Our executive officers and directors may sell stock in the future, either as part, or outside, of trading plans under Rule 10b5-1 under the Securities Exchange Act of 1934, as amended.
Item 1B. Unresolved Staff Comments
None.
Item 2. Properties
We have leased approximately 8,800 square feet of office space, located at 11975 El Camino Real, Suite 101, San Diego, California 92130. The location houses the Company's executive and administrative offices. The lease commenced in July 2010 and expires in June 2015. The monthly rental rate is approximately $25,000 over the remaining lease term, plus a pro rata share of certain building expenses. We believe these facilities will adequately meet our office needs for the foreseeable future.
Item 3. Legal Proceedings
None.
Item 4. Mine Safety Disclosures
Not applicable.
24
PART II
| Item 5. | Market for the Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Our common stock is listed on the Nasdaq Capital Market under the symbol "MEIP". The following table sets forth, for the periods indicated, the high and low sale prices of our common stock for each quarterly period within the two most recent fiscal years.
| Prices | ||||||||
| High $ | Low $ | |||||||
Year Ended June 30, 2014 | ||||||||
First Quarter | 12.45 | 6.52 | ||||||
Second Quarter | 11.31 | 7.30 | ||||||
Third Quarter | 13.98 | 7.71 | ||||||
Fourth Quarter | 11.50 | 5.51 | ||||||
Year Ended June 30, 2013 | ||||||||
First Quarter | 4.80 | 1.98 | ||||||
Second Quarter | 13.18 | 2.10 | ||||||
Third Quarter | 9.65 | 4.37 | ||||||
Fourth Quarter | 9.40 | 6.89 | ||||||
Holders
As of September 4, 2014, there were 21,607,296 shares of our common stock outstanding and 3,071 holders of record of our common stock. This number was derived from our shareholder records and does not include beneficial owners of our common stock whose shares are held in the name of various dealers, clearing agencies, banks, brokers and other fiduciaries.
For a discussion of outstanding warrants and other securities exercisable for or convertible into shares of our common stock, see Note 6 under Item 8 in this Annual Report.
Dividends
We have never declared or paid any cash dividends on our common stock and do not anticipate paying any cash dividends in the foreseeable future. We currently intend to retain all available funds and future earnings, if any, to support operations and finance the growth and development of our business. Any future determination related to our dividend policy will be made at the discretion of our board of directors.
25
Securities authorized for issuance under equity compensation plans
The table below shows, as of June 30, 2014, information for all equity compensation plans previously approved by stockholders and for all compensation plans not previously approved by stockholders.
Plan Category | Number of securities to be issued upon exercise of outstanding options, warrants and rights (a) | Weighted- average exercise price of outstanding options, warrants and rights (b) | Number
of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) ( c) | |||||||||
Equity compensation plans approved by security holders (1) | 1,128,521 | $ | 7.71 | 657,479 | ||||||||
Equity compensation plans not approved by security holders (2) | 66,333 | 14.91 | - | |||||||||
|
|
|
|
|
| |||||||
Total | 1,194,854 | $ | 8.11 | 657,479 | ||||||||
|
|
|
|
|
| |||||||
| (1) | Consists of stock options issuable under the MEI Pharma, Inc. Amended and Restated 2008 Stock Omnibus Equity Compensation Plan ("the Plan"), under which 2,186,000 shares of common stock are authorized for issuance. The Plan provides for the grant of options and/or other stock-based or stock-denominated awards to the Company's non-employee directors, officers, employees and advisors. |
| (2) | The Company granted options to purchase 36,730 shares of common stock to Dr. Daniel Gold under the terms of his employment agreement. The Company granted options to Dr. Gold to purchase 18,365 shares of common stock upon his appointment as President and Chief Executive Officer on April 23, 2010 and also granted options to purchase 18,365 shares of common stock on June 7, 2010. Of these two tranches of options, 25% vested in April 2011, and, thereafter, the remaining 75% of Dr. Gold's options vested in equal monthly installments over the following thirty-six months. The Company granted options to purchase 29,603 shares of common stock to Dr. Robert Mass on June 1, 2011 under the terms of his employment agreement. Of Dr. Mass's options, 25% vested in June 2012 and, thereafter, the remaining 75% of Dr. Mass's options vest in equal monthly installments over the following thirty-six months. These option grants took place outside of the Plan. |
Performance graph
The graph below compares the cumulative five-year total return on our common stock from July 1, 2009, through June 30, 2014, to the cumulative total return over such period for (i) the NASDAQ Composite Index and (ii) the NASDAQ Biotechnology Index. The graph assumes the investment of $100 on July 1, 2009, with the reinvestment of dividends, although dividends have not been declared on our common stock, and is calculated according to the Securities and Exchange Commission's methodology. We caution that the stock price performance shown in the graph may not be indicative of future stock price performance. The graph, including each of the graph lines, was provided by Zacks Investment Research, Inc.
This information, including the graph below, is not deemed to be "soliciting material" or to be "filed" with the Securities and Exchange Commission, or subject to the Securities and Exchange Commission's proxy rules, other than as provided in such rules, or to the liabilities of Section 18 of the Securities Exchange Act of 1934, and shall not be deemed incorporated by reference into any prior or subsequent filing by us under the Securities Act of 1933 or the Securities Exchange Act of 1934, except to the extent that we specifically incorporate it by reference into any such filing.
26
COMPARISON OF FIVE-YEAR CUMULATIVE TOTAL RETURN
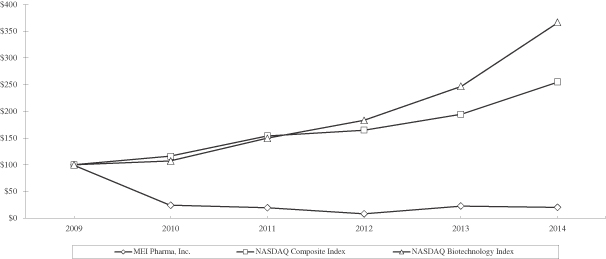
| Item 6. | Selected Financial Data |
The following Selected Financial Data should be read in conjunction with "Item 7. Management's Discussion and Analysis of Financial Condition and Results of Operations" and "Item 8. Financial Statements and Supplementary Data" included below in this Annual Report.
The following table presents our selected historical condensed financial data. The condensed statements of operations data for fiscal years ended June 30, 2014, 2013 and 2012 and the condensed balance sheet data as of June 30, 2014 and 2013 are derived from our audited financial statements included elsewhere in this Annual Report. The condensed statements of operations data for the fiscal years ended June 30, 2011 and 2010 and the condensed balance sheet data as of June 30, 2012, 2011 and 2010 are derived from our audited financial statements that are not included in this Form 10-K.
27
The Company's former wholly-owned subsidiary, Marshall Edwards Pty Ltd ("MEPL"), was legally dissolved in April 2012. As MEPL was the Company's only subsidiary, the financial statements as of and for the year ended, June 30, 2013 are no longer consolidated. In December 2012, the Company effected a 1-for-6 reverse stock split (the "2012 Reverse Stock Split") of the Company's common stock. As a result of the 2012 Reverse Stock Split, every six shares of the Company's issued and outstanding common stock were combined into one share of common stock. The 2012 Reverse Stock Split did not change the number of authorized shares of the Company's common stock, nor the common stock par value. All financial data and share information is presented on an as-adjusted basis to give effect to the 2012 Reverse Stock Split.
| Years Ended June 30, | ||||||||||||||||||||
| 2014 | 2013 | 2012 | 2011 | 2010 | ||||||||||||||||
| (In thousands, except share and per share data) | ||||||||||||||||||||
Statement of Operations Data: | ||||||||||||||||||||
Operating expenses: | ||||||||||||||||||||
Research and development | $ | (19,331 | ) | $ | (6,084 | ) | $ | (4,915 | ) | $ | (2,115 | ) | $ | (4,031 | ) | |||||
License fees | - | - | - | - | (1,500 | ) | ||||||||||||||
General and administrative | (7,897 | ) | (5,138 | ) | (3,479 | ) | (4,336 | ) | (2,448 | ) | ||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Total operating expenses | (27,228 | ) | (11,222 | ) | (8,394 | ) | (6,451 | ) | (7,979 | ) | ||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Loss from operations | (27,228 | ) | (11,222 | ) | (8,394 | ) | (6,451 | ) | (7,979 | ) | ||||||||||
Other income (expense), net | 80 | 36 | 871 | (330 | ) | 83 | ||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Net loss | $ | (27,148 | ) | $ | (11,186 | ) | $ | (7,523 | ) | $ | (6,781 | ) | $ | (7,896 | ) | |||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Net loss per share, basic and diluted | $ | (1.35 | ) | $ | (1.10 | ) | $ | (3.35 | ) | $ | (5.32 | ) | $ | (6.45 | ) | |||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Shares used to calculate net loss per share, basic and diluted | 20,061,387 | 10,160,835 | 2,247,709 | 1,273,901 | 1,224,387 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
| As of June 30, | ||||||||||||||||||||
| 2014 | 2013 | 2012 | 2011 | 2010 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
Balance Sheet Data: | ||||||||||||||||||||
Cash, cash equivalents and short-term investments | $ | 48,793 | $ | 35,573 | $ | 6,202 | $ | 3,858 | $ | 9,031 | ||||||||||
Total assets | 49,808 | 36,547 | 6,373 | 4,168 | 9,136 | |||||||||||||||
Total liabilities | 4,616 | 1,675 | 1,774 | 2,374 | 1,755 | |||||||||||||||
Accumulated deficit | (123,445 | ) | (96,297 | ) | (85,111 | ) | (77,588 | ) | (70,807 | ) | ||||||||||
Total stockholders' equity | 45,192 | 34,872 | 4,599 | 1,794 | 7,381 | |||||||||||||||
| Item 7. | Management's Discussion and Analysis of Financial Condition and Results of Operations. |
The following discussion and analysis should be read in conjunction with "Item 8. Financial Statements and Supplementary Data" included below in this Annual Report. Operating results are not necessarily indicative of results that may occur in future periods.
This discussion and analysis contains forward-looking statements that involve a number of risks, uncertainties and assumptions. Actual results may differ materially from those anticipated in the forward-looking statements as a result of many factors including, but not limited to, those set forth under "Cautionary Statement About Forward-Looking Statements" and "Risk Factors" in Item 1A. included above in this Annual Report. All forward-looking statements included in this Annual Report are based on the information available to us as of the time we file this Annual Report, and except as required by law, we undertake no obligation to update publicly or revise any forward-looking statements.
28
Overview and Recent Developments
Our business purpose is the development of drugs for the treatment of cancer. We are principally focused on the clinical development of our lead drug candidate, Pracinostat, which we are currently investigating in three Phase II clinical trials. Pracinostat is an orally available histone deacetylase (HDAC) inhibitor that is being developed for advanced hematologic diseases such as myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). In August 2012, we completed the acquisition of certain assets and intellectual property, including those related to Pracinostat, from S*Bio Pte Ltd ("S*Bio"). Our clinical development pipeline also includes our isoflavone-based mitochondrial inhibitor drug candidate, ME-344. Results from a Phase I clinical trial of ME-344 in patients with refractory solid tumors were presented in October 2013. A Phase Ib trial in patients with small cell lung and ovarian cancers is ongoing. In September 2013, we acquired PWT143, an oral inhibitor of phosphatidylinositide 3-kinase (PI3K) delta, a molecular target that has been shown to play a critical role in the proliferation and survival of hematologic cancer cells. We expect to complete the pre-clinical work required to support the filing of an IND application for PWT143 by the end of calendar year 2014.
We have an accumulated deficit of $123.4 million as of June 30, 2014, and may incur substantial net losses in the future as we advance our research and development programs. We have not generated any revenues from operations since inception and we expect to incur operating losses and generate negative cash flows from operations for the foreseeable future. We may need additional financing to fund our operations in the future, including the continued development of our lead drug candidates.
Clinical Developments
Pracinostat
In August 2014, we completed enrollment in a randomized, double-blind, placebo-controlled Phase II clinical trial of Pracinostat in combination with azacitidine in intermediate-2 or high-risk patients with previously untreated MDS. The multicenter trial enrolled 108 patients with a one-to-one randomization. We expect to unblind the study after a six month follow-up period and report topline data by the first quarter of 2015. The primary endpoint of the study is CR. Secondary endpoints include overall response rate, hematologic improvement, duration of response, progression-free survival, rate of leukemic transformation, overall survival and safety.
In addition, two open-label Phase II trials of Pracinostat are ongoing: one in combination with azacitidine or decitabine in patients with MDS who either failed to respond or maintain a response to their hypomethylating agent alone and the other in combination with azacitidine in elderly patients with newly diagnosed AML who are not suited for intensive chemotherapy. In June 2014, we announced preliminary data from the front line AML trial. Of the first nine patients enrolled in the study, three achieved a CR or CRi, each following one or two treatment cycles. In addition, three patients achieved a PR or PRi after their initial or second treatment evaluation, for an overall response rate of 67%. We expect to report additional data from this open-label study at a scientific conference in December 2014.
In February 2014, the FDA granted orphan drug designation to Pracinostat for the treatment of AML. The designation provides orphan status to drugs defined by the FDA as those intended for the safe and effective treatment, diagnosis or prevention of rare diseases that affect fewer than 200,000 people in the U.S. Orphan designation qualifies us for certain development incentives, including tax credits for qualified clinical testing, prescription drug user fee exemptions and seven-year marketing exclusivity upon FDA approval.
