UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2013
or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File Number: 000-50679
CORCEPT THERAPEUTICS INCORPORATED
(Exact Name of Corporation as Specified in Its Charter)
| Delaware | 77-0487658 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
149 Commonwealth Drive
Menlo Park, CA 94025
(Address of principal executive offices) (zip code)
(650) 327-3270
(Registrant's telephone number, including area code)
Securities registered pursuant to Section 12 (b) of the Act:
Title of Each Class: | Name of Each Exchange on which Registered: | |
| Common Stock, $0.001 par value | The NASDAQ Capital Market |
Securities registered pursuant to Section 12 (g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15 (d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No r
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the Registrant's knowledge, in definitive proxy or information statements incorporated by reference to Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See the definitions of "large accelerated filer", "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act.
Large accelerated filer | ¨ | Accelerated filer | x | |||
Non-accelerated filer | ¨ (Do not check if a smaller reporting company) | Smaller reporting company | ¨ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The aggregate market value of voting and non-voting common equity held by non-affiliates of the Registrant was $111,300,000 as of June 30, 2013 based upon the closing price on the NASDAQ Capital Market reported for such date. This calculation does not reflect a determination that certain persons are affiliates of the Registrant for any other purpose.
On March 3, 2014 there were 100,579,438 shares of common stock outstanding at a par value of $0.001 per share.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Registrant's definitive proxy statement for its 2014 Annual Meeting of Stockholders are incorporated by reference in Items 10, 11, 12, 13 and 14 of Part III.
TABLE OF CONTENTS
Form 10-K
For the year ended December 31, 2013
| Page | ||||||
PART I | ||||||
ITEM 1. | Business | 1 | ||||
ITEM 1A. | Risk Factors | 22 | ||||
ITEM 1B. | Unresolved Staff Comments | 46 | ||||
ITEM 2. | Properties | 46 | ||||
ITEM 3. | Legal Proceedings | 46 | ||||
ITEM 4. | Mine Safety Disclosures | 46 | ||||
PART II | ||||||
ITEM 5. | Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | 47 | ||||
ITEM 6. | Selected Financial Data | 49 | ||||
ITEM 7. | Management's Discussion and Analysis of Financial Condition and Results of Operations | 50 | ||||
ITEM 7A. | Quantitative and Qualitative Disclosures About Market Risk | 64 | ||||
ITEM 8. | Financial Statements and Supplementary Data | 64 | ||||
ITEM 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure | 64 | ||||
ITEM 9A. | Controls and Procedures | 64 | ||||
ITEM 9B. | Other Information | 66 | ||||
PART III | ||||||
ITEM 10. | Directors, Executive Officers and Corporate Governance | 67 | ||||
ITEM 11. | Executive Compensation | 67 | ||||
ITEM 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 67 | ||||
ITEM 13. | Certain Relationships and Related Transactions, and Director Independence | 67 | ||||
ITEM 14. | Principal Accounting Fees and Services | 67 | ||||
PART IV | ||||||
ITEM 15. | Exhibits, Financial Statement Schedules | 68 | ||||
| Signatures and Power of Attorney | 72 | |||||
PART I
This Annual Report on Form 10-K (Form 10-K) contains forward-looking statements within the meaning of Section 21E of the Securities Exchange Act of 1934, as amended (Exchange Act), and Section 27A of the Securities Act of 1933, as amended (Securities Act). All statements contained in this Form 10-K, other than statements of historical fact, are forward-looking statements. When used in this report or elsewhere by management from time to time, the words "believe," "anticipate," "intend," "plan," "estimate," "expect," "may," "will," "should," "seeks" and similar expressions are forward-looking statements. Such forward-looking statements are based on current expectations, but the absence of these words does not necessarily mean that a statement is not forward-looking. Forward-looking statements made in this Form 10-K include, but are not limited to, statements about:
| • | our ability to manufacture, market and sell Korlym ® (mifepristone) 300 mg Tablets; |
| • | our estimates regarding enrollment in and the dates by which we expect to report results of our clinical trials and the anticipated results of these trials; |
| • | the progress and timing of our research, development and clinical programs and the timing of regulatory activities for mifepristone for the treatment of the psychotic features of psychotic depression, triple-negative breast cancer or other indications; |
| • | our ability to realize the benefits of Orphan Drug Designation of Korlym in the United States; |
| • | the timing of the market introduction of future product candidates, including new uses for mifepristone and any compound in our families of selective glucocorticoid receptor II (GR-II) antagonists; |
| • | our ability to achieve marketing approval of mifepristone in the European Union (EU) (for which we have requested the brand name Corluxin ® ) and realize the benefits of Orphan Drug Designation there; |
| • | our ability to manufacture, market, commercialize and achieve market acceptance for our future product candidates, including mifepristone for the treatment of the psychotic features of psychotic depression, triple-negative breast cancer or any other indications and any compounds in our families of selective GR-II antagonists; |
| • | uncertainties associated with obtaining and enforcing patents; |
| • | our estimates for future performance, including revenue and profits; and |
| • | our estimates regarding our capital requirements. |
Forward-looking statements are not guarantees of future performance and involve risks and uncertainties. Actual events or results may differ materially from those discussed in the forward-looking statements as a result of various factors. For a more detailed discussion of such forward-looking statements and the potential risks and uncertainties that may impact upon their accuracy, see the "Risk Factors" section of this Form 10-K and the "Overview" and "Liquidity and Capital Resources" sections of the "Management's Discussion and Analysis of Financial Condition and Results of Operations" section of this Form 10-K. These forward-looking statements reflect our view only as of the date of this report. Except as required by law, we undertake no obligations to update any forward-looking statements. Accordingly, you should also carefully consider the factors set forth in other reports or documents that we file from time to time with the Securities and Exchange Commission (SEC).
| ITEM 1. | BUSINESS |
Overview
We are a pharmaceutical company engaged in the discovery, development and commercialization of drugs for the treatment of severe metabolic, psychiatric and oncologic disorders. Our focus is on those disorders that are associated with a steroid hormone called cortisol. Elevated levels and abnormal release patterns of cortisol have been implicated in a broad range of human disorders. Since our inception in 1998, we have been developing
1
mifepristone - a potent glucocorticoid receptor II (GR-II) antagonist that modulates the activity of cortisol - for the treatment of a number of severe metabolic, psychiatric and oncologic disorders. We have also discovered three series of novel selective GR-II antagonists and plan to move one or more of the new compounds from these series into clinical development in 2014.
In February 2012, the United States Food and Drug Administration (FDA) approved Korlym ® (mifepristone) 300 mg Tablets as a once-daily oral medication for the treatment of hyperglycemia secondary to hypercortisolism in adult patients with endogenous Cushing's syndrome who have type 2 diabetes mellitus or glucose intolerance and have failed surgery or are not candidates for surgery. FDA approval means that we can market the drug for the approved indication in the United States. We made Korlym available to patients in April 2012 and continue to develop the sales, marketing, medical affairs and logistical infrastructure needed to commercialize the drug.
We have an ongoing Phase 3 study of mifepristone, the active ingredient in Korlym, for the psychotic features of psychotic depression. We will perform an interim analysis of data from this study and expect to have results in the second quarter of 2014. If the results are positive, we believe they would support the submission of a supplemental New Drug Application (NDA) by the end of 2014. We have also begun a Phase 1 safety and efficacy study of mifepristone in combination with chemotherapy in the treatment of triple-negative breast cancer – a form of cancer with a particularly poor prognosis. There is no FDA-approved treatment for either of these illnesses.
Unless otherwise stated, all references in this document to "we," "us," "our," "Corcept," the "Company," "our company" and similar designations refer to Corcept Therapeutics Incorporated.
Cushing's Syndrome. Cushing's syndrome is a disorder caused by prolonged exposure of the body's tissues to high levels of the hormone cortisol. Sometimes called "hypercortisolism," it is relatively uncommon and most often affects adults aged 20 to 50. An estimated 10 to 15 of every one million people are newly diagnosed with this syndrome each year, resulting in approximately 3,000 new patients and an estimated prevalence of 20,000 patients with Cushing's syndrome in the United States.
We received Orphan Drug Designation from the FDA in July 2007 and in the EU in October 2011 for Korlym for the treatment of endogenous Cushing's syndrome. In the United States, Orphan Drug Designation is a special status granted by the FDA to encourage the development of treatments for diseases or conditions that affect fewer than 200,000 patients. Drugs that receive Orphan Drug Designation in the United States obtain seven years of marketing exclusivity for the approved indication from the date of drug approval, as well as tax credits for clinical trial costs, marketing application filing fee waivers and assistance from the FDA in the drug development process. Even after an orphan drug is approved for its orphan indication, the FDA can later approve a different drug for the same condition if the FDA concludes that the later drug is safer, more effective or makes a major contribution to patient care. In addition, the FDA may, during the orphan exclusivity period, approve the same drug for a different indication.
Orphan Drug Designation in the EU confers benefits similar to those in the U.S., but includes ten years of marketing exclusivity for the approved indication in all 28 Member States, free scientific advice during drug development, access to a centralized review process and a reduction or complete waiver of fees levied by the European Medicines Agency (EMA). In October 2013, we submitted a Marketing Authorization Application (MAA) to the EMA that, subject to review by the EMA, could serve as the basis for the approval of mifepristone in the EU.
Psychotic Depression. We are also developing mifepristone, Korlym's active ingredient, for treatment of the psychotic features of psychotic depression under an exclusive patent license from Stanford University. The FDA has granted "fast track" status to evaluate the safety and efficacy of mifepristone for the treatment of the psychotic features of psychotic depression.