ME-344
In October 2013, results from our first-in-human, single-agent Phase I clinical trial of ME-344 in patients with refractory solid tumors were presented at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics. The results indicated that eight of 21 evaluable patients (38%) treated with ME-344 achieved stable disease or better, including five who experienced progression-free
29
survival that was at least twice the duration of their last prior treatment before entry into the study. In addition, one of these patients, a heavily pre-treated patient with small cell lung cancer, achieved a confirmed partial response. This patient remains on study and has continued weekly dosing for more than two years (as of September 2014). ME-344 was generally well tolerated at doses equal to or less than 10 mg/kg delivered on a weekly schedule for extended durations. Dose limiting toxicities were observed at both the 15 mg/kg and 20 mg/kg dose levels, consisting primarily of Grade 3 peripheral neuropathy. Other medically significant adverse events observed in single patients included angina and QTc prolongation at the 10 mg/kg dose.
In May 2014, we initiated a Phase Ib clinical trial of ME-344 in combination with Hycamtin ® (topotecan) in patients with solid tumors, including small cell lung and ovarian cancers. The Phase Ib study is evaluating the safety and tolerability of intravenous ME-344 in combination with topotecan, a chemotherapy approved by the FDA for the treatment of small cell lung, ovarian and cervical cancers. The initial stage of the study will establish the MTD of ME-344 in combination with topotecan in up to 24 patients. Once the MTD has been determined, the study will enroll an additional 40 patients into two cohorts: relapsed/ refractory small cell lung cancer and platinum-refractory ovarian cancer.
Equity Transactions
Underwritten Registered Offerings
In October 2013, we completed an underwritten registered offering of 4,375,000 shares of our common stock at a price per share of $8.00 pursuant to a "shelf" registration statement previously filed and declared effective by the Securities and Exchange Commission. We received net proceeds of $32.7 million associated with the offering.
In April 2013, we completed an underwritten registered offering of 2,030,000 shares of our common stock at a price per share of $7.50 pursuant to a "shelf" registration statement previously filed and declared effective by the Securities and Exchange Commission. We received net proceeds of $14.2 million associated with the offering.
December 2012 Private Placement
In December 2012, we completed the sale (the "December 2012 private placement") of 9,166,665 shares (the "Initial Shares") of common stock and warrants (the "Warrants") to purchase an additional 6,416,665 shares (the "Warrant Shares" and, together with the Initial Shares, the "Shares") of common stock for an aggregate offering price of $27.5 million, pursuant to the terms a Securities Purchase Agreement, dated November 4, 2012, between us and certain accredited investors identified therein.
Reverse Stock Split
On December 18, 2012, we filed a Certificate of Amendment to our Restated Certificate of Incorporation in order to effect a 1-for-6 reverse stock split of our common stock effective on December 18, 2012. As a result of the 2012 Reverse Stock Split, every six shares of our issued and outstanding common stock were combined into one share of common stock. The 2012 Reverse Stock Split did not change the number of authorized shares of common stock. All financial data and share information in this Annual Report on Form 10-K has been presented on an as-adjusted basis to give effect to the 2012 Reverse Stock Split.
S*Bio Asset Purchase
In August 2012, we entered into a definitive asset purchase agreement with S*Bio, pursuant to which we agreed to acquire certain assets comprised of intellectual property and technology including rights to Pracinostat, in exchange for $500,000 of common stock. On August 22, 2012, we completed the asset purchase and issued 195,756 shares of common stock to S*Bio. We also agreed to make certain milestone payments to S*Bio based on the achievement of certain clinical, regulatory and net sales-based milestones, as well as to make
30
certain contingent earnout payments to S*Bio. Milestone payments will be made to S*Bio up to an aggregate amount of $75.2 million if certain U.S., E.U. and Japanese regulatory approvals are obtained and if certain net sales thresholds are met in North America, the E.U. and Japan. We will pay $500,000 of the first milestone payment in shares of common stock. S*Bio will be entitled to receive certain contingent earnout payments based on certain worldwide net sales thresholds on a product-by-product and country-by-country basis.
Rights Offering
In May 2012, we completed a rights offering ("Rights Offering") pursuant to which we distributed, at no charge, to holders of record as of March 30, 2012, subscription rights (the "Rights") to purchase up to 17,129,361 units for an aggregate purchase price of up to $7.6 million. The subscription period for the Rights Offering expired on May 11, 2012. Each unit consisted of 0.0833 shares of common stock and a warrant representing the right to purchase 0.04167 shares of our common stock at an exercise price of $7.14 per share. The exercise of one Right entitled holders to purchase one unit at a subscription price of $0.445 per unit, which represented the subscription price of $5.34 per whole share. Eligible participants in the Rights Offering exercised Rights to purchase an aggregate of 11,660,606 units; accordingly, we issued 971,700 shares of common stock and warrants to purchase an additional 485,857 shares of our common stock. The warrants are exercisable for a five-year period beginning on May 11, 2012. We received net proceeds of $4.8 million associated with the Rights Offering.
Waiver Agreement
In December 2012, the Company entered into an agreement (the "Waiver Agreement") with Novogen and Novogen Research Pty Limited, a wholly-owned subsidiary of Novogen (together, the "Novogen Parties"), Graham Kelly, an individual, and Andrew Heaton, an individual, pursuant to which the Company granted a limited waiver with respect to certain non-compete provisions contained in the Asset Purchase Agreement dated as of December 20, 2010, between the Company and the Novogen Parties. In consideration of the Company's grant of the limited waiver, upon the execution of the Waiver Agreement, Novogen surrendered to the Company for cancellation warrants held by Novogen for the purchase of 166,666 shares of Common Stock.
Securities Subscription Agreements – Novogen
In September 2011, we entered into a Securities Subscription Agreement with Novogen, pursuant to which we sold to Novogen 222,222 shares of our common stock, at a purchase price of $9.00 per share, for proceeds of $2.0 million. The offering closed on September 29, 2011. In December 2011, we entered into a Securities Subscription Agreement with Novogen, pursuant to which we sold to Novogen 323,625 shares of our common stock, at a purchase price of $6.18 per share, for proceeds of $2.0 million. The offering closed on December 29, 2011.
Corporate Developments
Board of Directors and Management
In April 2014, we announced the appointment of David Urso as Senior Vice President of Corporate Development and General Counsel. Mr. Urso joined MEI Pharma with more than two decades of experience in the life science industry, most recently as Chief Operating Officer and General Counsel at Tioga Pharmaceuticals, a company he co-founded in 2005 as a Principal at Forward Ventures.
Corporate Bylaws
In March 2014, the Board of Directors amended and restated the Company's bylaws. A new Section 3 of Article II was added to the bylaws to establish advance notice requirements for stockholders who propose nominations for the election of directors or propose other business to be brought before any annual or special meeting of stockholders of the Company.
31
Critical Accounting Policies and Management Estimates
Management's discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities, expenses and related disclosures. Actual results could differ from those estimates. We believe the following accounting policies to be critical to the judgments and estimates used in the preparation of our financial statements.
Clinical Trials Expenses
Estimates have been used in determining the expenses under certain clinical trial contracts where services have been performed but not yet invoiced. Generally, the costs associated with clinical trial contracts are based on the number of patients in each trial, the service contracts associated with clinical sites, service providers and drug development contracts. The length of time before actual amounts can be determined will vary, and are therefore estimated based on enrollment of subjects, the length of drug administration cycles, the completion of trials and other events.
Share-Based Compensation
Share-based compensation expense for employees and directors is recognized in the statement of operations based on estimated amounts, including the grant date fair value and the expected service period. For stock options, we estimate the grant date fair value using a binomial valuation model, which requires the use of multiple subjective inputs including estimated future volatility, expected forfeitures and the expected term of the awards. We estimate our expected future volatility based on our stock's historical price volatility. Our stock's future volatility may differ from our estimated volatility at the grant date. For restricted stock unit (RSU) equity awards, we estimate the grant date fair value using the Company's closing stock price on the date of grant. Share-based compensation recorded in our statement of operations is based on awards expected to ultimately vest and has been reduced for estimated forfeitures. Our estimated forfeiture rates may differ from actual forfeiture rates which would affect the amount of expense recognized during the period. We recognize the value of the awards over the awards' requisite service or performance periods. The requisite service period is generally the time over which our share-based awards vest. Unrecognized compensation expense as of June 30, 2014 related to non-vested stock options and RSUs totalled $3.3 million and $1.4 million, respectively. Such compensation expense is expected to be recognized over weighted-average periods of 3.1 years and 2.2 years, respectively.
32
Results of Operations
We are providing the following summary of our research and development expenses and general and administrative expenses to supplement the more detailed discussions below. The dollar values in the following tables are in thousands.
Research and development expenses | ||||||||||||
| Years Ended June 30, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
Clinical and drug development costs | $ | (14,875 | ) | $ | (4,201 | ) | $ | (3,523 | ) | |||
Salaries and benefits | (3,304 | ) | (1,091 | ) | (582 | ) | ||||||
Patent-related legal costs | (754 | ) | (487 | ) | (806 | ) | ||||||
Other | (398 | ) | (305 | ) | (4 | ) | ||||||
|
|
|
|
|
| |||||||
Total research and development expenses | $ | (19,331 | ) | $ | (6,084 | ) | $ | (4,915 | ) | |||
|
|
|
|
|
| |||||||
General and administrative expenses | ||||||||||||
| Years Ended June 30, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
Salaries and benefits | $ | (5,369 | ) | $ | (3,059 | ) | $ | (2,118 | ) | |||
Legal and professional fees | (1,079 | ) | (905 | ) | (616 | ) | ||||||
Other | (1,449 | ) | (1,174 | ) | (745 | ) | ||||||
|
|
|
|
|
| |||||||
Total general and administrative expenses | $ | (7,897 | ) | $ | (5,138 | ) | $ | (3,479 | ) | |||
|
|
|
|
|
| |||||||
Comparison of Years Ended June 30, 2014 and 2013
Research and Development: Research and development expenses consist primarily of clinical trial costs (including payments to CROs), pre-clinical study costs, cost to manufacture our drug candidates for non-clinical and clinical studies, and salaries and other personnel costs. Research and development expenses increased $13.2 million to $19.3 million for the year ended June 30, 2014 compared to $6.1 million for the year ended June 30, 2013. The increase was primarily due to costs associated with Phase II clinical trials for Pracinostat. Additionally, we incurred costs associated with a Phase I clinical trial for ME-344, as well as pre-clinical costs related to PWT143. Additionally, salaries and benefits costs, including share-based compensation of $1.5 million for the year ended June 30, 2014, increased due to hiring of additional employees and issuance of additional stock options to research and development personnel. We expect research and development expenses to increase during the fiscal year ending June 30, 2015 related primarily to our clinical trials.
General and Administrative: General and administrative expenses increased by $2.8 million to $7.9 million for the year ended June 30, 2013 compared to $5.1 million for the year ended June 30, 2013. The increase primarily relates to higher levels of salaries and benefits, including share-based compensation of $3.2 million for the year ended June 30, 2014 compared with $1.3 million for the year ended June 30, 2013.
Other income or expense: We received interest on cash, cash equivalents and short-term investments of $81,000 for the year ended June 30, 2014 and $37,000 for the year ended June 30, 2013. The increase was due to higher cash and short-term investment balances.
Comparison of Years Ended June 30, 2013 and 2012
Research and Development: Research and development expenses consist primarily of clinical trial costs (including payments to contract research organizations, or CROs), pre-clinical study costs, cost to manufacture our drug candidates for non-clinical and clinical studies, and salaries and other personnel costs.
33
Research and development expenses increased $1.2 million to $6.1 million for the year ended June 30, 2013 compared to $4.9 million for the year ended June 30, 2012. The increase was primarily due to costs associated with drug manufacturing and preparations for Phase II clinical trials for Pracinostat and costs associated with a Phase I clinical trial for ME-344. Additionally, salaries and benefits costs, including share-based compensation, increased due to hiring of additional employees and issuance of additional stock options to research and development personnel.
General and Administrative: General and administrative expenses increased by $1.6 million to $5.1 million for the year ended June 30, 2013 compared to $3.5 million for the year ended June 30, 2012. The increase primarily relates to higher levels of salaries and benefits, including share-based compensation of $1.3 million for the year ended June 30, 2013 compared with $0.4 million for the year ended June 30, 2012. In addition, we incurred legal fees and other costs associated with the issuance of common stock to S*Bio in conjunction with the purchase of Pracinostat, and professional and consulting expenses related to corporate matters including the Company's reverse stock split.
Other income or expense: We received interest on cash and cash equivalents of $37,000 for the year ended June 30, 2013 and $10,000 for the year ended June 30, 2012. The increase was due to higher cash balances. We also received dividends of $29,000 from a small investment in a privately-held company during the year ended June 30, 2012. We recognized a gain of $100,000 during the year ended June 30, 2012 from the sale of this investment.
Additionally, during the year ended June 30, 2011, we issued securities that were accounted for as derivative liabilities. As of June 30, 2012, our obligations related to these securities were contractually completed, resulting in the elimination of the derivative liabilities and a corresponding net decrease in their value of $1.1 million during the year ended June 30, 2012, which was recorded as non-operating income. In connection with the Supplemental Agreement entered into in September 2011 with the investors in the May 2011 private placement, we incurred financing costs in the amount of $0.4 million during the year ended June 30, 2012.
Recent Accounting Pronouncements
In June 2014, the Financial Accounting Standards Board issued Accounting Standards Update ("ASU") No. 2014-10, "Development Stage Entities." ASU 2014-10 removes the financial reporting distinction between development stage entities and other reporting entities in United States Generally Accepted Accounting Principles. The amendment therefore eliminates the requirements for development stage entities to (1) present inception-to-date information in the statements of income, cash flows, and shareholder equity, (2) label the financial statements as those of a development stage entity, (3) disclose a description of the development stage activities in which the entity is engaged, and (4) disclose in the first year in which the entity is no longer a development stage entity that in prior years it had been in the development stage. We early adopted ASU 2014-10 retrospectively during the year ended June 30, 2014; while the adoption of this standard impacted financial statement presentation and disclosure, it did not have a material impact on our financial condition, results of operations or cash flows.
In May 2014, the Financial Accounting Standards Board (FASB) issued ASU No. 2014-09, "Revenue from Contracts with Customers" (ASU 2014-09), which supersedes nearly all existing revenue recognition guidance under accounting principles generally accepted in the Unites States of America ("U.S. GAAP"). The core principle of ASU 2014-09 is to recognize revenues when promised goods or services are transferred to customers in an amount that reflects the consideration to which an entity expects to be entitled for those goods or services. ASU 2014-09 defines a five step process to achieve this core principle and, in doing so, more judgment and estimates may be required within the revenue recognition process than are required under existing U.S. GAAP.
34
The standard is effective for annual periods beginning after December 15, 2016, and interim periods therein, using either of the following transition methods: (i) a full retrospective approach reflecting the application of the standard in each prior reporting period with the option to elect certain practical expedients, or (ii) a retrospective approach with the cumulative effect of initially adopting ASU 2014-09 recognized at the date of adoption (which includes additional footnote disclosures). Currently the Company is not generating any revenue. Therefore, we have not yet determined the transition method by which we will adopt the standard in 2017.
Off-Balance Sheet Arrangements
We do not currently have any off-balance sheet arrangements.
Liquidity and Capital Resources
We have accumulated losses of $123.4 million since inception and expect to incur operating losses and generate negative cash flows from operations for the foreseeable future. As of June 30, 2014, we had $48.8 million in cash and cash equivalents and short-term investments. We believe our cash, cash equivalents and short-term investments will be sufficient to fund our operations for at least the next 12 months. Our current business operations are focused on continuing the clinical development of our lead drug candidate, Pracinostat. Our clinical development pipeline also includes ME-344 and PWT143. Changes to our research and development plans or other changes affecting our operating expenses may affect actual future use of existing cash resources. To date, we have obtained cash and funded our operations primarily through equity financings. In order to continue the development of our drug candidates, at some point in the future we expect to pursue one or more capital transactions, whether through the sale of equity securities or entry into strategic partnerships.
Sources and Uses of Our Cash
Net cash used in operations for the year ended June 30, 2014 was $19.5 million compared to $10.0 million in the year ended June 30, 2013 due to an increase in expenses incurred for research and development and general and administrative costs as described above.
Net cash used in investing activities for the year ended June 30, 2014 was $35.1 million compared to $38,000 in the year ended June 30, 2013. The increase was primarily due to the acquisition of short-term investments.
Net cash provided by financing activities was $32.7 million during the year ended June 30, 2014 compared with $39.5 million during the year ended June 30, 2013. Cash raised during the year ended June 30, 2014 reflected $32.7 million in net proceeds received associated with the issuance of common stock. Cash raised during the year ended June 30, 2013 reflected $39.5 million net proceeds received associated with the issuance of common stock.
Contractual Obligations
We have contracted with various consultants and third parties to assist us in pre-clinical research and development and clinical trials work for our leading drug compounds. The contracts are terminable at any time, but obligate us to reimburse the providers for any time or costs incurred through the date of termination. Additionally, we have employment agreements with certain of our current employees that provide for severance payments and accelerated vesting for share-based awards if their employment is terminated under specified circumstances.
We have leased approximately 8,800 square feet of office space, located at 11975 El Camino Real, Suite 101, San Diego, California 92130. The location houses the Company's executive and administrative offices. The lease commenced in July 2010 and expires in June 2015. The monthly rental rate is approximately
35
$25,000 over the remaining lease term, plus a pro rata share of certain building expenses. The remaining contractual obligation is $303,000. We believe these facilities will adequately meet our office needs for the foreseeable future.
License Agreement
On September 28, 2012, the Company entered into a license agreement with CyDex Pharmaceuticals, Inc. ("CyDex"). Under the license agreement, CyDex granted to the Company an exclusive, nontransferable license to intellectual property rights relating to Captisol ® for use with the Company's two isoflavone-based drug compounds. The Company agreed to pay to CyDex a non-refundable license issuance fee, future milestone payments, and royalties at a low, single-digit percentage rate on future sales of the Company's approved drugs utilizing Captisol. Contemporaneously with the license agreement, the Company and CyDex entered into a commercial supply agreement pursuant to which the Company agreed to purchase 100% of its requirements for Captisol from CyDex. The Company may terminate both the license agreement and the supply agreement for convenience at any time upon 90 days' prior written notice.
S*Bio Asset Purchase
In August 2012, we entered into a definitive asset purchase agreement with S*Bio, pursuant to which we agreed to acquire certain assets comprised of intellectual property and technology including rights to Pracinostat, in exchange for $500,000 of common stock. On August 22, 2012, we completed the asset purchase and issued 195,756 shares of common stock to S*Bio. We also agreed to make certain milestone payments to S*Bio based on the achievement of certain clinical, regulatory and net sales-based milestones, as well as to make certain contingent earnout payments to S*Bio. Milestone payments will be made to S*Bio up to an aggregate amount of $75.2 million if certain U.S., E.U. and Japanese regulatory approvals are obtained and if certain net sales thresholds are met in North America, the E.U. and Japan. We will pay $500,000 of the first milestone payment in shares of common stock. S*Bio will be entitled to receive certain contingent earnout payments based on certain worldwide net sales thresholds on a product-by-product and country-by-country basis.
| Item 7a. | Quantitative and Qualitative Disclosures about Market Risk |
Interest Rate Risk
Our exposure to market interest rates relates primarily to the investment of cash balances and short-term investments. We have cash reserves held in U.S. dollars and we place funds on deposit with financial institutions, which are readily available. Our short-term investments consist solely of U.S. government securities with a maturity of three to twelve months.
We place our cash deposits with high credit quality financial institutions and by policy limit the amount of credit exposure to any one corporation or bank. We are adverse to principal loss and we ensure the safety and preservation of our invested funds by limiting default risk, market risk and reinvestment risk. We seek to mitigate default risk by depositing funds with high credit quality financial institutions, by purchasing short-term investments consisting of U.S. government securities, and by positioning our portfolio to respond appropriately to a significant reduction in a credit rating of any such financial institution.
We do not consider the effects of interest rate movements to be a material risk to our financial condition.
36
| Item 8. | Financial Statements and Supplementary Data |
MEI Pharma, Inc.
Index to Financial Statements
Report of Independent Registered Public Accounting Firm | 38 | |||
Balance Sheets | 39 | |||
Statements of Operations | 40 | |||
Statement of Stockholders' Equity | 41 | |||
Statements of Cash Flows | 42 | |||
Notes to Financial Statements | 43 |
37
REPORT OF INDEPENDENT REGISTERED ACCOUNTING FIRM
Board of Directors and Stockholders
MEI Pharma, Inc.
San Diego, California
We have audited the accompanying balance sheets of MEI Pharma, Inc. as of June 30, 2014 and 2013 and the related statements of operations, stockholders' equity, and cash flows for each of the three years in the period ended June 30, 2014. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of MEI Pharma, Inc. at June 30, 2014 and 2013, and the results of its operations and its cash flows for each of the three years in the period ended June 30, 2014, in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), MEI Pharma, Inc.'s internal control over financial reporting as of June 30, 2014, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) and our report dated September 8, 2014 expressed an unqualified opinion thereon.
/s/ BDO USA, LLP
San Diego, California
September 8, 2014
38
MEI PHARMA, INC.
BALANCE SHEETS
(In thousands, except share and per share amounts)
| June 30, | ||||||||
| 2014 | 2013 | |||||||
| ASSETS | ||||||||
Current assets: | ||||||||
Cash and cash equivalents | $ | 13,777 | $ | 35,573 | ||||
Short-term investments | 35,016 | - | ||||||
Prepaid expenses and other current assets | 497 | 456 | ||||||
|
|
|
| |||||
Total current assets | 49,290 | 36,029 | ||||||
Intangible assets, net | 435 | 470 | ||||||
Property and equipment, net | 83 | 48 | ||||||
|
|
|
| |||||
Total assets | $ | 49,808 | $ | 36,547 | ||||
|
|
|
| |||||
| LIABILITIES AND STOCKHOLDERS' EQUITY | ||||||||
Current liabilities: | ||||||||
Accounts payable | $ | 1,708 | $ | 537 | ||||
Accrued liabilities | 2,908 | 1,138 | ||||||
|
|
|
| |||||
Total current liabilities | 4,616 | 1,675 | ||||||
Commitments and contingencies (Note 8) | ||||||||
Stockholders' equity: | ||||||||
Preferred stock, $0.01 par value; 100,000 shares authorized; none outstanding | - | - | ||||||
Common stock, $0.00000002 par value; 113,000,000 shares authorized; 21,607,296 and 17,116,571 shares issued and outstanding at June 30, 2014 and 2013, respectively | - | - | ||||||
Additional paid-in-capital | 168,637 | 131,169 | ||||||
Accumulated deficit | (123,445 | ) | (96,297 | ) | ||||
|
|
|
| |||||
Total stockholders' equity | 45,192 | 34,872 | ||||||
|
|
|
| |||||
Total liabilities and stockholders' equity | $ | 49,808 | $ | 36,547 | ||||
|
|
|
| |||||
See accompanying notes to financial statements.
39
MEI PHARMA, INC.
STATEMENTS OF OPERATIONS
(In thousands, except share and per share amounts)
| Years Ended June 30, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
Operating expenses: | ||||||||||||
Research and development | $ | (19,331 | ) | $ | (6,084 | ) | $ | (4,915 | ) | |||
General and administrative | (7,897 | ) | (5,138 | ) | (3,479 | ) | ||||||
|
|
|
|
|
| |||||||
Total operating expenses | (27,228 | ) | (11,222 | ) | (8,394 | ) | ||||||
|
|
|
|
|
| |||||||
Loss from operations | (27,228 | ) | (11,222 | ) | (8,394 | ) | ||||||
Other income (expense): | ||||||||||||
Interest and dividend income | 81 | 37 | 39 | |||||||||
Adjustments to fair value of derivatives | - | - | 1,139 | |||||||||
Financing costs | - | - | (406 | ) | ||||||||
Gain on sale of investment | - | - | 100 | |||||||||
Income tax expense | (1 | ) | (1 | ) | (1 | ) | ||||||
|
|
|
|
|
| |||||||
Net loss | $ | (27,148 | ) | $ | (11,186 | ) | $ | (7,523 | ) | |||
|
|
|
|
|
| |||||||
Net loss per share, basic and diluted | $ | (1.35 | ) | $ | (1.10 | ) | $ | (3.35 | ) | |||
|
|
|
|
|
| |||||||
Weighted average shares outstanding – basic and diluted | 20,061,387 | 10,160,835 | 2,247,709 | |||||||||
|
|
|
|
|
| |||||||
See accompanying notes to financial statements.
40
MEI PHARMA, INC.
STATEMENTS OF STOCKHOLDERS' EQUITY
(In thousands, except share amounts)
| Series A Preferred Shares | Series B Preferred Shares | Common Shares | Additional paid in capital | Accumulated Deficit | Total Stockholders' Equity | |||||||||||||||||||
Balance at June 30, 2011 | 1,000 | - | 1,480,181 | $ | 79,382 | $ | (77,588 | ) | $ | 1,794 | ||||||||||||||
Net loss | - | - | - | - | (7,523 | ) | (7,523 | ) | ||||||||||||||||
Issuance of common stock | - | - | 1,936,310 | 9,831 | - | 9,831 | ||||||||||||||||||
Amendment of warrant terms | - | - | - | (14 | ) | - | (14 | ) | ||||||||||||||||
Share-based compensation expense | - | - | - | 511 | - | 511 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||
Balance at June 30, 2012 | 1,000 | - | 3,416,491 | 89,710 | (85,111 | ) | 4,599 | |||||||||||||||||
Net loss | - | - | - | - | (11,186 | ) | (11,186 | ) | ||||||||||||||||
Issuance of common stock | - | - | 12,699,824 | 39,453 | - | 39,453 | ||||||||||||||||||
Conversion of Series A preferred stock | (1,000 | ) | - | 804,500 | - | - | - | |||||||||||||||||
Issuance of common stock for purchase of intangible assets | - | - | 195,756 | 500 | - | 500 | ||||||||||||||||||
Share-based compensation expense | - | - | - | 1,506 | - | 1,506 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||
Balance at June 30, 2013 | - | - | 17,116,571 | 131,169 | (96,297 | ) | 34,872 | |||||||||||||||||
Net loss | - | - | - | - | (27,148 | ) | (27,148 | ) | ||||||||||||||||
Issuance of common stock | - | - | 4,375,000 | 32,695 | - | 32,695 | ||||||||||||||||||
Exercise of warrants | - | - | 115,725 | 26 | - | 26 | ||||||||||||||||||
Share-based compensation expense | - | - | - | 4,747 | - | 4,747 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||
Balance at June 30, 2014 | - | - | 21,607,296 | $ | 168,637 | $ | (123,445 | ) | $ | 45,192 | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||
See accompanying notes to financial statements.