2
In 2008, we began enrollment in Study 14, our ongoing Phase 3 trial in psychotic depression. The protocol for this trial incorporates what we have learned from our three previously completed Phase 3 trials. It attempts to address the established relationship between increased drug plasma levels and clinical response and attempts to decrease the random variability observed in the results of the psychometric instruments used to measure efficacy. In one of the previously completed Phase 3 trials, Study 06, we prospectively tested and confirmed that patients whose plasma levels rose above a predetermined threshold statistically separated from both those patients whose plasma levels were below the threshold and those patients who received placebo; this threshold was established from data produced in earlier studies.
As expected, the group of patients who took 1200 milligrams (mg) of mifepristone in Study 06 developed higher drug plasma levels than did the groups of patients who received lower doses. Further, there was no discernible difference in the incidence of adverse events between patients who received placebo in Study 06 and those who received 300 mg, 600 mg or 1200 mg of mifepristone in that study. In August 2011, we published our analysis of these data in The Journal of Clinical Psychopharmacology . Based on this information, we are using a mifepristone dose of 1200 mg once per day for seven days in Study 14.
In addition, we are utilizing a third party centralized rating service to independently evaluate patients for entry into the study as well as to evaluate their level of response throughout their participation. We believe the centralization of this process will improve the consistency of rating across clinical trial sites and reduce the background noise that was experienced in earlier studies and is endemic to psychopharmacologic studies. We believe that this change in dose, as well as the other modifications to the protocol, should allow us to demonstrate the efficacy of mifepristone in the treatment of the psychotic symptoms of psychotic depression. We will perform an interim analysis of data from this study and expect to have results in the second quarter of 2014. If the results are positive, we believe they would support the submission of a supplemental NDA by the end of the year.
Triple-Negative Breast Cancer. In January 2014 we began a Phase 1 study of mifepristone in combination with the chemotherapy drug eribulin in the treatment of triple-negative breast cancer.
Triple-negative breast cancer is a form of the disease in which the three receptors that fuel most breast cancer growth – estrogen, progesterone, and the HER-2/neu gene – are not present. Since the tumor cells lack these receptors, common treatments, such as hormone therapy and drugs that target estrogen, progesterone, and HER-2, are ineffective. In 2013, approximately 40,000 women were diagnosed with triple-negative breast cancer. There is no FDA-approved treatment and neither a targeted treatment nor a preferred standard chemotherapy regimen for relapsed triple-negative breast cancer patients exists.
We plan to conduct our study in two phases. The first phase will determine in up to 20 patients with metastatic breast cancer the recommended dose for the second phase of the study. In the second phase, 20 patients with GR-II positive triple-negative breast cancer will be dosed to determine a preliminary estimate of efficacy. Mifepristone will be administered orally with food once daily and eribulin will be administered intravenously. We expect to have initial results in the first half of 2015.
Antipsychotic-Induced Weight Gain Mitigation. In 2005, we published the results of studies in rats that demonstrated that mifepristone both reversed the weight gain associated with the ongoing use of olanzapine and mitigated the weight gain associated with the initiation of treatment with olanzapine (the active ingredient in Zyprexa ® ). The results from this study were published in the journal Brain Behavioral Research in 2006. The study was paid for by Eli Lilly and Company (Eli Lilly).
During 2007 we announced positive results from our clinical proof-of-concept study in lean healthy male volunteers evaluating the ability of mifepristone to mitigate weight gain associated with the use of Zyprexa. The results show a statistically significant reduction in weight gain in those subjects who took Zyprexa plus mifepristone compared to those who took Zyprexa plus placebo. Also, the addition of mifepristone to treatment with Zyprexa had a beneficial impact on secondary metabolic measures such as fasting insulin, triglycerides and
3
abdominal fat, as indicated by waist circumference. Eli Lilly provided Zyprexa and financial support for this study and its results were published in the journal Advances in Therapy in 2009. In January 2009, we announced positive results from a similar proof-of-concept study evaluating the ability of mifepristone to mitigate weight gain associated with the use of Johnson & Johnson's Risperdal ® . This study confirmed and extended the earlier results seen with mifepristone and Zyprexa, demonstrating a statistically significant reduction in weight and secondary metabolic endpoints of fasting insulin, triglycerides and abdominal fat, as indicated by waist circumference. The results from the study of mifepristone and Risperdal were presented at several scientific conferences, including the American Diabetes Association meeting in June 2009, and were published in the journal Obesity in 2010.
The combination of Zyprexa or Risperdal and mifepristone is not approved for any indication. The purpose of these studies was to explore the hypothesis that GR-II antagonists, such as mifepristone and our next generation of selective GR-II antagonists, would mitigate weight gain associated with antipsychotic medications. The group of medications known as second generation antipsychotic medication, including Zyprexa, Risperdal, Clozaril ® and Seroquel ® , are widely used to treat schizophrenia and bipolar disorder. All medications in this group are associated with treatment-emergent weight gain of varying degrees and carry a warning in their labels relating to treatment-emergent hyperglycemia and diabetes mellitus.
Selective GR-II Receptor Antagonists. In 2003, we initiated a discovery research program to identify and patent selective GR-II antagonists. Our intent is to develop a pipeline of products for proprietary use. Three distinct series of selective GR-II antagonists have been identified. These compounds, like the active ingredient in our lead product Korlym, potently block a cortisol receptor (GR-II) but do not appear to block the PR (progesterone), ER (estrogen), AR (androgen) or GR-I (mineralocorticoid) receptors. Both the United States Patent & Trademark Office (USPTO) and the European Patent Office (EPO) have issued to us composition of matter patents in each of the three series. One additional composition of matter patent application is pending. See "Business - Intellectual Property."
Several of our new compounds have demonstrated positive results in animal or in vitro models for the prevention and reversal of alcohol dependence; amyotrophic lateral sclerosis (Lou Gehrig's disease); Alzheimer's disease; anti-psychotic-induced weight gain; breast, ovarian and prostate cancer (in combination with a chemotherapeutic agent); electroconvulsive-induced retrograde amnesia; metabolic syndrome; muscular dystrophy; obesity; prevention of glucocorticoid-induced neurological damage in premature infants; and post-traumatic stress disorder. One of these new compounds, CORT 108297, has completed Phase 1 trials and we may explore its potential use in psychiatric and other central nervous system disorders. We intend to continue our discovery research program with the goal of identifying new selective GR-II antagonists, to manufacture and conduct pre-clinical development of one or more of these compounds and to study the most promising of them in humans. We expect to begin Phase 1 studies in one or more of our new compounds during 2014.
The Role of Cortisol in Disease
Cortisol is a steroid hormone that plays a significant role in the way the body reacts to stressful conditions and is essential for survival. Cortisol significantly influences metabolism, exerts a clinically useful anti-inflammatory effect and contributes to emotional stability. Insufficient levels of cortisol may lead to dehydration, hypotension, shock, fatigue, low resistance to infection, trauma, stress and hypoglycemia. Excessive levels of cortisol may lead to impaired glucose tolerance, diabetes, obesity, depressed mood, psychosis, wasting of the arms and legs, edema, fatigue, hypertension, and other problems. Pre-clinical and clinical data suggest that cortisol activity at GR-II may protect certain cancer cells from the effects of chemotherapy.
Elevated levels and abnormal release patterns of cortisol have also been linked to a broad range of conditions, such as weight gain, diabetes, hypertension, mood changes, psychosis and cognitive impairment.
4
While excess cortisol may play a role in numerous diseases, Cushing's syndrome (sometimes called "hypercortisolism") is the archetypal disease of excess cortisol, as Cushing's syndrome patients have tumors that produce excess levels of cortisol or adrenocorticotropic hormone (ACTH), which stimulates the production of cortisol. Exposure to high levels of cortisol can result in weight gain, diabetes, hypertension, infections, severe fatigue and psychosis.
Many studies have shown that patients with psychotic depression have elevated levels and abnormal release patterns of cortisol. This abnormal cortisol activity is not usually present in patients with nonpsychotic depression. More than 20 years ago, one of our scientific co-founders postulated that elevated levels of cortisol in patients with psychotic depression lead to elevated levels of dopamine, an important chemical substance found in the brain. Elevated levels of dopamine have been implicated in both delusional thinking and hallucinations. This hypothesis led to the concept that, by regulating the level and release patterns of cortisol, one could normalize dopamine levels in the brain, which may, in turn, ameliorate the symptoms of psychotic depression. In addition to cortisol's effect on dopamine levels, research has shown that prolonged elevated cortisol may also play a direct role in causing the symptoms of psychotic depression.
The challenge in regulating levels of cortisol is that cortisol is needed for natural processes in the human body. Destroying the ability of the body to make cortisol or to drastically reduce its presence would result in serious detrimental effects. To have a viable therapeutic effect, a compound must be able to selectively modulate cortisol's effects.
Glucocorticoid Receptor Antagonists
Cortisol is produced by the adrenal glands and is carried via the bloodstream throughout the body, including to the brain, where it directly influences neuronal function. In the brain, cortisol binds to two receptors, Glucocorticoid Receptor I and Glucocorticoid Receptor II, also known as GR-I and GR-II. GR-I is a high-affinity receptor that is involved in the routine functions of cortisol in the brain. It has approximately ten times the affinity of GR-II for cortisol and its binding sites are filled with cortisol nearly all the time. In general, GR-II binding sites do not fill until levels of cortisol become elevated. Short-term activation of GR-II has benefits, which include helping the individual to be more alert and better able to function under stressful conditions. Long-term activation of GR-II, however, has been shown to have significant toxicity and appears to be linked to multiple metabolic, psychiatric and oncologic disease states, such as Cushing's syndrome and psychotic depression. Cortisol activity also appears to suppress the effect of chemotherapy in triple-negative breast cancer as well as ovarian and prostate cancer. The action of cortisol can be moderated by the use of blockers, or antagonists, that prevent the binding of the hormone to its receptors. These antagonists, referred to as glucocorticoid or cortisol receptor antagonists, may prevent the undesirable effects of elevated levels and abnormal release patterns of cortisol.