41
MEI PHARMA, INC.
STATEMENTS OF CASH FLOWS
(In thousands)
| Year Ended June 30, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
Cash flows from operating activities: | ||||||||||||
Net loss | $ | (27,148 | ) | $ | (11,186 | ) | $ | (7,523 | ) | |||
Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||
Share-based compensation | 4,747 | 1,506 | 511 | |||||||||
Gain on adjustment to fair value of derivatives | - | - | (1,139 | ) | ||||||||
Financing costs | - | - | 406 | |||||||||
Depreciation and amortization | 50 | 45 | 13 | |||||||||
Changes in operating assets and liabilities: | ||||||||||||
Prepaid expenses and other current assets | (41 | ) | (310 | ) | 126 | |||||||
Accounts payable | 1,171 | (57 | ) | 266 | ||||||||
Accrued liabilities | 1,770 | (42 | ) | 259 | ||||||||
|
|
|
|
|
| |||||||
Net cash used in operating activities | (19,451 | ) | (10,044 | ) | (7,081 | ) | ||||||
|
|
|
|
|
| |||||||
Cash flows from investing activities: | ||||||||||||
Purchases of property and equipment | (50 | ) | (38 | ) | - | |||||||
Acquisition of short-term investments, net | (35,016 | ) | - | - | ||||||||
|
|
|
|
|
| |||||||
Net cash used in investing activities | (35,066 | ) | (38 | ) | - | |||||||
|
|
|
|
|
| |||||||
Cash flows from financing activities: | ||||||||||||
Net proceeds from issuance of common stock | 32,721 | 39,453 | 9,831 | |||||||||
Financing costs | - | - | (406 | ) | ||||||||
|
|
|
|
|
| |||||||
Net cash provided by financing activities | 32,721 | 39,453 | 9,425 | |||||||||
|
|
|
|
|
| |||||||
Net (decrease) increase in cash and cash equivalents | (21,796 | ) | 29,371 | 2,344 | ||||||||
Cash and cash equivalents at beginning of the period | 35,573 | 6,202 | 3,858 | |||||||||
|
|
|
|
|
| |||||||
Cash and cash equivalents at end of the period | $ | 13,777 | $ | 35,573 | $ | 6,202 | ||||||
|
|
|
|
|
| |||||||
Supplemental cash flow information: | ||||||||||||
Income taxes paid | $ | (1 | ) | $ | (1 | ) | $ | (1 | ) | |||
|
|
|
|
|
| |||||||
Issuance of common stock for purchase of intangible assets | $ | - | $ | 500 | $ | - | ||||||
|
|
|
|
|
| |||||||
See accompanying notes to financial statements.
42
MEI PHARMA, INC.
NOTES TO FINANCIAL STATEMENTS
June 30, 2014
Note 1. The Company and Summary of Significant Accounting Policies
The Company
MEI Pharma, Inc. (formerly Marshall Edwards, Inc.), or the Company, is an oncology company focused on the clinical development of novel therapeutics for cancer. The Company's common stock is listed on the Nasdaq Capital Market under the symbol "MEIP". The Company was incorporated in December 2000 as a wholly-owned subsidiary of Novogen Limited ("Novogen"). In December 2012, Novogen distributed to its shareholders substantially all of its MEI Pharma common stock.
The Company's business purpose is the development of drugs for the treatment of cancer. The Company is principally focused on the clinical development of its lead drug candidate, Pracinostat, which it is currently investigating in three Phase II clinical trials. Pracinostat is an orally available histone deacetylase (HDAC) inhibitor that is being developed for advanced hematologic diseases such as myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). In August 2012, the Company acquired certain assets and intellectual property, including those related to Pracinostat, from S*Bio Pte Ltd ("S*Bio"). The Company's clinical development pipeline also includes its isoflavone-based mitochondrial inhibitor drug candidate, ME-344. Results from a Phase I clinical trial of ME-344 in patients with refractory solid tumors were presented in October 2013. The Company initiated a Phase Ib trial in patients with small cell lung cancer and ovarian cancer in May 2014. In September 2013, the Company acquired PWT143, an oral inhibitor of phosphatidylinositide 3-kinase (PI3K) delta, a molecular target that has been shown to play a critical role in the proliferation and survival of hematologic cancer cells. The Company expects to complete the pre-clinical work required to support the filing of an Investigational New Drug (IND) application for PWT143 by the end of calendar year 2014.
The results of pre-clinical studies and completed clinical trials are not necessarily predictive of future results, and the Company's current drug candidates may not have favorable results in later studies or trials. The commercial opportunity will be reduced or eliminated if competitors develop and market products that are more effective, have fewer side effects or are less expensive than the Company's drug candidates. The Company will need substantial additional funds to progress the clinical trial program for the drug candidates Pracinostat, ME-344 and PWT143, and to develop new compounds. The actual amount of funds that will be needed are determined by a number of factors, some of which are beyond the Company's control. Negative U.S. and global economic conditions may pose challenges to the Company's business strategy, which relies on funding from the financial markets or collaborators.
Reverse Stock Split and Presentation
On December 18, 2012, the Company effected a 1-for-6 reverse stock split (the "2012 Reverse Stock Split") of the Company's common stock. As a result of the 2012 Reverse Stock Split, every six shares of the Company's issued and outstanding common stock were combined into one share of common stock. The 2012 Reverse Stock Split did not change the number of authorized shares of the Company's common stock, nor the common stock par value. All financial data and share information is presented on an as-adjusted basis to give effect to the 2012 Reverse Stock Split.
Recent Accounting Pronouncements
In June 2014, the Financial Accounting Standards Board issued Accounting Standards Update ("ASU") No. 2014-10, "Development Stage Entities." ASU 2014-10 removes the financial reporting distinction between development stage entities and other reporting entities in United States Generally Accepted Accounting Principles. The amendment therefore eliminates the requirements for development stage entities to (1) present
43
inception-to-date information in the statements of income, cash flows, and shareholder equity, (2) label the financial statements as those of a development stage entity, (3) disclose a description of the development stage activities in which the entity is engaged, and (4) disclose in the first year in which the entity is no longer a development stage entity that in prior years it had been in the development stage. We early adopted ASU 2014-10 retrospectively during the year ended June 30, 2014; while the adoption of this standard impacted financial statement presentation and disclosure, it did not have a material impact on our financial condition, results of operations or cash flows.
In May 2014, the Financial Accounting Standards Board ("FASB") issued ASU No. 2014-09, "Revenue from Contracts with Customers" (ASU 2014-09), which supersedes nearly all existing revenue recognition guidance under accounting principles generally accepted in the Unites States of America ("U.S. GAAP"). The core principle of ASU 2014-09 is to recognize revenues when promised goods or services are transferred to customers in an amount that reflects the consideration to which an entity expects to be entitled for those goods or services. ASU 2014-09 defines a five step process to achieve this core principle and, in doing so, more judgment and estimates may be required within the revenue recognition process than are required under existing U.S. GAAP.
The standard is effective for annual periods beginning after December 15, 2016, and interim periods therein, using either of the following transition methods: (i) a full retrospective approach reflecting the application of the standard in each prior reporting period with the option to elect certain practical expedients, or (ii) a retrospective approach with the cumulative effect of initially adopting ASU 2014-09 recognized at the date of adoption (which includes additional footnote disclosures). Currently the Company is not generating any revenue. Therefore, the Company has not yet determined the transition method by which they will adopt the standard in 2017.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the amounts reported in the financial statements and disclosures made in the accompanying notes to the financial statements. The Company uses estimates for certain accruals including clinical and pre-clinical study fees and expenses, share-based compensation, and valuations of derivative liabilities, among others. Actual results could materially differ from those estimates.
Cash and Cash Equivalents
Cash and cash equivalents consist of cash and highly liquid investments with remaining maturities of three months or less when purchased.
Short-Term Investments
Investments that have maturities of greater than three months but less than one year at the time of purchase are classified as short-term investments. Short-term investments are comprised of money market funds and U.S. government securities and are recorded at their amortized cost.
Fair Value of Financial Instruments
The carrying amounts of financial instruments such as cash equivalents and other current liabilities approximate the related fair values due to the short-term maturities of these instruments. The Company invests its excess cash in financial instruments which are readily convertible into cash, consisting of money market funds and U.S. government securities. Cash is deposited in financial institutions that are FDIC insured; these deposits are in excess of the FDIC insurance limits. Financial instruments include cash and cash equivalents, short-term investments and accounts payable, which approximate fair value because of their short maturities.
44
The fair value of financial assets and liabilities is measured under a three-tier fair value hierarchy as follows: Level 1 fair value is determined from observable, quoted prices in active markets for identical assets or liabilities. Level 2 fair value is determined from quoted prices for similar items in active markets or quoted prices for identical or similar items in markets that are not active. Level 3 fair value is determined using the entity's own assumptions about the inputs that market participants would use in pricing an asset or liability. Cash equivalents, where applicable, and short-term investments are classified as Level 1 as defined by the fair value hierarchy.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentrations of credit risk consist primarily of cash in financial institutions (which exceeds federally insured limits), and cash equivalents and short-term investments (comprised of U.S. government securities). However, management believes that the Company is not exposed to significant credit risk due to the financial positions of the depository institutions in which these deposits are held.
Intangible Assets
Intangible assets consist of patents acquired from S*Bio in August 2012, relating to a family of heterocyclic compounds that inhibit HDACs. Capitalized amounts are amortized on a straight-line basis over the expected life of the intellectual property of 14 years. The carrying values of intangible assets are periodically reviewed to determine if the facts and circumstances suggest that a potential impairment may have occurred. Results of operations for the years ended June 30, 2014 and 2013 do not reflect any write-downs associated with the potential impairment of intangible assets.
Property and Equipment
Property and equipment are stated at cost and depreciated over the estimated useful lives of the assets (generally three to seven years) using the straight-line method. Leasehold improvements are stated at cost and are amortized over the shorter of the estimated useful lives of the assets or the lease term.
Research and Development Costs
Research and development costs are expensed as incurred and include costs paid to third-party contractors to perform research, conduct clinical trials and develop and manufacture drug materials. Clinical trial costs, including costs associated with third-party contractors, are a significant component of research and development expenses. The Company accrues research and development costs based on work performed. In determining the amount to accrue, management relies on estimates of total costs based on contract components completed, the enrollment of subjects, the completion of trials, and other events. Costs incurred related to the purchase of in-process research and development for early-stage products or products that are not commercially viable and ready for use, or have no alternative future use, are charged to expense in the period incurred.
License Fees
Costs incurred related to the licensing of products that have not yet received regulatory approval to be marketed, or that are not commercially viable and ready for use, or have no alternative future use, are charged to expense in the period incurred.
Share-based Compensation
Share-based compensation expense for employees and directors is recognized in the statement of operations based on estimated amounts, including the grant date fair value and the expected service period. For stock options, the Company estimates the grant date fair value using a binomial valuation model, which requires the use of multiple subjective inputs including estimated future volatility, expected forfeitures and the expected
45
term of the awards. The Company estimates the expected future volatility based on the stock's historical price volatility. The stock's future volatility may differ from our estimated volatility at the grant date. For restricted stock unit (RSU) equity awards, the Company estimates the grant date fair value using the Company's closing stock price on the date of grant. Share-based compensation recorded in the statement of operations is based on the awards expected to ultimately vest and has been reduced for estimated forfeitures. The estimated forfeiture rates may differ from actual forfeiture rates which would affect the amount of expense recognized during the period. The Company recognizes the value of the awards over the awards' requisite service or performance periods. The requisite service period is generally the time over which the share-based awards vest.
Interest and Dividend Income
Interest on cash balances is recognized when earned. Dividend income is recognized when the right to receive the payment is established.
Income Taxes
The Company's income tax expense consists of current and deferred income tax expense or benefit. Current income tax expense or benefit is the amount of income taxes expected to be payable or refundable for the current year. A deferred income tax asset or liability is recognized for the future tax consequences attributable to tax credits and loss carryforwards and to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets are reduced by a valuation allowance when, in the opinion of management, it is more likely than not that some portion or all of the deferred tax assets will not be realized. As of June 30, 2014 and 2013, the Company has established a valuation allowance to fully reserve its net deferred tax assets. Tax rate changes are reflected in income during the period such changes are enacted. Changes in ownership of the Company may limit the amount of net operating loss carry-forwards that can be utilized in the future to offset taxable income.
The FASB Topic on Income Taxes prescribes a recognition threshold and measurement attribute criteria for the financial statement recognition and measurement of tax positions taken or expected to be taken in a tax return. For those benefits to be recognized, a tax position must be more-likely-than-not to be sustained upon examination by taxing authorities. An uncertain income tax position will not be recognized if it has less than a 50% likelihood of being sustained. There were no unrecognized tax benefits as of June 30, 2014 and 2013.
Derivative Liabilities
The Company accounts for its warrants and other derivative financial instruments as either equity or liabilities based upon the characteristics and provisions of each instrument. Warrants classified as equity are recorded at fair value as additional paid-in capital on our balance sheet and are not marked to market. Warrants classified as derivative liabilities and other derivative financial instruments that require separate accounting as liabilities are recorded on the Company's balance sheet at their fair value on the date of issuance and are revalued on each subsequent balance sheet date until such instruments are exercised, amended to remove features that result in derivative liability classification, or expire, with any changes in the fair value between reporting periods recorded as other income or expense. Management estimates the fair value of the Company's derivative liabilities using option pricing models and assumptions that are based on the individual characteristics of the warrants or instruments on the valuation date, as well as assumptions for expected volatility, expected life, yield, and risk-free interest rate. All instruments creating derivative liability accounting treatment were settled during the year ended June 30, 2012. There were no derivative liabilities as of June 30, 2014 or June 30, 2013.
Net Loss Per Share
Basic and diluted net loss per share are computed using the weighted-average number of shares of common stock outstanding during the period, less any shares subject to repurchase or forfeiture. There were no shares of common stock subject to repurchase or forfeiture for the years ended June 30, 2014, 2013 and 2012.
46
Net loss per share was determined as follows (in thousands, except share and per share amounts):
| Years ended June 30, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
Numerator | ||||||||||||
Net loss | $ | (27,148 | ) | $ | (11,186 | ) | $ | (7,523 | ) | |||
|
|
|
|
|
| |||||||
Denominator | ||||||||||||
Weighted average common shares outstanding | 20,061,387 | 10,160,835 | 2,247,709 | |||||||||
|
|
|
|
|
| |||||||
Basic and diluted net loss per share | $ | (1.35 | ) | $ | (1.10 | ) | $ | (3.35 | ) | |||
|
|
|
|
|
| |||||||
Because the Company is in a net loss position, it has excluded stock options, warrants, unvested restricted stock units and convertible preferred stock from its calculation of diluted net loss per share, and the Company's diluted net loss per share is the same as the Company's basic net loss per share. For the years ended June 30, 2014, 2013 and 2012, the Company did not have any items that would be classified as other comprehensive income or losses. The table below presents the potentially dilutive securities that would have been included in the Company's calculation of diluted net loss per share allocable to common stockholders as if they were not antidilutive as of June 30, 2014, 2013 and 2012.
| Year ended June 30, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
Anti-dilutive securities not included in diluted loss per share: | ||||||||||||
Stock options | 1,194,854 | 635,094 | 143,927 | |||||||||
Warrants | 4,900,999 | 5,062,000 | 937,295 | |||||||||
Restricted stock units | 400,000 | 400,000 | - | |||||||||
Convertible preferred shares | - | - | 804,500 | |||||||||
|
|
|
|
|
| |||||||
Total anti-dilutive securities not included in diluted net loss per share | 6,495,853 | 6,097,094 | 1,885,722 | |||||||||
|
|
|
|
|
| |||||||
Note 2. Intangible Assets
Intangible assets consisted of the following, in thousands:
| June 30, | ||||||||
| 2014 | 2013 | |||||||
S*Bio Patents – Gross | $ | 500 | $ | 500 | ||||
S*Bio Patents – Accumulated amortization | (65 | ) | (30 | ) | ||||
|
|
|
| |||||
Intangible assets, net | $ | 435 | $ | 470 | ||||
|
|
|
| |||||
Amortization expense of intangible assets for the years ended June 30, 2014 and 2013 was $35,000 and $30,000, respectively, with no amortization expense during the year ended June 30, 2012. We expect to record amortization of $35,000 per year through 2026 for our S*Bio patents.
47
Note 3. Property and Equipment
Property and equipment consisted of the following, in thousands:
| June 30, | ||||||||
| 2014 | 2013 | |||||||
Furniture and equipment | $ | 107 | $ | 67 | ||||
Leasehold improvements | 32 | 22 | ||||||
|
|
|
| |||||
| 139 | 89 | |||||||
Less: accumulated depreciation | (56 | ) | (41 | ) | ||||
|
|
|
| |||||
Property and equipment, net | $ | 83 | $ | 48 | ||||
|
|
|
| |||||
Depreciation expense of property and equipment for the years ended June 30, 2014, 2013 and 2012 was $15,000, $15,000 and $13,000, respectively.
Note 4. Accrued Liabilities
Accrued liabilities consisted of the following, in thousands:
| June 30, | ||||||||
| 2014 | 2013 | |||||||
Accrued pre-clinical and clinical trial expenses | $ | 1,899 | $ | 279 | ||||
Accrued compensation and benefits | 800 | 604 | ||||||
Accrued legal and professional services expenses | 102 | 145 | ||||||
Other | 107 | 110 | ||||||
|
|
|
| |||||
Total accrued liabilities | $ | 2,908 | $ | 1,138 | ||||
|
|
|
| |||||
Note 5. Related Party Transactions
Novogen was the Company's majority shareholder from the Company's inception through December 3, 2012. On such date, Novogen completed the distribution of substantially all of its MEI Pharma common stock to its shareholders. Historically, the Company licensed from Novogen the rights to Novogen patents and applications for the Company's isoflavone-based drug candidates, as well as other compounds. Additionally, Novogen historically provided research and development services and administrative and finance services to the Company under service agreements. The Company's license agreements with Novogen were terminated in May 2011 in conjunction with the Isoflavone Transaction. The service agreements with Novogen were terminated in December 2010.
Rights Offering
In March 2012, the Company distributed one subscription right for each share of common stock and each Series A warrant exercisable for a share of common stock to holders of record as of March 30, 2012. Each subscription right entitled the holder to purchase one Unit, which consisted of 0.0833 shares of our common stock and a warrant representing the right to purchase 0.04167 shares of the Company's common stock. In connection with the rights offering, in May 2012, Novogen purchased 8,988,675 units consisting of 749,056 shares of common stock and warrants to purchase an additional 374,528 shares of common stock. The warrants are exercisable for a five-year period beginning on May 11, 2012 at an exercise price of $7.14 per share. See further discussion regarding the Rights Offering in Note 6 "Stockholders' Equity".
48
Waiver Agreement
In December 2012, the Company entered into an agreement (the "Waiver Agreement") with Novogen and Novogen Research Pty Limited, a wholly-owned subsidiary of Novogen (together, the "Novogen Parties"), Graham Kelly, an individual, and Andrew Heaton, an individual, pursuant to which the Company granted a limited waiver with respect to certain non-compete provisions contained in the Asset Purchase Agreement dated as of December 20, 2010, between the Company and the Novogen Parties. In consideration of the Company's grant of the limited waiver, upon the execution of the Waiver Agreement, Novogen surrendered to the Company for cancellation warrants held by Novogen for the purchase of 166,666 shares of Common Stock.
Securities Subscription Agreements
In September 2011, the Company entered into a Securities Subscription Agreement with Novogen, pursuant to which the Company sold to Novogen 222,222 shares of common stock, at a purchase price of $9.00 per share, for proceeds of $2.0 million. The offering closed on September 29, 2011. In December 2011, the Company entered into a Securities Subscription Agreement with Novogen, pursuant to which the Company sold to Novogen 323,625 shares of common stock, at a purchase price of $6.18 per share, for proceeds of $2.0 million. The offering closed on December 29, 2011.
Note 6. Stockholders' Equity
Equity Transactions
Underwritten Registered Offerings
In October 2013, the Company completed an underwritten registered offering of 4,375,000 shares of its common stock at a price per share of $8.00 pursuant to a "shelf" registration statement previously filed and declared effective by the Securities and Exchange Commission. The Company received net proceeds of $32.7 million associated with the offering.
In April 2013, the Company completed an underwritten registered offering of 2,030,000 shares of its common stock at a price per share of $7.50 pursuant to a "shelf" registration statement previously filed and declared effective by the Securities and Exchange Commission. The Company received net proceeds of $14.2 million associated with the offering.
Private Placement
In December 2012, the Company completed the sale (the "December 2012 private placement") of 9,166,665 shares of common stock and warrants to purchase an additional 6,416,665 shares of common stock for an aggregate offering price of $27.5 million, pursuant to the terms of the Securities Purchase Agreement, dated November 4, 2012, between the Company and certain accredited investors identified therein. The Company received net proceeds of $25.3 million associated with the Private Placement. In the period from December 2012 through June 2014, the investors exercised, on a cashless basis, warrants representing the right to purchase 2,046,871 shares of common stock. The Company issued 1,496,018 shares of common stock in conjunction with the exercise of the warrants.
S*Bio Asset Purchase
In August 2012, the Company acquired from S*Bio certain assets comprised of intellectual property and technology including rights to Pracinostat, in exchange for 195,756 shares of common stock valued at $0.5 million (see Note 8).
49
Rights Offering
In May 2012, the Company completed a rights offering ("Rights Offering") pursuant to which the Company distributed, at no charge, to holders of record as of March 30, 2012, subscription rights (the "Rights") to purchase up to 17,129,361 units for an aggregate purchase price of up to $7.6 million. The subscription period for the Rights Offering expired on May 11, 2012. Each unit consisted of 0.0833 shares of common stock and a warrant representing the right to purchase 0.04167 shares of common stock at an exercise price of $7.14 per share. The exercise of one Right entitled holders to purchase one unit at a subscription price of $0.445 per unit, which represented the subscription price of $5.34 per whole share. Eligible participants in the Rights Offering exercised Rights to purchase an aggregate of 11,660,606 units; accordingly, the Company issued 971,700 shares of common stock and warrants to purchase an additional 485,857 shares of common stock. The warrants are exercisable for a five-year period beginning on May 11, 2012. The Company received net proceeds of $4.8 million associated with the Rights Offering. In December 2012, upon the execution of the Waiver Agreement, Novogen surrendered to the Company for cancellation warrants acquired by Novogen in the Rights Offering for the purchase of 166,666 shares of common stock. In the period from March 2013 through June 2014, holders exercised warrants acquired in the Rights Offering representing the right to purchase 3,707 shares of common stock.
Private Placements with Novogen
In September 2011, the Company entered into a Securities Subscription Agreement with Novogen, pursuant to which the Company sold to Novogen 222,222 shares of the Company's common stock, at a purchase price of $9.00 per share, for proceeds of $2.0 million. The offering closed on September 29, 2011. In December 2011, the Company entered into a Securities Subscription Agreement with Novogen, pursuant to which the Company sold to Novogen 323,625 shares of our common stock, at a purchase price of $6.18 per share, for proceeds of $2.0 million. The offering closed on December 29, 2011.
May 2011 Private Placement
In May 2011, the Company entered into an Amended and Restated Securities Purchase Agreement (the "Amended Securities Purchase Agreement") with certain accredited investors pursuant to which the Company agreed to issue and sell to the investors certain shares of the Company's common stock, and warrants to purchase additional shares of common stock. Pursuant to the Amended Securities Purchase Agreement, in May 2011 the Company issued to the investors: (i) 139,203 shares (the "Initial Shares") of common stock, at a purchase price of $8.00 per share; (ii) series A warrants (the "Series A warrants") which initially represented the right to purchase up to 104,402 shares of common stock, up to a maximum of 375,094 shares; and (iii) series B warrants (the "Series B warrants") which initially represented the right to purchase up to 360,922 shares of common stock. In addition, the Company agreed to issue certain additional shares of common stock (the "Adjustment Shares") to the extent the price of the common stock is below $8.00 per share, but greater than or equal to $4.50 per share, on certain dates ("Adjustment Dates") during the period ending June 26, 2012, including as a result of a subsequent offering by the Company of its securities at a price below the purchase price of the Initial Shares. The number of Adjustment Shares issuable was initially limited to 108,207, subject to proportionate increases to the extent the Series B warrants have been exercised prior to the applicable Adjustment Date, up to a maximum of 388,764 shares. If the trading price of the Company's common stock is below $4.50 per share on any Adjustment Date, the Company will, in addition to issuing the applicable number of Adjustment Shares, refund to the investors an amount per share of common stock received by the investors in the transaction equal to the difference between $4.50 and the price of the common stock on such Adjustment Date. The transactions contemplated by the Amended Securities Purchase Agreement are referred to as the May 2011 private placement. Upon the closing of the May 2011 private placement, the Company also issued warrants to the placement agent for the purchase of up to 35,008 shares of common stock, which warrants were exercisable on the same terms as the Series A warrants.
On December 29, 2011, the Company issued an aggregate of 111,212 Adjustment Shares to the investors in accordance with the calculation of the applicable price, based on the trading price of the Company's
50
common stock, with respect to the first Adjustment Date. Additionally, on December 29, 2011, the Company issued an aggregate of 40,950 Adjustment Shares to the investors in connection with the private placement of common stock to Novogen that closed on December 29, 2011.
Terms of Series A and Series B Warrants
The Series A warrants became exercisable on the six month anniversary of the May 18, 2011 closing of the May 2011 private placement. The Series A warrants will expire on the fifth anniversary of the date on which the Series A warrants first became exercisable. Prior to the amendment of the warrant terms in September 2011 in conjunction with the Supplemental Agreement, as defined and described below, the Series A warrants were initially exercisable at an exercise price of $9.42 per share, subject to adjustment as provided in the Series A warrant agreements. Under the terms of the warrant agreements, the number of shares of common stock issuable upon exercise of the Series A warrants would be increased by an amount equal to 75% of the number of shares of common stock issued upon each exercise of the Series B warrants.
Prior to the amendment of the warrant terms in September 2011 in conjunction with the Supplemental Agreement, as described below, the initial exercise price per share of the Series B warrants was equal to the lower of (i) $8.00, and (ii) 85% of the arithmetic average of the lowest eight weighted average prices of the common stock during the 20 consecutive trading day period in the case of a voluntary exercise by the holders, ending on the trading day immediately preceding the date of delivery of a notice of exercise.