Mifepristone, the active ingredient in Korlym, works by selectively blocking the binding of cortisol to GR-II. It is neither an antagonist nor agonist of GR-I. It also blocks the binding of progesterone to the progesterone receptor (PR). Because of its selective affinity, we believe that mifepristone can have a therapeutic benefit by modulating the effects of abnormal levels and release patterns of cortisol without compromising the necessary normal functions of cortisol. We have also discovered three series of additional compounds that, like mifepristone, potently block the GR-II receptor, but do not block the progesterone receptor. One of these compounds, CORT 108297, has successfully completed Phase 1 trials and we believe is a potential therapy for several indications. We plan to advance one or more compounds to Phase 1 studies in 2014.
Overview of Cushing's Syndrome
Endogenous Cushing's syndrome is caused by prolonged exposure of the body's tissues to high levels of the hormone cortisol produced by a tumor or tumors. In endogenous Cushing's syndrome, the excess cortisol is stimulated or directly produced by pituitary, adrenal or ectopic tumors. Cushing's syndrome is an orphan
5
indication which most commonly affects adults aged 20 to 50. An estimated 10 to 15 of every one million people are newly diagnosed with this syndrome each year, resulting in approximately 3,000 new patients in the United States. An estimated 20,000 patients in the United States have been diagnosed with Cushing's syndrome. Symptoms vary, but most people have one or more of the following manifestations: high blood sugar, diabetes, high blood pressure, upper body obesity, rounded face, increased fat around the neck, thinning arms and legs, severe fatigue and weak muscles. Irritability, anxiety, cognitive disturbances and depression are also common. Cushing's syndrome can affect every organ system in the body and can be lethal if not treated effectively.
The preferred treatment for Cushing's syndrome patients is surgery, which if successful can cure the disease. Depending on the type of tumor, surgery can result in a range of complications and has varying rates of success. In approximately half of the patients, surgery is not successful, either because the tumor cannot be removed completely or the disease returns.
Commercialization of Korlym
Korlym is the first approved therapy for patients with endogenous Cushing's syndrome. In February 2012, the FDA approved Korlym for the treatment of hyperglycemia secondary to hypercortisolism in adult patients with endogenous Cushing's syndrome who have type 2 diabetes mellitus or glucose intolerance and have failed surgery or are not candidates for surgery. As indicated in the medicine's prescribing information, physicians prescribing Korlym may determine the appropriate dose for each patient by assessing tolerability and degree of improvement in manifestations of Cushing's syndrome. In the first six weeks, these manifestations may include changes in glucose control, anti-diabetic medication requirements, insulin levels and psychiatric symptoms. After two months, assessment may also be based on improvements in cushingoid appearance, acne, hirsutism, striae and decreased body weight, along with further changes in glucose control.
We first made Korlym available to patients in April 2012 and have been marketing it in the United States without a partner because we believe that the market is concentrated and accessible. Following the drug's approval, we hired a small number of experienced medical science liaisons (MSLs), supported by medical affairs and other infrastructure, to educate health care providers about Korlym. To reach more physicians, in October 2012 we deployed a small force of experienced field sales personnel. We intend for our MSLs and sales representatives to target the approximately 1,500 endocrinologists who care for a large portion of the Cushing's syndrome population. We also reach patients directly through web-based initiatives and interactions with patient groups. We use a specialty pharmacy, a specialty distributor, and a third-party logistics company to distribute Korlym and provide logistical support and have also engaged a contract sales organization.
A large percentage of the people who suffer from Cushing's syndrome remain undiagnosed or inadequately treated. We intend to develop programs to educate the medical community and patients about early diagnosis of this syndrome and to increase awareness regarding the role of GR-II antagonists for this syndrome. We have retained a vendor to help patients with the reimbursement process and to administer our financial assistance programs for uninsured or under-insured patients. We donate money to the National Organization for Rare Disorders (NORD), an independent charitable foundation that helps Cushing's syndrome patients who satisfy its financial criteria pay for their Cushing's syndrome care.
Both the FDA and the European Commission have granted Orphan Drug Designation for Korlym. In 2014, we plan to commence a study of Korlym in pediatric Cushing's syndrome patients. If we complete the study and submit the data to the FDA pursuant to a pediatric written request and to the EMA pursuant to an approved pediatric investigation plan that we may agree upon with these agencies, our Orphan Drug marketing exclusivity period will be extended by six months in the United States and two years in the EU.
Additional Trials and Preclinical Studies
As part of its approval for Korlym, the FDA has required us to study the interactions, if any, between Korlym and ketoconazole, an anti-fungal agent that is sometimes used to treat Cushing's syndrome, although it is
6
not approved by the FDA for that purpose. Further, the FDA has required us to perform a drug utilization study to better characterize the reporting rates of adverse events associated with the long-term use of Korlym. On our own initiative, we conducted a long-term extension study in patients who completed the Phase 3 trial to assess safety of chronic dosing. Upon the approval of Korlym, we transitioned study patients to commercial product and terminated the study.
Overview of Psychotic Depression
Psychotic depression is a serious psychiatric disease in which a patient suffers from severe depression accompanied by delusions, hallucinations or both. These psychotic features typically develop after the onset of a depressed mood, but may develop concurrently as well. Once psychotic symptoms occur, they usually reappear with each subsequent depressive episode. Of particular importance, when the patient's mood returns to normal the psychosis also resolves.
Data from the National Institute of Mental Health published in 2005 indicate that depressive disorders affect an estimated 9.5% of adults in the United States, or about 19 million people each year. Of these 19 million people, many published studies show that approximately 15-20%, or about three million people, have psychotic depression. Most patients with psychotic depression suffer their first episode of major depression between the ages of 30 and 40 and the majority will experience more than one episode in their lifetime. People with psychotic depression are approximately 70 times more likely to commit suicide in their lifetime than the general population and often require lengthy and expensive hospital stays.
Current Treatments for Psychotic Depression
There are two treatment approaches for the psychotic features of psychotic depression currently used by psychiatrists: electroconvulsive therapy (ECT) and combination drug therapy, which is a combination of antidepressant and antipsychotic medication. Neither of these treatments has been approved by the FDA for the psychotic features of psychotic depression and both approaches almost always have a slow onset of action, which may result in lengthy and costly hospitalization. Each of these treatments can have debilitating side effects. Of the two treatments, ECT is generally considered to be more effective.
| • | ECT involves passing an electrical current through the brain until the patient has a seizure. At least 100,000 patients receive ECT each year in the United States, with each patient requiring approximately six to twelve procedures over a period of three to five weeks. |
| • | Combination drug therapy is an alternative treatment for the psychotic features of psychotic depression that involves taking antipsychotic drugs such as olanzapine, haloperidol or chlorpromazine in combination with antidepressant drugs, such as fluoxetine, imipramine or venlafaxine. Patients on combination drug therapy often require three weeks or more to show improvement in their symptoms and treatment can take months before the symptoms are resolved entirely. Antipsychotic drugs can cause significant adverse side effects, including weight gain, diabetes, sedation, permanent movement disorders and sexual dysfunction. |
Mifepristone for the Psychotic Features of Psychotic Depression
We are developing mifepristone as an oral medication to treat the psychotic features of psychotic depression. As a GR-II antagonist, mifepristone appears to mitigate the effects of the elevated and abnormal release patterns of cortisol in patients suffering from psychotic depression. We intend mifepristone to be a once-daily treatment given to patients with psychotic depression over seven consecutive days in a controlled setting, such as a hospital or physician's office.
7
We believe that mifepristone may significantly reduce psychotic symptoms of psychotic depression in many patients within one week and allow patients to be more easily maintained on antidepressant therapy alone without the need for ECT or antipsychotic medication. We believe that mifepristone may be superior to currently available treatments because we believe that mifepristone will enable patients with psychotic depression to improve their quality of life more quickly and with fewer side effects than with ECT or combination drug therapy.
Completed Clinical Trials of Mifepristone for Psychotic Depression
We have completed seven prior clinical trials evaluating mifepristone for treatment of the psychotic features of psychotic depression, in addition to our ongoing Phase 3 trial. The trials include three Phase 3 trials conducted from 2004 through 2007, in addition to four earlier stage clinical trials with mifepristone. These completed trials generated important data confirming the safety profile of mifepristone (alone and in combination with commonly prescribed antipsychotic and antidepressant medications), demonstrated positive efficacy trends, and provided insights into the design of future clinical trials which might improve the probability of clinical success.
Completed Phase 3 Clinical Trials : In addition to Phase 1 and 2 studies, we have completed three randomized, double-blind, placebo-controlled Phase 3 clinical trials to further assess the safety and efficacy of mifepristone for the treatment of the psychotic features of psychotic depression. Two of these trials (Study 06 and Study 07) were conducted primarily in the United States. The third trial (Study 09) was conducted in Eastern Europe.
The primary endpoint for Study 06 and Study 07 was the proportion of patients with at least a 50% improvement in the Brief Psychiatric Rating Scale Positive Symptom Subscale (BPRS PSS) at both Day 7 and Day 56. The primary endpoint for Study 09 was the proportion of patients with at least a 50% improvement in the BPRS PSS, at both Day 7 and Day 28, with Day 56 as a secondary endpoint. Patients must have had at least mild psychotic symptoms (BPRS PSS ³ 12) to enter the studies and were hospitalized if clinically necessary.
| • | Study 07: The first of these trials enrolled 257 patients randomized one-to-one to either treatment or placebo. Patients in the treatment arm received 600 mg of mifepristone once daily for seven days. Patients did not take any antidepressant or antipsychotic medication for at least one week before beginning the seven day treatment period. After the seven days of mifepristone treatment, all patients received antidepressant therapy through Day 56. Treatment with antipsychotic medications or ECT was not allowed at any time during the study. |
In this study patients receiving mifepristone did not have a statistically significant difference in response rate at the primary endpoint than did the patients receiving placebo. A retrospective analysis of the data showed that patients achieving drug plasma levels higher than 1,800 nanograms per milliliter (ng/ml) had a statistically significant greater response rate than placebo. There was also a statistically significant site by treatment effect in this trial. Among the 20 sites that participated from the trial onset, patients who were given mifepristone had a significantly higher response rate than patients who received placebo. Among the sites added later in the trial, there was no significant difference in response rate between mifepristone and placebo patients. These findings were published in 2009 by Contemporary Clinical Trials.
| • | Study 09 : This study was a randomized, double-blind, placebo-controlled study in which 247 patients were enrolled at sites in Eastern Europe. Patients in the treatment arm received 600 mg of mifepristone once daily for seven days. The primary endpoint was the proportion of patients with at least a 50% improvement in the BPRS PSS score at both Day 7 and Day 28. The study did not demonstrate a significant difference in response between patients receiving mifepristone and patients receiving placebo as measured by the primary endpoint. The results at the two key secondary endpoints of Study 09 also were not statistically significant. Study 09 had an extremely high placebo response rate. |
8
| • | Study 06: This trial enrolled 443 patients. These patients were randomly assigned to three active dose groups (300 mg, 600 mg and 1200 mg) or a placebo group, with patients receiving once daily dosing for a period of seven days. The three dosing levels responded to the FDA's request to supplement data on a range of doses to augment the data provided by our open label dose ranging study completed in 2001. |
The study did not achieve statistical significance with respect to the primary endpoint. However, there was a statistically significant correlation between plasma levels and clinical outcome achieved during treatment. Response rates for patients whose plasma levels rose above a predetermined threshold of 1661 ng/ml were statistically different than those patients whose plasma levels were below the threshold and those patients who received placebo. Further, the incidence of serious adverse events did not differ between placebo and any of the three mifepristone dose groups. In August 2011, we published an analysis of these results in The Journal of Clinical Psychopharmacology .
Ongoing Phase 3 trial – Study 14: We believe that the confirmation of a correlation between drug concentration and clinical response, as well as other observations from Study 06 and our two other completed Phase 3 clinical trials, served as a strong basis for the design of our ongoing Phase 3 study, which commenced in March 2008. The protocol for this trial incorporates information learned from the three completed Phase 3 trials in that it addresses the established relationship between increased drug plasma levels and clinical response, and it attempts to decrease the random variability observed in the results of the psychometric instruments used to confirm diagnosis and measure efficacy.
| • | Increased "Signal": In this trial we are administering a mifepristone dose of 1200 mg once per day for seven days instead of 600 mg once per day for seven days. |
| • | Decreased "Noise": We also are utilizing a third party centralized rating service to independently evaluate the patient's diagnosis prior to entry into the study as well as to assess response. We believe the centralization of this process will improve the accuracy of diagnosis and the consistency of rating across clinical trial sites and reduce the background noise that is endemic to psychopharmacologic studies and clearly visible in our earlier studies. |
We believe that these changes in the protocol should allow us to establish the efficacy of mifepristone in the treatment of the psychotic features of psychotic depression. Given the serious nature of psychotic depression, the lack of any approved drugs for the disorder and the data from our first clinical trial, the FDA granted a fast track designation for mifepristone for the treatment of the psychotic features of psychotic depression. In addition, the FDA has indicated that mifepristone will receive a priority review if no other treatment is approved for the psychotic features of psychotic depression at the time we submit our NDA.
Enrollment in Study 14 is ongoing. We will perform an interim analysis of the first 226 patients and expect to have the results of that analysis in the second quarter of 2014. If the outcome is positive, we plan to stop the study and submit an NDA by the end of the year.
Mifepristone for the Treatment of Triple-Negative Breast Cancer
We are studying the safety and efficacy of mifepristone in combination with chemotherapy as a treatment for triple-negative breast cancer, a particularly deadly form of cancer. In January 2014, we began our Phase 1 study of mifepristone in combination with the chemotherapy drug eribulin. Our study follows positive findings of a Phase 1 study conducted by researchers at the University of Chicago in which 5 of 8 patients with triple-negative breast cancer had partial or complete responses to treatment with a combination of mifepristone and nab-paclitaxel. The University of Chicago results were announced at the San Antonio Breast Cancer Symposium in December 2013.
9
We plan to conduct our study in two phases. The first phase will determine in up to 20 patients with metastatic breast cancer the recommended dose for the second phase of the study. In the second phase, 20 patients with GR-II-positive, triple-negative breast cancer will be dosed to determine a preliminary estimate of efficacy. Mifepristone will be administered orally with food once daily and eribulin will be administered intravenously. We expect to have initial results in the first half of 2015.
Clinical Trial Agreements
Many of our clinical trials are conducted through the use of clinical research organizations (CROs.) At our request, these organizations oversee clinical trials at various institutions to test the safety and efficacy of our product candidates for the targeted indications. Our ongoing Phase 3 clinical trial, Study 14, evaluating mifepristone for the treatment of the psychotic features of psychotic depression is being conducted under an agreement with ICON Clinical Research, LP (ICON). We may terminate this agreement with 60 days notice to ICON, or sooner based on mutual agreement of the parties. In addition, we entered into an agreement with MedAvante, Inc. (MedAvante) to provide the centralized psychiatric diagnosis and rating services for patients being screened and enrolled in Study 14. We may terminate this agreement with 30 days notice to MedAvante. Our Phase 1 trial for the study of mifepristone in the treatment of triple-negative breast cancer is being conducted under an agreement with Ockham Development Group Inc. (Ockham). This agreement may be terminated with 60 days notice to Ockham or sooner if the parties agree to do so.
Discovery Research: Next-Generation Selective GR-II Antagonists
In 2003, we initiated a discovery research program to identify and patent selective GR-II antagonists at a contract research organization in the United Kingdom. Through this program we have identified and filed patent applications for three distinct series of selective GR-II antagonists. These compounds appear to be as potent as our lead product mifepristone in blocking cortisol but do not appear to block the progesterone or other steroid receptors. Currently, we are investigating several compounds in our research programs. We plan to submit Investigational New Drug (IND) applications for such additional compounds as our research indicates may be promising and as we deem appropriate.
We have assembled a patent portfolio covering both a broad range of uses and the composition of our new chemical entities.
| • | We have six United States composition of matter patents containing claims relating to three distinct series of novel selective GR-II antagonists, with an additional such patent pending. Three of these patents have issued in Europe, with applications for three more pending. The expiration dates of these U.S. and European patents range from 2026 to 2032. |
| • | We also have a portfolio of patents describing the use of drugs that competitively antagonize the GR-II receptor for the treatment of metabolic, psychiatric and oncologic disorders. In addition to psychotic depression, we own or have exclusively licensed issued patents for the use of GR-II antagonists for treatment and / or prevention of: |
| • | triple-negative breast cancer; |
| • | weight gain following treatment with antipsychotic medications; |
| • | mild cognitive impairment; |
| • | stress disorders; |
| • | early dementia, including early Alzheimer's disease; |
| • | migraine headaches; |
| • | delirium; |
| • | gastroesophageal reflux disease; |
| • | cognitive deterioration in adults with Down's Syndrome; |
| • | psychosis associated with Interferon-Alpha therapy; |
10
| • | psychosis associated with cocaine addiction; |
| • | catatonia; and |
| • | increased therapeutic response to ECT. |
We also own a method of use patent for the optimization of mifepristone levels in plasma serum in patients suffering from mental disorders.
See "Business – Intellectual Property."
Proof-of-Concept Studies with Next-Generation Selective GR-II Antagonists
Prevention and Reversal of Antipsychotic-Induced Weight Gain
In January 2009, we announced results from two preclinical studies of our first next-generation selective GR-II receptor antagonist, CORT 108297, for the prevention and reversal of weight gain caused by olanzapine, a medication marketed by Eli Lilly as Zyprexa. Using the same experimental rat model used previously with mifepristone, the preclinical studies demonstrated that CORT 108297 (1) reversed and (2) prevented the weight gain caused by olanzapine in rats. Eli Lilly provided olanzapine and funded the cost of the studies. The results of these two experiments replicated the findings from previous animal studies of mifepristone, and were also consistent with results from randomized trials conducted in humans. The results were published in the peer-reviewed journal, Diabetes Obesity and Metabolism in 2010. A third study in the rat further evaluated the dose response relationship of CORT 108297 in preventing olanzapine induced weight gain with doses from 2 mg/kg to 20 mg/kg.
At the American Diabetes Association conference in 2009 there was a presentation of preclinical data from a study which demonstrated that CORT 108297 suppresses body weight gain and improves insulin sensitivity in healthy mice fed a 60% fat diet and high sucrose liquid. In 2011, these study results were published in the peer-reviewed publication, The Journal of Nutrition and Metabolism .
Other Disorders
We have collaborated with researchers investigating the utility of our proprietary selective GR-II antagonists in pre-clinical studies in a wide range of disorders, including Cushing's syndrome, alcoholism, post-traumatic stress disorder, Alzheimer's disease, amyotrophic lateral sclerosis (ALS), muscular dystrophy, the metabolic syndrome, ovarian cancer, castration-resistance prostatic cancer and triple-negative breast cancer.