In July and August 2011, the investors exercised an aggregate of Series B warrants representing the right to purchase 215,667 shares of common stock. The Company received net proceeds of $1.1 million in conjunction with the exercise of the Series B warrants. Pursuant to the terms of the Amended Securities Purchase Agreement, an additional 161,750 Series A warrants became exercisable as a result of these Series B warrant exercises.
Supplemental Agreement
In September 2011, the Company entered into a Supplemental Agreement (the "Supplemental Agreement") with each of the investors party to the Amended Securities Purchase Agreement.
Pursuant to the Supplemental Agreement, each of the Series A warrants and the Series B warrants issued pursuant to the Amended Securities Purchase Agreement were amended and restated (the "Amended Series A Warrants" and "Amended Series B Warrants", respectively). The exercise price of each of the Series A warrants and Series B warrants was reduced to $6.00 per share. As amended, the exercise price of the Amended Series A Warrants is no longer subject to further adjustment upon the occurrence of certain events, including the subsequent sale or deemed sale by the Company of shares of common stock at a price per share below the exercise price of the Amended Series A Warrants; however, the Amended Series A Warrants continue to provide for certain customary anti-dilution adjustments.
The Series B warrants were amended to permit the exercise of such warrants on a cashless basis. Pursuant to the terms of the Supplemental Agreement, the investors exercised, on a cashless basis, the Amended Series B Warrants for all of the remaining shares of common stock for which such Amended Series B Warrants were exercisable, resulting in the exercise of Series B Warrants representing the right to purchase 145,256 shares of common stock and the issuance by the Company of an aggregate of 50,934 shares of common stock. Pursuant to the terms of the Amended Securities Purchase Agreement, additional Series A warrants to purchase 108,942 shares of common stock became exercisable as a result of these Series B warrant exercises. As of September 28, 2011, there were no remaining outstanding Series B warrants.
In December 2012, the investors exercised, on a cashless basis, Series A warrants representing the right to purchase an aggregate of 194,381 shares of common stock. The Company issued 119,158 shares of common stock in conjunction with the exercise of the Series A warrants.
51
The Supplemental Agreement also effected certain amendments to the Amended Securities Purchase Agreement, including the extension, through September 28, 2013, of the period during which the investors had the right to participate in subsequent equity offerings of the Company. In connection with the amendments described above, the Company made cash payments to the investors in an aggregate amount of $365,000, which, together with $41,000 that the Company paid in other expenses related to the Supplemental Agreement, have been classified as ‘Financing Costs' in the Statement of Operations.
Derivative Liabilities
The Company accounted for the Series A and B warrants and the Adjustment Shares feature pursuant to the Amended Securities Purchase Agreement in accordance with accounting guidance for derivatives. As a result of the Company's completion of its contractual obligations under the Amended Securities Purchase Agreement related to the issuance of Adjustment Shares during December 2011, the Company had no remaining derivative liabilities as of June 30, 2014 or June 30, 2013.
On the closing date of the May 2011 private placement, the derivative liabilities were initially recorded at their estimated fair values of $1.2 million. The fair value of the derivative liabilities exceeded the proceeds of the private placement of $0.7 million, and accordingly, no net amounts were allocated to the common stock. The $508,000 amount by which the recorded liabilities exceeded the proceeds was charged to other expense. On June 30, 2011, the total value of the derivative liabilities was $1.1 million, resulting in other income of $49,000 classified as ‘Adjustments to Fair Value of Derivatives' in the Statement of Operations. Such decrease in the estimated fair value was primarily due to the decrease in the Company's common stock price and updates to the assumptions used in the option pricing models. The completion of the Company's obligations related to the derivative liabilities during the year ended June 30, 2012 resulted in extinguishment of the derivative liabilities; accordingly, the Company recorded other income of $1.1 million, classified as ‘Adjustments to Fair Value of Derivatives' in the Statement of Operations, associated with the decrease in fair value of the derivative liabilities. Additionally, during the year ended June 30, 2012, the Company recorded a gain of $14,000 in conjunction with amending the Series A warrant terms, based on the fair value of the Amended Series A Warrants, classified as ‘Adjustments to Fair Value of Derivatives' in the Statement of Operations.
Shelf Registration Statement
In April 2014, the Company filed a shelf registration statement on Form S-3 with the SEC (the "new shelf registration statement"). The new shelf registration statement was declared effective by the SEC in April 2014. The new shelf registration statement permits the Company to sell, from time to time, up to $150 million of common stock, preferred stock and warrants. Pursuant to SEC regulations, if the market value of the Company's public float is below $75 million, the Company cannot sell securities from the shelf registration statement which represent more than one third of the market value of the Company's non-affiliated public float during any 12-month period.
In April 2011, the Company filed a shelf registration statement on Form S-3 with the SEC (the "shelf registration statement"). The shelf registration statement was declared effective by the SEC in May 2011 and expired in April 2014. The shelf registration statement permits the Company to sell, from time to time, up to $50 million of common stock, preferred stock and warrants. In April 2013, the Company completed an underwritten registered offering of 2,030,000 shares of its common stock at a price per share of $7.50 pursuant to the shelf registration statement. In October 2013, the Company completed an underwritten registered offering of 4,375,000 shares of its common stock at a price per share of $8.00 pursuant to the shelf registration statement.
Description of Capital Stock
The Company's total authorized share capital is 113,100,000 shares consisting of 113,000,000 shares of common stock, $0.00000002 par value per share, and 100,000 shares of preferred stock, $0.01 par value per share.
52
Common Stock
The holders of common stock are entitled to one vote per share. In the event of a liquidation, dissolution or winding up of the Company's affairs, holders of the common stock will be entitled to share rateably in all the Company's assets that are remaining after payment of the Company's liabilities and the liquidation preference of any outstanding shares of preferred stock. All outstanding shares of common stock are fully paid and non-assessable. The rights, preferences and privileges of holders of common stock are subject to any series of preferred stock that the Company has issued or that the Company may issue in the future. The holders of common stock have no pre-emptive rights and are not subject to future calls or assessments by the Company.
Preferred Stock
The Company's Board of Directors has the authority to issue up to 100,000 shares of preferred stock with par value of $.01 per share in one or more series and to fix the rights, preferences, privileges and restrictions in respect of that preferred stock, including dividend rights, dividend rates, conversion rights, voting rights, terms of redemption (including sinking fund provisions), redemption prices and liquidation preferences, and the number of shares constituting such series and the designation of any such series, without future vote or action by the stockholders. Therefore, the board without the approval of the stockholders could authorize the issue of preferred stock with voting, conversion and other rights that could affect the voting power, dividend and other rights of the holders of shares or that could have the effect of delaying, deferring or preventing a change of control. There were no shares of preferred stock outstanding as of June 30, 2014 or 2013.
Warrants
As of June 30, 2014, there were outstanding warrants to purchase 315,484 shares of the Company's common stock at an exercise price of $7.14 per share, which expire in May 2017, issued in conjunction with the Rights Offering; outstanding Series A warrants and warrants issued to the Company's placement agent for the May 2011 private placement to purchase up to 215,721 shares of common stock at an exercise price of $6.00 per share, which expire in November 2016, and warrants to purchase 4,369,794 shares of the Company's common stock at an exercise price of $3.12 per share, which expire in December 2017, issued in conjunction with the December 2012 private placement.
Note 7. Share-based Compensation
The Company uses equity-based compensation programs to provide long-term performance incentives for its employees. These incentives consist primarily of stock options and restricted stock units (RSUs). In December 2008, the Company adopted the MEI Pharma, Inc. 2008 Stock Omnibus Equity Compensation Plan (the "2008 Plan"), as amended and restated in 2011 and 2013, under which 2,186,000 shares of common stock are authorized for issuance. The 2008 Plan provides for the grant of options and/or other stock-based or stock-denominated awards to the Company's non-employee directors, officers, employees and advisors. As of June 30, 2014, there were 657,479 shares available for future grant under the 2008 Plan.
Total share-based compensation expense for all stock awards consists of the following, in thousands:
| Years Ended June 30, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
Research and development | $ | 1,549 | $ | 213 | $ | 65 | ||||||
General and administrative | 3,198 | 1,293 | 446 | |||||||||
|
|
|
|
|
| |||||||
Total share-based compensation | $ | 4,747 | $ | 1,506 | $ | 511 | ||||||
|
|
|
|
|
| |||||||
53
Stock Options
As of June 30, 2014, there were a total of 1,194,854 options outstanding, including options representing the right to purchase a total of 66,333 shares of common stock which were granted to two of the Company's officers outside of the 2008 Plan.
A summary of the Company's stock option activity and related data follows:
| Number of Options | Weighted-Average Exercise Price | Weighted-Average Remaining Contractual Term (in years) | Aggregate Intrinsic Value | |||||||||||||
Outstanding at June 30, 2013 | 635,094 | $ | 8.26 | |||||||||||||
Granted | 568,593 | 7.97 | ||||||||||||||
Expired | (8,833 | ) | 10.33 | |||||||||||||
|
|
|
|
|
|
|
| |||||||||
Outstanding at June 30, 2014 | 1,194,854 | $ | 8.11 | 3.7 | $ | 340,397 | ||||||||||
|
|
|
|
|
|
|
| |||||||||
Vested and exercisable at June 30, 2014 | 326,363 | $ | 8.94 | 2.7 | $ | 173,574 | ||||||||||
|
|
|
|
|
|
|
| |||||||||
No stock option exercises occurred during the years ended June 30, 2014, 2013 or 2012. As of June 30, 2014, the aggregate intrinsic value of outstanding options is calculated as the difference between the exercise price of the underlying options and the closing price of the Company's common stock of $6.34 on that date. The total fair value of options that vested during the years ended June 30, 2014, 2013, and 2012 was $1.2 million, $0.9 million, and $0.3 million, respectively.
Unrecognized compensation expense related to non-vested stock options totalled $3.3 million as of June 30, 2014. Such compensation expense is expected to be recognized over a weighted-average period of 3.1 years. As of June 30, 2014, the Company expects all outstanding options to vest.
The Company uses a binomial valuation model to estimate the grant date fair value of stock options. To calculate these fair values, the following weighted-average assumptions were used:
| Years ended June 30, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
Risk-free interest rate | 1.5 | % | 0.7 | % | 1.2 | % | ||||||
Expected life (years) | 5.0 | 5.0 | 5.0 | |||||||||
Expected volatility | 145.9 | % | 159.3 | % | 146.5 | % | ||||||
Dividend yield | 0.0 | % | 0.0 | % | 0.0 | % | ||||||
Grant date fair value | $ | 6.85 | $ | 6.57 | $ | 8.56 | ||||||
Restricted Stock Units
On March 29, 2013, the Compensation Committee of the Board of Directors granted 400,000 RSUs to the Company's Chief Executive Officer, Dr. Daniel P. Gold. Each RSU represents the contingent right to receive one share of the Company's common stock. One third of the RSUs will vest on each of August 30, 2014, August 30, 2015 and August 30, 2016. The shares underlying the RSUs will be delivered to Dr. Gold on the earliest to occur of (i) March 29, 2018, (ii) Dr. Gold's death, disability or separation from service from the Company for any reason, or (iii) a change in control involving the Company.
The fair value of the RSUs on the date of grant was $3.5 million. The grant date fair value per unit was $8.63. As of June 30, 2014, unrecognized compensation expense related to the unvested portion of the Company's RSUs was approximately $1.4 million and is expected to be recognized over approximately 2.2 years.
54
Note 8. Commitments and Contingencies
The Company has contracted with various consultants and third parties to assist it in pre-clinical research and development and clinical trials work for its leading drug compounds. The contracts are terminable at any time, but obligate the Company to reimburse the providers for any time or costs incurred through the date of termination. The Company also has employment agreements with certain of its current employees that provide for severance payments and accelerated vesting for share-based awards if their employment is terminated under specified circumstances.
The Company currently leases approximately 8,800 square feet of office space for the Company's executive and administrative offices. The monthly rental rate ranges from $25,077 to $25,957 during the remaining term of the lease, plus a pro-rata share of certain building expenses. The lease expires in June 2015. Future minimum payments under the lease are $303,000 as of June 30, 2014.
Asset Purchase Agreement
On August 7, 2012, the Company entered into a definitive asset purchase agreement with S*Bio, pursuant to which the Company agreed to acquire certain assets comprised of intellectual property and technology including rights to Pracinostat, in exchange for $500,000 of common stock. On August 22, 2012, the Company completed the asset purchase and issued 195,756 shares of common stock to S*Bio. The Company has also agreed to make certain milestone payments to S*Bio based on the achievement of certain clinical, regulatory and net sales-based milestones, as well as to make certain contingent earnout payments to S*Bio. Milestone payments will be made to S*Bio up to an aggregate amount of $75.2 million if certain U.S., E.U. and Japanese regulatory approvals are obtained and if certain net sales thresholds are met in North America, the E.U. and Japan. The first milestone payment of $200,000 plus shares of the Company's common stock having a value of $500,000 will be due upon the first dosing of a patient in a Phase III clinical trial or other pivotal trial, for any indication. Subsequent milestone payments will be due upon certain regulatory approvals. S*Bio will be entitled to receive certain contingent earnout payments based on certain worldwide net sales thresholds on a product-by-product and country-by-country basis. As of June 30, 2014, the Company has not accrued any amounts for potential future payments.
License Agreement
On September 28, 2012, the Company entered into a license agreement with CyDex Pharmaceuticals, Inc. ("CyDex"). Under the license agreement, CyDex granted to the Company an exclusive, nontransferable license to intellectual property rights relating to Captisol ® for use with the Company's two isoflavone-based drug compounds. The Company agreed to pay to CyDex a non-refundable license issuance fee, future milestone payments, and royalties at a low, single-digit percentage rate on future sales of the Company's approved drugs utilizing Captisol. Contemporaneously with the license agreement, the Company and CyDex entered into a commercial supply agreement pursuant to which the Company agreed to purchase 100% of its requirements for Captisol from CyDex. The Company may terminate both the license agreement and the supply agreement for convenience at any time upon 90 days' prior written notice. As of June 30, 2014, the Company has not accrued any amounts for potential future payments.
Note 9. Segment Information
The Company has one operating segment, the development of pharmaceutical compounds. The Company's business contained two geographic segments, the United States of America and Australia, from inception until MEPL's legal dissolution in April 2012. For the year ended June 30, 2012, net losses attributable to Australia were immaterial. All of the Company's assets and liabilities were located in the United States of America as of June 30, 2014 and June 30, 2013.
55
Note 10. Income Taxes
Pre-tax loss consists of the following jurisdictions (in thousands):
| Years ended June 30, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
Domestic | $ | (27,147 | ) | $ | (11,185 | ) | $ | (8,546 | ) | |||
Foreign | - | - | 1,024 | |||||||||
|
|
|
|
|
| |||||||
Pre-tax loss | $ | (27,147 | ) | $ | (11,185 | ) | $ | (7,522 | ) | |||
|
|
|
|
|
| |||||||
The reconciliation of income tax computed at the U.S. federal statutory tax rates to income tax expense is as follows (in thousands):
| Years Ended June 30, | ||||||||||||||||||||||||
| 2014 | 2013 | 2012 | ||||||||||||||||||||||
| $ | % | $ | % | $ | % | |||||||||||||||||||
Tax benefit at U.S. statutory rates | $ | 9,230 | 34 | % | $ | 3,803 | 34 | % | $ | 2,557 | 34 | % | ||||||||||||
State tax | 1,583 | 6 | % | 652 | 6 | % | 474 | 6 | % | |||||||||||||||
Australian tax | - | 0 | % | - | 0 | % | 41 | 1 | % | |||||||||||||||
Expiration of foreign tax losses | - | 0 | % | - | 0 | % | (28,202 | ) | -375 | % | ||||||||||||||
(Increase)/ decrease in valuation allowance | (10,814 | ) | -40 | % | (4,456 | ) | -40 | % | 25,129 | 334 | % | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||
| $ | (1 | ) | 0 | % | $ | (1 | ) | 0 | % | $ | (1 | ) | 0 | % | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||
Deferred tax liabilities and assets are comprised of the following (in thousands):
| June 30, | ||||||||
| 2014 | 2013 | |||||||
Deferred tax liabilities: | ||||||||
Change in accounting method adjustments | $ | (804 | ) | $ | (1,607 | ) | ||
|
|
|
| |||||
Total deferred tax liabilities | (804 | ) | (1,607 | ) | ||||
Deferred tax assets: | ||||||||
Tax carried forward losses | 2,947 | 2,463 | ||||||
Share-based payments | 2,948 | 1,057 | ||||||
Consultant and other accruals | 39 | 29 | ||||||
Fixed and intangible assets | 19,858 | 12,315 | ||||||
Compensation accruals | 319 | 240 | ||||||
Capital loss carryforward | 26,382 | 26,382 | ||||||
|
|
|
| |||||
Total deferred tax assets | 52,493 | 42,486 | ||||||
Valuation allowance for deferred tax assets | (51,689 | ) | (40,879 | ) | ||||
|
|
|
| |||||
Net deferred tax assets and liabilities | $ | - | $ | - | ||||
|
|
|
| |||||
Management evaluates the recoverability of the deferred tax assets and the amount of the required valuation allowance. Due to the uncertainty surrounding the realization of the tax deductions in future tax returns, the Company has recorded a valuation allowance against its net deferred tax assets as of June 30, 2014 and 2013. At such time as it is determined that it is more likely than not that the deferred tax assets will be realized, the valuation allowance would be reduced.
The Company had federal and state net operating loss carryforwards of approximately $7.7 million and $5.4 million as of June 30, 2014. The federal and state net operating losses will begin to expire in 2022 and 2029, respectively. Due to the dissolution of the Company's foreign subsidiary, all foreign tax losses expired unutilized during the year ended June 30, 2012.
56
The Company's ability to utilize its net operating loss carryforwards may be substantially limited due to ownership changes that have occurred or that could occur in the future under Section 382 of the Internal Revenue Code and similar state laws. The Company has not completed a study to determine whether one or more ownership changes have occurred.
The Company did not previously record a deferred tax asset for any basis difference in its subsidiary because the Company intended to permanently reinvest any subsidiary earnings. However, in the year ended June 30, 2011, the Company determined that it might wind up its subsidiary. As such, the Company recorded a deferred tax asset for this difference. The Company realized this loss for tax purposes during the year ended June 30, 2012, which resulted in a capital loss carryforward of $66.2 million. This capital loss will expire in 2017.
None of the Company's prior income tax returns have been selected for examination by a major taxing jurisdiction; however, the statutes of limitations for various filings remain open. The oldest filings subject to potential examination for federal, state, and foreign purposes are 2011, 2010, and 2010, respectively. If the Company utilizes a net operating loss related to a closed year, the statute for that year would re-open. The Company has not reduced any tax benefit on its financial statements due to uncertain tax positions as of June 30, 2014 and it is not aware of any circumstance that would significantly change this result through the end of fiscal year 2015. To the extent the Company incurs income-tax related penalties or interest, the Company recognizes them as additional income tax expense.
Note 11. Selected Quarterly Financial Information (Unaudited)
The following table presents the Company's unaudited quarterly results of operations for the years ended June 30, 2014 and 2013 (in thousands, except per share amounts).
| Quarters Ended | Year Ended June 30, 2014 | |||||||||||||||||||
| June 30, 2014 | March 31, 2014 | December 31, 2013 | September 30, 2013 | |||||||||||||||||
Net loss | $ | (8,541 | ) | $ | (7,387 | ) | $ | (6,324 | ) | $ | (4,896 | ) | $ | (27,148 | ) | |||||
Basic and diluted loss per share | $ | (0.40 | ) | $ | (0.34 | ) | $ | (0.32 | ) | $ | (0.29 | ) | $ | (1.35 | ) | |||||
| Quarters Ended | Year Ended June 30, 2013 | |||||||||||||||||||
| June 30, 2013 | March 31, 2013 | December 31, 2012 | September 30, 2012 | |||||||||||||||||
Net loss | $ | (3,215 | ) | $ | (2,753 | ) | $ | (2,754 | ) | $ | (2,464 | ) | $ | (11,186 | ) | |||||
Basic and diluted loss per share | $ | (0.19 | ) | $ | (0.18 | ) | $ | (0.50 | ) | $ | (0.70 | ) | $ | (1.10 | ) | |||||
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
None.
| Item 9A. | Controls and Procedures |
(a) Disclosure Controls and Procedures
At the end of the period covered by this Annual Report on Form 10-K, the Company's management, with the participation of the Company's Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of the Company's disclosure controls and procedures. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by an issuer in the reports that it files or submits under the Act is accumulated and communicated to the issuer's management, including its principal executive and principal financial officers, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure. Based on that evaluation, the Company's Chief Executive Officer and Chief Financial Officer have concluded that the Company's disclosure controls and procedures are effective to ensure that the information required to be disclosed by the Company in reports that it files or submits under the Securities Exchange Act of 1934 ("Exchange Act") is recorded, processed, summarized and reported within the time periods specified in the SEC's rules and forms.
57
A control system no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues, if any, within the Company are detected. Accordingly, our disclosure controls and procedures are designed to provide reasonable, not absolute, assurance that the objectives of our disclosure control system are met and, as set forth above, our Chief Executive Officer and Chief Financial Officer have concluded, based on their evaluation as of the end of the period covered by this Annual Report on Form 10-K, that our disclosure controls and procedures were effective to provide reasonable assurance that the objectives of our disclosure control system were met.
(b) Management's Annual Report on Internal Controls Over Financial Reporting
The Company's management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a – 15(f) under the Exchange Act. The Company's internal control was designed to provide reasonable assurance to the Company's management and Board of Directors regarding the preparation and fair presentation of published financial statements. All internal control systems, no matter how well designed, have inherent limitations. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation.
Management maintains a comprehensive system of controls intended to ensure that transactions are executed in accordance with management's authorization, assets are safeguarded and financial records are reliable. Management also takes steps to ensure that information and communication flows are effective, and to monitor performance, including performance of internal control procedures.
Management assessed the effectiveness of the Company's internal control over financial reporting as of June 30, 2014, based on the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control-Integrated Framework (2013 Framework). Based on this assessment, management believes that the Company's internal control over financial reporting is effective as of June 30, 2014.
There were no changes in internal control over financial reporting during the quarter ended June 30, 2014, that have materially affected, or are reasonably likely to materially affect, the Company's internal control over financial reporting.
The effectiveness of our internal control over financial reporting as of June 30, 2014 has been audited by BDO USA, LLP, an independent registered public accounting firm, as stated in their report which is included herein.
58
Report of Independent Registered Public Accounting Firm
Board of Directors and Stockholders
MEI Pharma, Inc.
San Diego, California
We have audited MEI Pharma, Inc.'s internal control over financial reporting as of June 30, 2014, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). MEI Pharma, Inc.'s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying, "Item 9A, Management's Annual Report on Internal Control Over Financial Reporting". Our responsibility is to express an opinion on the company's internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, MEI Pharma, Inc. maintained, in all material respects, effective internal control over financial reporting as of June 30, 2014, based on the COSO criteria .
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the balance sheets of MEI Pharma, Inc. as of June 30, 2014 and 2013 and the related statements of operations, stockholders' equity, and cash flows for each of the three years in the period ended June 30, 2014 and our report dated September 8, 2014, expressed an unqualified opinion thereon.
/s/ BDO USA, LLP
San Diego, California
September 8, 2014
Item 9B. Other Information
Not applicable.
59
PART III
Item 10. Directors and Executive Officers of the Registrant
Code of Ethics
We have adopted a Code of Business and Ethics policy that applies to our directors and employees (including our principal executive officer and our principal financial officer), and have posted the text of our policy on our website (www.meipharma.com). In addition, we intend to promptly disclose (i) the nature of any amendment to the policy that applies to our principal executive officer and principal financial officer and (ii) the nature of any waiver, including an implicit waiver, from a provision of the policy that is granted to one of these specified individuals, the name of such person who is granted the waiver and the date of the waiver on our website in the future.
The other information required by this item is incorporated herein by reference to our proxy statement for the fiscal year ended June 30, 2014 (the "Proxy Statement").
Item 11. Executive Compensation
The information required by this item is incorporated herein by reference to the Proxy Statement.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required by this item is incorporated herein by reference to the Proxy Statement.
Item 13. Certain Relationships and Related Transactions
The information required by this item is incorporated herein by reference to the Proxy Statement.
Item 14. Principal Accountant Fees and Services.
The information required by this item is incorporated herein by reference to the Proxy Statement.
60
PART IV
Item 15. Exhibits and Financial Statement Schedules
| (a) | 1. Financial Statements |
Reference is made to the Financial Statements under Item 8 in Part II hereof.
| 2. Financial Statement Schedules |
The Financial Statement Schedules have been omitted either because they are not required or because the information has been included in the financial statements or the notes thereto included in this Annual Report on Form 10-K.