Proof-of-Concept Studies with Mifepristone
Metabolic Disorders
In 2005, we announced results from two preclinical studies conducted in a rat model of olanzapine-induced weight gain. These studies demonstrated that mifepristone's GR-II antagonist action has the potential to both reverse the weight gain associated with olanzapine and to prevent the weight gain associated with the initiation of treatment with olanzapine, which led to our studies in humans.
In 2007, we announced results of our human clinical proof-of-concept study evaluating the ability of mifepristone to mitigate weight gain associated with the administration of Eli Lilly's Zyprexa (olanzapine). The results indicated a statistically significant reduction in weight gain in those subjects who took Zyprexa plus mifepristone compared to those who took Zyprexa plus placebo. Eli Lilly provided Zyprexa and financial support for this study. During 2009, we announced results from another proof-of-concept study evaluating the ability of mifepristone to mitigate weight gain associated with the administration of Johnson & Johnson's Risperdal
11
(risperidone). The results indicated a statistically significant reduction in weight gain in those subjects who took Risperdal plus mifepristone compared to those who took Risperdal plus placebo. Both Zyprexa and Risperdal are indicated for the treatment of schizophrenia and bipolar disorder.
In the study of mifepristone and Zyprexa, 57 lean, healthy men (body mass index of 25 or less) were randomized to receive either Zyprexa plus placebo (n=22), Zyprexa plus mifepristone (n=24) or mifepristone plus placebo (n=11). This study took place in an institutional setting where daily weights were recorded and a range of metabolic parameters were measured. In the two week study, subjects in the Zyprexa plus placebo group gained an average of 7.0 pounds and subjects in the Zyprexa plus mifepristone group gained an average of 4.4 pounds; which is a statistically significant difference (p<.001). Subjects in the mifepristone plus placebo group gained an average of 4.4 pounds. The difference in weight gain trajectory was apparent in the first days of the study, reaching statistical significance during the first week. The increase in waist circumference, a surrogate for abdominal fat, in subjects who received Zyprexa plus placebo was also significantly greater than subjects who received Zyprexa plus mifepristone (p<.01). The study was not designed to enroll a sufficient number of patients to have statistical power to detect significant effects on metabolic measures; however, the effect of mifepristone in this model was greater than expected. In addition to the finding about waist circumference, notable additional non-statistically significant group differences were observed. Patients taking Zyprexa plus placebo experienced greater increases from baseline to end of study in both triglycerides and fasting insulin compared to patients taking Zyprexa plus mifepristone. No unexpected study drug related adverse events were observed. These results were published in Advances in Therapy in 2009.
In the study of mifepristone and Risperdal, 75 lean, healthy men (body mass index of 23 or less) were randomized to receive either Risperdal plus placebo (n=30), Risperdal plus mifepristone (n=30) or mifepristone plus placebo (n=15). This study also took place in an institutional setting where daily weights were recorded and a range of metabolic parameters were measured. In this four-week randomized double-blind controlled study, subjects in the Risperdal plus placebo group gained an average of 9.2 pounds, compared to a gain of 5.1 pounds in the Risperdal plus mifepristone group. This difference was statistically significant (p<0.0001). Additional important metabolic parameters, including fasting insulin, triglycerides and abdominal fat, as reflected by waist circumference, were also measured. The addition of mifepristone to Risperdal resulted in a statistically significant reduction in fasting insulin levels, triglyceride levels, and abdominal fat (as measured by waist circumference). Consistent with prior studies, mifepristone appeared to be well tolerated. These results were published in Obesity in 2010.
The combinations of Zyprexa and mifepristone or Risperdal and mifepristone are not approved for any indication. The purpose of these studies was to explore the hypothesis that GR-II antagonists would mitigate weight gain and other metabolic effects associated with antipsychotic medications. The group of medications sometimes referred to as "atypical antipsychotics," including Zyprexa, Risperdal, Clozaril (clozapine) and Seroquel (quetiapine), are widely used to treat schizophrenia and bipolar disorder. All medications in this group are associated with treatment-emergent weight gain of varying degrees and carry a warning in the label relating to treatment-emergent hyperglycemia and diabetes mellitus.
Other Disorders
We have collaborated with researchers investigating the utility of mifepristone in pre-clinical and human proof-of-concept studies in a wide range of disorders, including alcoholism, post-traumatic stress disorder, Alzheimer's disease, central serous chorioretinopathy, triple-negative breast cancer, castration resistant prostatic cancer, and ovarian cancer.
Research and Development
We incurred $20.5 million, $14.1 million and $21.0 million of research and development expenses in the years ended December 31, 2013, 2012 and 2011, respectively, which accounted for 39%, 36% and 65% of our
12
total operating expenses in these respective fiscal years. For a further discussion, see Part II, Item 7, Management's Discussion and Analysis of Financial Conditions and Results of Operations – Results of Operations.
Manufacturing Korlym
As a drug discovery, development and commercialization company, we intend to continue to utilize our financial resources to commercialize Korlym and advance other product candidates rather than diverting resources to establishing our own manufacturing facilities.
We intend to continue to rely on experienced contract manufacturers to produce our product candidates. We have entered into a manufacturing agreement with one contract manufacturer, Produits Chimiques Auxiliaires et de Synthese SA (PCAS), to produce the active pharmaceutical ingredient (API) for Korlym. The FDA approved our commercial use of material produced by PCAS as part of our NDA submission for Korlym. The agreement with PCAS, which was executed in November 2006, was for an initial period of five years with an automatic extension for one additional year and has been extended to March 31, 2014. We are pursuing discussions to continue the relationship thereafter. The agreement calls for us to purchase from PCAS at least 75 percent of our requirements until the expiration of the agreement. If PCAS is unable to manufacture the product for a consecutive six-month period, we have the right to terminate the agreement, without penalty.
We have one tablet manufacturer for Korlym with an operational facility – AAI Pharma Services Corp. (AAI), which was approved by the FDA in November 2012 for the manufacture of our commercial tablets, subject to the successful manufacture of validation batches. We plan to undertake this validation process at AAI early in 2014 and are currently in negotiations for a long-term commercial manufacturing agreement with AAI.
Our original tablet manufacturer, Formex LLC (formerly PharmaForm, L.L.C. and referred to herein as PharmaForm), has temporarily suspended manufacturing operations while it relocates to a new facility. We have not taken steps to qualify PharmaForm's new facility for the manufacture of Korlym.
Competition for Korlym
Korlym competes with established treatments, including surgery, radiation, and approved medicines prescribed "off-label." Korlym also competes with Novartis' drug, Signifor ® (pasireotide) Injection, which the FDA approved in December 2012 for the treatment of adult patients with Cushing's disease (a subset of Cushing's syndrome) who are not candidates for pituitary surgery or for whom surgery did not work. In April 2012, Signifor received marketing approval in the EU. It has Orphan Drug designation in the United States and the EU. Signifor is a somatostatin analogue that inhibits ACTH production by the pituitary, which leads to reduced cortisol production in some patients. In the Phase 3 study that served as the basis for Novartis' NDA, the drug normalized cortisol levels in 26 percent of patients. Sixty-seven percent of patients developed hyperglycemia or diabetes. Signifor must be taken twice daily, by injection. Novartis has also announced that it is undertaking an investigational study of an experimental compound (LC1699) to determine whether it can safely reduce the level of urinary free cortisol in patients with Cushing's disease.
Korlym may also experience competition from compounds under development for Cushing's syndrome. We are aware that Laboratoire HRA Pharma (HRA) has received an Orphan Drug Designation in the United States and EU for the use of mifepristone to treat a subtype of Cushing's syndrome. HRA began a Phase 2 trial in Europe and the United States for this indication, which has been terminated. We are also aware that Exelgyn Laboratories, which operates as a subsidiary of Medi Challenge (Pty) Ltd., received Orphan Drug Designation for endogenous Cushing's syndrome in the EU, but they have stated that they have not yet conducted any clinical trials.
13
Many colleges, universities and public and private research organizations are also active in the human health care field. While these entities focus on education, they may develop or acquire proprietary technology that we may require for the development of our product candidates. We may attempt to obtain licenses to this proprietary technology.
Our ability to compete successfully will be based on our ability to develop proprietary products, attract and retain scientific personnel, obtain patent or other protection for our product candidates, obtain required regulatory approvals and manufacture and successfully market Korlym and our future products either alone or through outside parties.
Intellectual Property
Patents and other proprietary rights are important to our business. It is our policy to seek patent protection for our inventions, and to rely upon trade secrets, know-how, continuing technological innovations and licensing opportunities to develop and maintain our competitive position.
Psychotic Depression
Under an agreement with Stanford University, we have licensed exclusive rights to the following issued U.S. patents and any corresponding foreign patents:
U.S. Patent Number | Subject Matter | Expiration Date | ||||
| 6,150,349 | Use of GR-II antagonists in the treatment of psychotic major depression | October 5, 2018 | ||||
| 6,362,173 | Use of GR-II antagonists in the treatment of cocaine-induced psychosis | October 5, 2018 | ||||
| 6,369,046 | Use of GR-II antagonists in the treatment of early dementia | February 4, 2019 | ||||
The corresponding foreign patents expire in 2018.
We are required to make milestone payments and pay royalties to Stanford University on sales of products commercialized under any of the above patents. We are currently in compliance with our obligations under the agreement. If Stanford University were to terminate any of our exclusive licenses due to breach of the license on our part, we would not be able to commercialize mifepristone for the treatment of the psychotic features of psychotic depression, cocaine-induced psychosis or early dementia.