| 3. Exhibits |
Exhibit Index
| 3.1 | Restated Certificate of Incorporation (incorporated by reference to Exhibit 3.1 to the Registrant's Registration Statement on Form S-1 filed on September 25, 2003 (Reg. No. 333-109129)). | |
| 3.2 | Certificate of Amendment to the Restated Certificate of Incorporation (incorporated by reference to Exhibit 3.1.1 to the Registrant's Current Report on Form 8-K filed on March 31, 2010 (File No. 000-50484)). | |
| 3.3 | Certificate of Amendment to the Restated Certificate of Incorporation (incorporated by reference to Exhibit 3.1 to the Registrant's Current Report on Form 8-K filed on December 19, 2012 (File No. 000-50484)). | |
| 3.4 | Certificate of Ownership and Merger (incorporated by reference to Exhibit 3.1 to the Registrant's Current Report on Form 8-K filed on July 2, 2012 (File No. 000-50484)). | |
| 3.5 | Certificate of Designation of Series A Convertible Preferred Stock of Marshall Edwards, Inc. (incorporated by reference to Exhibit 3.1 to the Registrant's Current Report on Form 8-K filed on May 11, 2011 (File No. 000-50484)) | |
| 3.6 | Certificate of Designation of Series B Preferred Stock of Marshall Edwards, Inc. (incorporated by reference to Exhibit 10.1 to the Registrant's Current Report on Form 8-K filed on March 18, 2011 (File No. 000-50484)) | |
| 3.8 | Amended and Restated Bylaws (incorporated by reference to Exhibit 3.2 to the Registrant's Current Report on Form 8-K filed on December 19, 2012 (File No. 000-50484)). | |
| 4.1 | Specimen Stock Certificate (incorporated by reference to Exhibit 4.1 to Amendment No. 1 to the Registrant's Registration Statement on Form S-1 filed on October 31, 2003 (Reg. No. 333-109129)). | |
| 4.2 | Specimen Warrant Certificate (incorporated by reference to Exhibit 4.2 to the Registrant's Registration Statement on Form S-3 filed on August 9, 2006 (Reg. No. 333-136440). | |
| 4.3 | Specimen Warrant Certificate (incorporated by reference to Exhibit 4.4 to the Registrant's Annual Report on Form 10-K filed on September 27, 2007 (File No. 000-50484)). | |
| 4.4 | Form of Warrant Agreement (incorporated by reference to Exhibit 10.3 to the Registrant's Current Report on Form 8-K filed on July 12, 2006 (File No. 000-50484)). | |
| 4.5 | Warrant Agreement (incorporated by reference to Exhibit 10.3 to the Registrant's Current Report on Form 8-K filed on August 6, 2007 (File No. 000-50484)). | |
61
| 4.6 | Amended and Restated Warrant Agreement (incorporated by reference to Exhibit 10.1 to the Registrant's Current Report on Form 8-K/A filed on September 27, 2007 (File No. 000-50484)). | |
| 4.7 | Form of Warrant (incorporated by reference to Exhibit 10.4 to the Registrant's Current Report on Form 8-K filed on July 12, 2006 (File No. 000-50484)). | |
| 4.8 | Form of Warrant (incorporated by reference to Exhibit 10.4 to the Registrant's Current Report on Form 8-K filed on August 6, 2007 (File No. 000-50484)). | |
| 4.9 | Form of Warrant (incorporated by reference to Exhibit 10.3 to the Registrant's Current Report on Form 8-K/A filed on September 27, 2007 (File No. 000-50484)). | |
| 4.10 | Warrant dated July 30, 2008 issued to Mr John O'Connor (incorporated by reference to Exhibit 4.1 to the Registrant's Current Report on Form 8-K filed on July 30, 2008 (File No. 000-50484)). | |
| 4.11 | Form of Amended and Restated Series A and Series B Warrants (incorporated by reference to Exhibits 4.1 and 4.2 to the Registrant's Current Report on Form 8-K filed on September 29, 2011 (File No. 000-50484)). | |
| 4.12 | Form of Subscription Agent Agreement between Marshall Edwards, Inc. and Computershare, Inc. (incorporated by reference to Exhibit 4.12 to Amendment No. 1 to the Registrant's Registration Statement on Form S-1 filed on March 20, 2012 (File No. 333-179590)). | |
| 4.13 | Form of Information Agent Agreement between the Company and Georgeson, Inc. (incorporated by reference to Exhibit 4.13 to Amendment No. 1 to the Registrant's Registration Statement on Form S-1 filed on March 20, 2012 (File No. 333-179590)). | |
| 4.14 | Form of Subscription Rights Certificate (incorporated by reference to Exhibit 4.14 to Amendment No. 1 to the Registrant's Registration Statement on Form S-1 filed on March 20, 2012 (File No. 333-179590)). | |
| 4.15 | Form of Warrant Agreement between the Company and Computershare, Inc. (incorporated by reference to Exhibit 4.15 to Amendment No. 1 to the Registrant's Registration Statement on Form S-1 filed on March 20, 2012 (File No. 333-179590)). | |
| 4.16 | Form of Warrant (incorporated by reference to Exhibit 4.16 to Amendment No. 1 to the Registrant's Registration Statement on Form S-1 filed on March 20, 2012 (File No. 333-179590)). | |
| 4.17 | Form of Warrant (incorporated by reference to Exhibit 4.1 to the Registrant's Current Report on Form 8-K filed on November 5, 2012 (File No. 000-50484)). | |
| 10.1 | Employment letter dated April 23, 2010, between Marshall Edwards, Inc. and Daniel Gold (incorporated by reference to Exhibit 10.1 to the Registrant's Current Report on Form 8-K filed on April 26, 2010 (File No. 000-50484)). | |
| 10.2 | Employment letter dated June 18, 2010, between Marshall Edwards, Inc. and Thomas Zech (incorporated by reference to Exhibit 10.1 to the Registrant's Current Report on Form 8-K filed on June 23, 2010 (File No. 000-50484)). | |
| 10.3 | Employment letter dated June 1, 2011, between Marshall Edwards, Inc. and Robert D. Mass (incorporated by reference to Exhibit 10.1 to the Registrant's Current Report on Form 8-K filed on June 2, 2011 (File No. 000-50484)). | |
| 10.4 | Registration Rights Agreement, dated July 11, 2006 by and among Marshall Edwards, Inc. and the investors signatory thereto (incorporated by reference to Exhibit 10.2 to the Registrant's Current Report on Form 8-K filed on July 12, 2006 (File No. 000-50484)). | |
| 10.5 | Registration Rights Agreement, dated as of August 6, 2007 by and among Marshall Edwards, Inc. and the purchasers signatory thereto (incorporated by reference to Exhibit 10.2 to the Registrant's Current Report on Form 8-K filed on August 6, 2007 (File No. 000-50484)). | |
62
| 10.6 | Registration Rights Agreement, dated as of September 26, 2007 by and among Marshall Edwards, Inc. and Blue Trading, LLC (incorporated by reference to Exhibit 10.2 to the Registrant's Current Report on Form 8-K/A filed on September 27, 2007 (File No. 000-50484)). | |
| 10.7 | Amended & Restated Registration Rights Agreement, dated as of May 16, 2011, between Marshall Edwards, Inc. and certain investors signatory thereto (incorporated by reference to Exhibit 10.2 to the Registrant's Current Report on Form 8-K filed on May 16, 2011 (File No. 000-50484). | |
| 10.8 | MEI Pharma, Inc. Amended and Restated 2008 Stock Omnibus Equity Compensation Plan (incorporated by reference to Exhibit 10.1 to the Registrant's Current Report on Form 8-K filed on March 29, 2013 (File No. 000-50484)). | |
| 10.9 | Asset Purchase Agreement, dated as of December 21, 2010, between Marshall Edwards, Inc. and Novogen Limited and Novogen Pty Limited (incorporated by reference to Exhibit 10.1 to the Registrant's Current Report on Form 8-K filed on December 22, 2010 (File No. 000-50484)). | |
| 10.10 | At Market Issuance Sales Agreement, dated February 7, 2011, between Marshall Edwards, Inc. and McNicoll, Lewis & Vlak LLC (incorporated by reference to Exhibit 10.1 to the Registrant's Current Report on Form 8-K filed on February 7, 2011 (File No. 000-50484)). | |
| 10.11 | Stock Purchase Agreement, dated March 17, 2011, between Marshall Edwards, Inc. and Ironridge Global IV, Ltd., including the form of Certificate of Designations of Preferences, Rights and Limitations of Series B Preferred Stock attached as Exhibit 4 thereto (incorporated by reference to Exhibit 10.1 to the Registrant's Current Report on Form 8-K filed on March 18, 2011 (File No. 000-50484)). | |
| 10.12 | Amended and Restated Securities Purchase Agreement, dated as of May 16, 2011, between Marshall Edwards, Inc. and the investors signatory thereto (incorporated by reference to Exhibit 10.1 to the Registrant's Current Report on Form 8-K filed on May 16, 2011 (File No. 000-50484)). | |
| 10.13 | Form of Indemnification Agreement (incorporated by reference to Exhibit 10.1 to the Registrant's Current Report on Form 8-K filed on August 29, 2011 (File No. 000-50484)). | |
| 10.14 | Securities Subscription Agreement, dated as of September 27, 2011, between Marshall Edwards, Inc. and Novogen Limited (incorporated by reference to Exhibit 10.1 to the Registrant's Current Report on Form 8-K filed on September 29, 2011 (File No. 000-50484)). | |
| 10.15 | Securities Subscription Agreement, dated as of December 28, 2011, between Marshall Edwards, Inc. and Novogen Limited (incorporated by reference to Exhibit 10.1 to the Registrant's Current Report on Form 8-K filed on December 29, 2011 (File No. 000-50484)). | |
| 10.16 | Letter, dated September 28, 2011, from Novogen Limited to Marshall Edwards, Inc. (incorporated by reference to Exhibit 10.2 to the Registrant's Current Report on Form 8-K filed on September 29, 2011 (File No. 000-50484)). | |
| 10.17 | Form of Supplemental Agreement between Marshall Edwards, Inc. and each of the investors party to that certain Amended and Restated Securities Purchase Agreement, dated as of May 16, 2011, by and among Marshall Edwards, Inc. and such investors (incorporated by reference to Exhibit 10.3 to the Registrant's Current Report on Form 8-K filed on September 29, 2011 (File No. 000-50484)). | |
| 10.18 | Asset Purchase Agreement, dated as of August 7, 2012, between MEI Pharma, Inc. and S*Bio Pte Ltd. (incorporated by reference to Exhibit 2.1 to the Registrant's Current Report on Form 8-K filed on August 8, 2012 (File No. 000-50484)). | |
| 10.19 | Form of Registration Rights Agreement between the Company and S*Bio Pte Ltd. (incorporated by reference to Exhibit 10.1 to the Registrant's Current Report on Form 8-K filed on August 8, 2012 (File No. 000-50484)). | |
63
| 10.20** | License Agreement, dated September 28, 2012, between Cydex Pharmaceuticals, Inc. and the Company (incorporated by reference to Exhibit 10.1 to the Registrant's Quarterly Report on Form 10-Q filed on November 13, 2012 (File No. 000-50484)). | |
| 10.21** | Supply Agreement, dated September 28, 2012, between Cydex Pharmaceuticals, Inc. and the Company (incorporated by reference to Exhibit 10.2 to the Registrant's Quarterly Report on Form 10-Q filed on November 13, 2012 (File No. 000-50484)). | |
| 10.22 | Securities Purchase Agreement, dated as of November 4, 2012, by and among the Company, Vivo Ventures Fund VII, L.P., Vivo Ventures VII Affiliates Fund, L.P., and New Leaf Ventures II, L.P., and certain other accredited investors identified in Exhibit A thereto (incorporated by reference to Exhibit 10.1 to the Registrant's Current Report on Form 8-K filed on November 5, 2012 (File No. 000-50484)). | |
| 10.23 | Form of Governance Agreement between the Company and Vivo Ventures Fund VII, L.P., Vivo Ventures VII Affiliates Fund, L.P., and New Leaf Ventures II, L.P. (incorporated by reference to Exhibit 10.2 to the Registrant's Current Report on Form 8-K filed on November 5, 2012 (File No. 000-50484)). | |
| 10.24 | Form of Registration Rights Agreement between the Company and Vivo Ventures Fund VII, L.P., Vivo Ventures VII Affiliates Fund, L.P., and New Leaf Ventures II, L.P. (incorporated by reference to Exhibit 10.3 to the Registrant's Current Report on Form 8-K filed on November 5, 2012 (File No. 000-50484)). | |
| 10.25 | Agreement, dated December 5, 2012, between MEI Pharma, Inc., Novogen Limited, Novogen Research Pty Ltd., Graham Kelly and Andrew Heaton (incorporated by reference to Exhibit 10.1 to the Registrant's Current Report on Form 8-K filed on December 7, 2012 (File No. 000-50484)). | |
| 23.1 | Consent of Independent Registered Accounting Firm* | |
| 31.1 | Certification of Chief Executive Officer pursuant to Rule 13a-14(A) promulgated under the Securities Exchange Act of 1934* | |
| 31.2 | Certification of Chief Financial Officer pursuant to Rule 13a-14(A) promulgated under the Securities Exchange Act of 1934* | |
| 32.1 | Certification of Chief Executive Officer and Chief Financial Officer pursuant to 18 U.S.C. Section 1350 and Rule 13a-14(B) promulgated under the Securities Exchange Act of 1934* | |
| 101.INS | XBRL Instance Document* | |
| 101.SCH | XBRL Taxonomy Extension Schema Document* | |
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document* | |
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document* | |
| 101.LAB | XBRL Taxonomy Extension Label Linkbase Document* | |
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document* | |
| (*) | Filed herewith. |
| (**) | Portions of this exhibit have been redacted pursuant to a confidential treatment request filed with the Securities and Exchange Commission. |
64
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Exchange Act, the registrant has duly caused this Annual Report on Form 10-K to be signed on its behalf by the undersigned, thereunto duly authorized, on September 8, 2014.
MEI PHARMA, INC. A Delaware Corporation | ||
By: | /s/ Daniel P. Gold | |
Daniel P. Gold Chief Executive Offer | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this Annual Report on Form 10-K has been signed below by the following persons on behalf of the registrant and in the capacities indicated on September 8, 2014.
Signatures | Title | |||
By: | /s/ Daniel P. Gold Daniel P. Gold | President, Chief Executive Officer and Director (Principal Executive Officer) | ||
By: | /s/ Thomas M. Zech Thomas M. Zech | Secretary, Chief Financial Officer (Principal Financial and Accounting Officer) | ||
By: | /s/ Christine A. White Christine A. White | Lead Director | ||
By: | /s/ Leah Rush Cann Leah Rush Cann | Director | ||
By: | /s/ William D. Rueckert William D. Rueckert | Director | ||
By: | /s/ Charles V. Baltic Charles V. Baltic | Director | ||
By: | /s/ Thomas C. Reynolds Thomas C. Reynolds | Director | ||
By: | /s/ Nicholas R. Glover Nicholas Glover | Director | ||
65