Oncology
Under an agreement with University of Chicago, we have licensed exclusive rights to the claims contained in U.S. Patent Application No. 13/071,363 "Methods and Compositions Related to Glucocorticoid Receptor Antagonists and Breast Cancer." On August 8, 2013, the U.S. Patent and Trademark Office notified the University that certain claims in the Application had been allowed, although the patent has not yet issued.
While we only recently began a Phase 1 study of mifepristone in combination with the chemotherapy drug eribulin in the treatment of triple-negative breast cancer, in the event any products are commercialized under any of the above claims we would be required to make milestone payments and pay royalties to the University of Chicago on sales of such products. If the University of Chicago were to terminate any of our exclusive licenses due to breach of the license on our part, we would not be able to commercialize mifepristone for the treatment of triple-negative breast cancer.
14
Proprietary GR-II Antagonists
We also own issued U.S. patents for the use of GR-II antagonists in the treatment of mild cognitive impairment, the treatment of weight gain following treatment with antipsychotic medication, the prevention and treatment of stress disorders, improving the therapeutic response to ECT, the treatment of delirium, the treatment of catatonia, the treatment of gastroesophageal reflux disease, the treatment of migraine headaches, the treatment of psychosis with Interferon-Alpha therapy and for inhibiting cognitive deterioration in adults with Down's Syndrome. We also own a method of use patent for optimizing mifepristone levels in plasma serum in patients suffering from mental disorders. The expiration dates of these patents and their foreign counterparts range from 2020 to 2032.
In addition, we have six U.S. method-of-use applications covering certain GR-II antagonists, including the treatment of:
| • | patients suffering from mental disorders by optimizing mifepristone absorption; |
| • | neurological damage in premature infants; |
| • | muscular dystrophy; |
| • | diseases using combination steroid and glucocorticoid receptor antagonist therapy; |
| • | depression in patients taking Interleukin-2 (IL-2); and |
| • | amyotrophic lateral sclerosis (ALS). |
The approximate expiration dates of the patents that could issue from these applications and their foreign counterparts range from 2023 to 2032.
We have six United States composition of matter patents containing claims relating to three distinct series of novel selective GR-II antagonists, with an additional such patent pending. Three of these patents have issued in Europe, with applications for three more pending. The expiration dates of these U.S. and European patents range from 2026 to 2032.
We have also filed, where we deemed appropriate, foreign patent applications corresponding to our U.S. patents and applications. However, we cannot assure you that any of our patent applications will result in the issuance of patents, that any issued patent will include claims of the breadth sought in these applications, or that competitors will not successfully challenge or circumvent our patents if they are issued.
Although three of our patents have claims directed to the composition of compounds, we do not have a patent with claims directed to the composition of mifepristone. Our rights under our issued patents related to mifepristone cover only the use of that compound in the treatment of specific diseases.
Mifepristone
The composition of matter patent covering mifepristone has expired. The only previously FDA-approved use of mifepristone is to terminate pregnancy. The FDA has imposed significant restrictions on the use of mifepristone to terminate pregnancy. To protect our market for Korlym we plan to rely on (1) the exclusive marketing rights conferred as a benefit of Orphan Drug Designation in the United States and EU, (2) the restrictions imposed by the FDA on the use of mifepristone to terminate pregnancy, (3) the different patient populations, administering physicians and treatment settings between the use of mifepristone to terminate pregnancy and to treat Cushing's syndrome and (4) our method of use patents described above.
Competition
The patent positions of companies in the pharmaceutical industry are highly uncertain, involve complex legal and factual questions and have been and continue to be the subject of much litigation. Our product candidates may give rise to claims that we infringe on the products or proprietary rights of others. If it is
15
determined that our drug candidates infringe on others' patent rights, we may be required to obtain licenses to those rights. If we fail to obtain licenses when necessary, we may experience delays in commercializing our product candidates while attempting to design around other patents, or determine that we are unable to commercialize our product candidates at all. If we do become involved in intellectual property litigation, we are likely to incur considerable costs in defending or prosecuting the litigation. We believe that we do not currently infringe any third party's patents or other proprietary rights, and we are not obligated to pay royalties relating to the use of intellectual property to any third party other than Stanford University and The University of Chicago.
License Agreements
Under our exclusive license agreement with Stanford University to patents covering the use of mifepristone to treat the psychotic features of psychotic depression and for the treatment of early dementia, we are required to make milestone payments and pay royalties to Stanford University on sales of products commercialized under any of the above patents. These milestone payments are creditable against future royalties. This license agreement expires upon expiration of the related patents or upon notification by us to Stanford relating to the use of intellectual property. See "Intellectual Property."
In November 2013, we licensed from the University of Chicago exclusive rights to the claims contained in the University's U.S. Patent Application No. 13/071,363 "Methods and Compositions Related to Glucocorticoid Receptor Antagonists and Breast Cancer" (the "Application"). On August 8, 2013 the U.S. Patent and Trademark Office notified the University that certain claims in the Application had been allowed, although the patent has not yet issued. In exchange for the license, we have agreed to pay the University customary milestone fees and royalties on sales of any products commercialized under any of the claims. We have recently begun a Phase 1 study of mifepristone in combination with the chemotherapy drug eribulin in the treatment of triple-negative breast cancer.
Government Regulation
Prescription pharmaceutical products are subject to extensive pre- and post-approval regulation, including regulations that govern the testing, manufacturing, safety, efficacy, labeling, storage, record keeping, advertising, and promotion of the products under the Federal Food, Drug and Cosmetic Act. All of our product candidates will require regulatory approval by government agencies prior to commercialization. The process required by the FDA before a new drug may be marketed in the United States generally involves the following: completion of preclinical laboratory and animal testing; submission of an IND, which must become effective before clinical trials may begin; performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug or biologic's intended use; and, in the case of a new drug, approval by the FDA of an NDA. The process of complying with these and other federal and state statutes and regulations in order to obtain the necessary approvals and subsequently complying with federal and state statutes and regulations involves significant time and expense.
Preclinical studies are generally conducted in laboratory animals to evaluate the potential safety and the efficacy of a product. Drug developers submit the results of preclinical studies to the FDA as a part of an IND, which must be approved before beginning clinical trials in humans. Typically, human clinical trials are conducted in three sequential phases that may overlap.
| • | Phase 1. Clinical trials are conducted with a small number of subjects to determine the early safety profile, maximum tolerated dose and pharmacokinetics of the product candidate in human volunteers. |
| • | Phase 2. Clinical trials are conducted with groups of patients afflicted with a specific disease to determine preliminary efficacy, optimal dosages and expanded evidence of safety. |
| • | Phase 3. Large-scale, multi-center, comparative trials are conducted with patients afflicted with a target disease to establish the overall risk/benefit ratio of the drug and to provide enough data to demonstrate with substantial evidence the efficacy and safety of the product, as required by the FDA. |
16
The FDA and the Institutional Review Boards closely monitor the progress of each of the three phases of clinical trials that are conducted in the United States and may reevaluate, alter, suspend or terminate the testing at any time for various reasons, including a belief that the subjects are being exposed to an unacceptable health risk. The FDA may also require that additional studies be conducted, such as studies demonstrating that the drug being tested does not cause cancer.
After Phase 3 trials are completed, drug developers submit the results of preclinical studies, clinical trials, formulation studies and data supporting manufacturing to the FDA in the form of an NDA for approval to commence commercial sales. The FDA reviews all NDAs submitted before it accepts them for filing. The FDA may request additional information rather than accept an NDA for filing. If the FDA accepts an NDA for filing, it may grant marketing approval, request additional information or deny the application if it determines that the application does not meet regulatory approval criteria. Once an NDA has been accepted for filing, by law the FDA has 180 days to examine the application and respond to the applicant. However, the review process is often significantly extended by FDA requests for additional information or clarification. Under the Prescription Drug User Fee Act, the FDA has a goal of responding to NDAs within ten months of the filing date for standard review, and 6 months for priority review if a sponsor shows that its drug candidate provides a significant improvement compared to marketed drugs. The FDA has indicated to us that it will grant us a priority review of our NDA of mifepristone for the treatment of the psychotic features of psychotic depression if no other medications have been approved for this indication at the time of our submission. FDA approvals may not be granted on a timely basis, or at all.
If the FDA approves an NDA, the subject drug becomes available for physicians to prescribe in the United States. Once approved, the FDA may withdraw the product approval if compliance with pre- and post-approval regulatory standards is not maintained. The drug developer must submit periodic reports to the FDA. Adverse experiences with the product must be reported to the FDA and could result in the imposition of marketing restrictions through labeling changes or product removal. Product approvals may be withdrawn if problems with safety or efficacy occur after the product reaches the marketplace. In addition, the FDA may require post-marketing studies, referred to as Phase 4 studies, to monitor the effect of approved products, and may limit further marketing of the product based on the results of these post-approval studies.
Facilities used to manufacture drugs are subject to periodic inspection by the FDA and other authorities where applicable, and must comply with current Good Manufacturing Practices regulations (cGMP). Failure to comply with the statutory and regulatory requirements subjects the manufacturer to possible legal or regulatory action, such as suspension of manufacturing, seizure of product or voluntary recall of a product.
With respect to post-approval product advertising and promotion, the FDA imposes a number of complex regulations on entities that advertise and promote pharmaceuticals, which include, among others, standards and regulations for direct-to-consumer advertising, off-label promotion, industry sponsored scientific and educational activities, and promotional activities involving the Internet. The FDA has very broad enforcement authority under the Federal Food, Drug and Cosmetic Act, and failure to abide by these regulations can result in penalties including the issuance of a warning letter directing a company to correct deviations from FDA standards, a requirement that future advertising and promotional materials be pre-cleared by the FDA, and state and federal civil and criminal investigations and prosecutions.
In addition to studies requested by the FDA after approval, a drug developer may conduct other studies to explore use of the approved compound for treatment of new indications. The purpose of these trials and studies and related publications is to broaden the application and use of the drug and its acceptance in the medical community. Data supporting the use of a drug for these new indications must be submitted to the FDA in a new or supplemental NDA that must be approved by the FDA before the drug can be marketed for the new indications.
17
Orphan Drug Designation. We have received Orphan Drug designation for Korlym for the treatment of endogenous Cushing's syndrome in both the United States and the EU. In the United States, Orphan Drug designation provides special status to a product to treat a rare disease or condition providing that the product meets certain criteria. Orphan designation qualifies the sponsor of the product for the tax credit and marketing incentives of the Orphan Drug Act, including seven years of exclusive marketing rights for the specific drug for the orphan indication, if it receives the first regulatory approval for that indication, with limited exceptions. A marketing application for a prescription drug product that has been designated as a drug for a rare disease or condition is not subject to a prescription drug user fee unless the application includes an indication for other than a rare disease or condition. Orphan Drug designation does not prevent competitors from developing or marketing different drugs for an indication. It also does not convey an advantage in, or shorten the duration of, the review and approval process for a drug by the applicable regulatory authority.
Benefits of Orphan Drug Designation in the EU are similar to those in the U.S., but include ten years of marketing exclusivity in all 28 Member States, free scientific advice during drug development, access to a centralized review process and a reduction or complete waiver of fees levied by the European Medicines Agency (EMA).
Approvals outside the United States. Other than applying for and receiving Orphan Drug Designation for Korlym for Cushing's syndrome in the EU and submitting our MAA for that indication, we have not started the regulatory approval process in any jurisdiction other than the United States. We, or our potential future partners, will have to complete an approval process similar to the U.S. approval process in foreign target markets for our product candidates before we can commercialize our product candidates in those countries. The approval procedure and the time required for approval vary from country to country and can involve additional testing. Foreign approvals may not be granted on a timely basis, or at all. Regulatory approval of pricing is required in most countries other than the United States. The prices approved may be too low to generate an acceptable return to us.
Fast Track Designation. The FDA sometimes grants "fast track" status under the Food and Drug Administration Modernization Act of 1997. The fast track mechanism is intended to facilitate the development and approval of new drugs intended for the treatment of serious or life-threatening diseases or conditions and which demonstrate the potential to address unmet medical needs for the disease or condition. The fast track process includes scheduling of meetings to seek FDA input into development plans, the option of submitting an NDA serially in sections rather than submitting all components simultaneously, the option to request evaluation of studies using surrogate endpoints, and the potential for a priority review.
We have been granted fast track status for mifepristone for the treatment of the psychotic features of psychotic depression. However, the fast track designation may be withdrawn by the FDA at any time. The fast track designation does not guarantee that we will qualify for or be able to take advantage of the expedited review procedures and does not increase the likelihood that mifepristone will receive regulatory approval.
Priority Review . The FDA has indicated to us that it will grant us a priority review of our NDA of mifepristone for the treatment of the psychotic features of psychotic depression if no other medications have been approved for this indication at the time of our submission.
Coverage and Reimbursement . Sales of our products will depend, in part, on the extent to which our products will be covered by third-party payors, such as government health care programs, commercial insurance and managed healthcare organizations. Although this trend has not had a material impact on the amount or timing of our revenues, these third-party payors are increasingly limiting coverage and reducing reimbursements for medical products and services. In addition, the U.S. government, state legislatures and foreign governments have continued implementing cost-containment programs, including price controls, restrictions on coverage and reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and
18
measures, could limit our net revenue and results. Decreases in third-party reimbursement for our products or a decision by a third-party payor to not cover our products could reduce physician utilization of our products and have a material adverse effect on our sales, results of operations and financial condition.
Other Healthcare Laws. We are subject to healthcare regulation and enforcement by the federal government and the states and foreign governments in which we conduct our business. These laws include, without limitation, state and federal anti-kickback, fraud and abuse, false claims, privacy and security and physicians sunshine laws and regulations.
The federal Anti-Kickback Statute prohibits, among other things, any person from knowingly and willfully offering, soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual, for an item or service or the purchasing or ordering of a good or service, for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs. The Anti-Kickback Statute is subject to evolving interpretations. In the past, the government has enforced the Anti-Kickback Statute to reach large settlements with healthcare companies based on sham consulting and other financial arrangements with physicians. Further, the recently enacted Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the PPACA, among other things, amends the intent requirement of the federal Anti-Kickback Statute and the criminal statute governing healthcare fraud statutes. A person or entity no longer needs to have actual knowledge of these statutes or specific intent to violate them. In addition, the PPACA provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act or federal civil money penalties statute. The majority of states also have anti-kickback laws which establish similar prohibitions and in some cases may apply to items or services reimbursed by any third-party payor, including commercial insurers.
Additionally, the civil False Claims Act prohibits knowingly presenting or causing the presentation of a false, fictitious or fraudulent claim for payment to the U.S. government. Actions under the False Claims Act may be brought by the Attorney General or as a qui tam action by a private individual in the name of the government. Violations of the False Claims Act can result in very significant monetary penalties and treble damages. The federal government is using the False Claims Act, and the accompanying threat of significant liability, in its investigation and prosecution of pharmaceutical and biotechnology companies throughout the country, for example, in connection with the promotion of products for unapproved uses and other sales and marketing practices. The government has obtained multi-million and multi-billion dollar settlements under the False Claims Act in addition to individual criminal convictions under applicable criminal statutes. Given the significant size of actual and potential settlements, it is expected that the government will continue to devote substantial resources to investigating healthcare providers' and manufacturers' compliance with applicable fraud and abuse laws.
In addition, there has been a recent trend of increased federal and state regulation of payments made to physicians and other healthcare providers. The PPACA, among other things, imposes new reporting requirements on drug manufacturers for payments made by them to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Failure to submit required information may result in civil monetary penalties of up to an aggregate of $150,000 per year (or up to an aggregate of $1 million per year for "knowing failures"), for all payments, transfers of value or ownership or investment interests that are not timely, accurately and completely reported in an annual submission. Drug manufacturers were required to begin collecting data on August 1, 2013 and submit reports to the government by March 31, 2014 and the 90 th day of each subsequent calendar year. Certain states also mandate implementation of commercial compliance programs, impose restrictions on drug manufacturer marketing practices and/or require the tracking and reporting of gifts, compensation and other remuneration to physicians.
The shifting commercial compliance environment and the need to build and maintain robust and expandable systems to comply with different compliance and/or reporting requirements in multiple jurisdictions increase the possibility that a healthcare company may violate one or more of the requirements. If our operations are found to
19
be in violation of any of such laws or any other governmental regulations that apply to us, we may be subject to penalties, including, without limitation, civil and criminal penalties, damages, fines, the curtailment or restructuring of our operations, exclusion from participation in federal and state healthcare programs and imprisonment, any of which could adversely affect our ability to operate our business and our financial results.
Executive Officers
The following table sets forth, as of March 3, 2014, information about our executive officers:
Name | Age | Position | ||||
Joseph K. Belanoff, M.D. | 56 | Chief Executive Officer, President and Director | ||||
G. Charles Robb | 51 | Chief Financial Officer and Secretary | ||||
Steven Lo | 46 | Senior Vice President and Chief Commercial Officer | ||||
Anne M. LeDoux | 66 | Vice President, Controller and Chief Accounting Officer | ||||
Joseph K. Belanoff, M.D. is a co-founder of our company, has served as a member of our Board and as our Chief Executive Officer since 1999 and as our President since January 2014. Dr. Belanoff is currently a clinical faculty member and has held various positions in the Department of Psychiatry and Behavioral Sciences at Stanford University since 1992. Dr. Belanoff received his B.A. from Amherst College and his M.D. from Columbia University's College of Physicians & Surgeons. Our Board selected Dr. Belanoff to serve as a director because, as our Chief Executive Officer, he brings expertise and knowledge regarding our business and operations to our Board of Directors. Dr. Belanoff also has expertise in clinical medicine and psychopharmacology.
G. Charles Robb has served as Chief Financial Officer since September 2011 and as our Secretary since January 2014. Mr. Robb has more than 25 years of experience in executive management, operations and finance. From April 2005 through August 2011 Mr. Robb served as the Senior Vice President of Operations, Administration and Finance of Fitness Anywhere, Inc. (FAI), a private fitness equipment and training company with operations in the United States, Europe and Asia. From 2003 to 2005, Mr. Robb was engaged in the private practice of law. From 2000 to 2002 he was Senior Vice President of Citadon, Inc. He also held positions in business development for Normura Asset Capital Corporation from 1998 to 1999 and in sales and marketing for Legal Research Network, Inc. from 1996 to 1998. From 1992 to 1996 Mr. Robb practiced law at Howard, Rice, Nemerovski, Canady, Falk & Rabkin. Mr. Robb earned a B.A. in English and Political Philosophy from Yale and a J.D. from Harvard Law School, where he was a member of the Harvard Law Review.
Steven Lo joined us as Vice President of Commercial Operations in September 2010 and was promoted to the position of Senior Vice President and Chief Commercial Officer in November 2013. Mr. Lo has more than 19 years of commercial experience in the pharmaceutical and biotechnology industry. From 1997 to 2010, Mr. Lo held various positions in marketing, sales and managed markets at Genentech, Inc., a biotechnology company that became a member of the Roche Group in March 2009, most recently as Franchise Head, leading that company's endocrinology marketing and sales organization. Mr. Lo received his B.S. degree from the University of California, Davis and his Master of Health Administration degree from the University of Southern California.
Anne M. LeDoux joined us as Controller in 2004 and was promoted to the position of Vice President, Controller and Chief Accounting Officer in April 2007. Ms. LeDoux has over 20 years of financial and accounting management experience with public pharmaceutical and biotechnology companies. Prior to joining Corcept in 2004, Ms. LeDoux served in various financial positions at Aviron, Roche Biosciences and Syntex Corporation. She was also Vice President and Chief Financial Officer at the Northern California Health Center and Vice President, Finance for the Children's Hospital of San Francisco. Ms. LeDoux is a Certified Public Accountant with over 13 years of experience in public accounting, primarily at Coopers and Lybrand. Ms. LeDoux received her Bachelor of Arts degree in Business from the University of Massachusetts and a law degree from Western New England College, School of Law.
20
Employees
We are managed by a core group of experienced pharmaceutical executives with a track record of bringing new drugs to market. To facilitate advancement of development programs, we also enlist the expertise of associates and advisors with extensive pharmaceutical development experience.
As of December 31, 2013, we had 44 full-time employees, six part-time employees, a contracted sales force of 13 sales representatives and 14 long-term contract staff. Four of our employees have M.D.s. We consider our employee relations to be good. None of our employees is covered by a collective bargaining agreement.
General
We were incorporated in the State of Delaware on May 13, 1998. Our registered trademarks include Corcept ® , Korlym ® and CORLUX ® . Corluxin ® is a registered trademark in the EU; the application for this trademark is pending in the United States. Other service marks, trademarks and trade names referred to in this document are the property of their respective owners.
Available Information
We are subject to the information requirements of the Securities Exchange Act of 1934, as amended, and we therefore file periodic reports, proxy statements and other information with the SEC relating to our business, financial statements and other matters. The reports, proxy statements and other information we file may be inspected and copied at prescribed rates at the SEC's Public Reference Room, 100 F Street, N.E., Washington, D.C. 20549, on official business days during the hours of 10:00 A.M. to 3:00 P.M. You may obtain information on the operation of the SEC's Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC also maintains an Internet site that contains reports, proxy statements and other information regarding issuers like us that file electronically with the SEC. The address of the SEC's Internet site is www.sec.gov . For more information about us, please visit our website at www.corcept.com . You may also obtain a free copy of our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports on the day the reports or amendments are filed with or furnished to the SEC by visiting our website at www.corcept.com . The information found on, or otherwise accessible through, our website, is not incorporated information, and does not form a part of, this Form 10-K.
21
| ITEM 1A. | RISK FACTORS |
An investment in our common stock involves significant risks. You should carefully consider the risks described below and the other information in this Annual Report on Form 10-K, including our financial statements and related notes, before you decide to invest in our common stock. If any of the following risks or uncertainties actually occurs, our business, results of operations or financial condition could be materially harmed, the trading price of our common stock could decline and you could lose all or part of your investment. The risks and uncertainties described below are those that we currently believe may materially affect us; however, they may not be the only ones that we face. Additional risks and uncertainties of which we are unaware or currently deem immaterial may also become important factors that may harm our business. Except as required by law, we undertake no obligations to update any risk factors.
Risks Related to the Commercialization of Korlym
and Development of Mifepristone and Our Other Proprietary GR-II Antagonists
We depend heavily on the success of Korlym, which we began to sell in the United States in April 2012. If we are unable to increase revenues of Korlym to the levels that investors expect, or experience significant delays in doing so, our stock price will likely decline.
We anticipate that for the foreseeable future our ability to generate meaningful revenues and achieve profitability will be solely dependent on the successful commercialization of Korlym. Many factors could harm our efforts to commercialize Korlym, including:
| • | an inability to generate meaningful revenue due to low product usage, inadequate coverage and reimbursement or other factors; |
| • | competition from Novartis's Signifor and from other companies with greater financial, technical and marketing resources than ours; |
| • | an inability to manufacture Korlym or the active ingredient in Korlym in commercial quantities and at an acceptable cost; |
| • | political concerns relating to other uses of mifepristone, or RU-486, that could limit the market acceptance of Korlym; |
| • | negative, inconclusive or otherwise unfavorable results from any post-approval studies we conduct; |
| • | previously unknown, serious side effects that may be identified; and |
| • | rapid technological change making Korlym obsolete. |
Even if we are able to commercialize Korlym successfully, we cannot predict the rate at which success will occur.
As our current ability to generate revenue is wholly dependent upon the commercialization of Korlym, its rate of sale will directly and materially affect our results of operations. There are inherent difficulties in predicting the volumes of Korlym that will be sold, which are heightened by our limited experience commercializing Korlym or other products. Failure of our revenue to meet the expectations of investors could cause our stock price to decline. See also the discussion below under "If our operating and financial performance in any given period does not meet the guidance that we provide to the public, estimates published by research analysts or other investor expectations, our stock price may decline."
Physicians may accept Korlym slowly or may never accept it, which would adversely affect our financial results.
Many factors may affect the market acceptance and commercial success of Korlym.
Even though the FDA has approved Korlym, physicians may not adopt it as a treatment for their eligible patients. Physicians will prescribe Korlym only if they determine, based on experience, clinical data, side effect
22
profiles and other factors, that it is preferable to other products or treatments currently in use, even if those products are not approved for Cushing's syndrome. Because Cushing's syndrome is rare, most physicians are inexperienced in the care of patients with the illness and it may be difficult to persuade them to prescribe a newer treatment, such as Korlym, even with clinical trial results that suggest it may be a compelling treatment for them to consider.
Other factors that may affect the market acceptance and commercial success of Korlym include:
| • | the effectiveness of Korlym, including any side effects, as compared to alternative treatment methods; |
| • | the rate of adoption of Korlym by physicians and by target patient populations; |
| • | the possible preference of some physicians for more familiar, long-standing off-label treatments for Cushing's syndrome or for Novartis' drug, Signifor, for the treatment of Cushing's disease; |
| • | the cost-effectiveness of Korlym and the availability of third-party insurance coverage and reimbursement, in particular from government payors such as Medicare and Medicaid, for patients using Korlym; |
| • | the product labeling required by the FDA for Korlym; |
| • | the extent and success of our efforts to manufacture, commercialize, market, distribute and sell Korlym; and |
| • | negative publicity concerning Korlym, RU-486, Mifeprex ® or mifepristone. |
The failure of Korlym to achieve market acceptance would prevent us from generating meaningful revenue.
We will face competition from companies that attempt to develop mifepristone or other compounds for the treatment of Cushing's syndrome, which could limit our future revenues from the commercialization of Korlym and which could have a negative impact on future revenues from the commercialization of Korlym for any indication. These companies may have significantly more resources than we do. The Orphan Drug Designation for Korlym may not provide protection from competition and other benefits as anticipated.
In 2007, we received Orphan Drug Designation from the FDA for Korlym for the treatment of hyperglycemia secondary to hypercortisolism in adult patients with endogenous Cushing's syndrome who have type 2 diabetes mellitus or glucose intolerance and have failed surgery or are not candidates for surgery. Drugs that receive Orphan Drug Designation are eligible to obtain seven years of marketing exclusivity for the approved indication from the date of drug approval, with limited exceptions, as well as tax credits for clinical trial costs, marketing application filing fee waivers and assistance from the FDA in the drug development process.
In 2011, the European Commission granted us Orphan Drug Designation for mifepristone for the treatment of endogenous Cushing's syndrome (hypercortisolism) in the EU. Benefits of Orphan Drug Designation in the EU are similar to those in the United States, but include ten years of marketing exclusivity for the approved indication in all 28 Member States, free scientific advice during drug development, access to a centralized review process and a reduction or complete waiver of fees levied by the EMA. The EMA has accepted our plan to study the use of Korlym in children with Cushing's syndrome which we expect will, upon completion, extend our period of marketing exclusivity by two years in the EU. We submitted our Marketing Authorization Application request to the EMA in October 2013.
Although we have received Orphan Drug Designation in both the United States and the EU, we cannot be assured that we will recognize the potential benefits of these designations. Even after an orphan drug is approved for its orphan indication, the FDA can subsequently approve a different drug for the same condition if the FDA concludes that the later drug is safer, more effective or makes a major contribution to patient care. In addition, the FDA may, during the seven-year orphan drug exclusivity period, approve the same drug for a different indication or different drug for the same indication.
23
Notwithstanding Korlym's Orphan Drug Designation in both the United States and the EU, in 2012 Novartis received approval in both jurisdictions to market its somatostatin analogue Signifor for adult patients with Cushing's disease (a subset of Cushing's syndrome that afflicts approximately 70 percent of all Cushing's syndrome patients) for whom pituitary surgery is not an option or has not been curative. Novartis also announced that is undertaking an investigational study of an experimental compound (LC1699) to determine whether it can safely reduce the level of urinary free cortisol in patients with Cushing's disease and to examine the compound's safety and efficacy. Novartis has substantially more resources and experience than we do and may provide significant competition.
Further, we are aware that Laboratoire HRA Pharma has received Orphan Drug Designation in the United States and the EU for the use of mifepristone to treat a subtype of Cushing's syndrome. HRA had begun a Phase 2 clinical trial in Europe and the United States for this indication, which has been terminated. We are also aware that Exelgyn Laboratories, which operates as a subsidiary of Medi Challenge (Pty) Ltd., received Orphan Drug Designation for Cushing's syndrome in the EU, but it has stated that it has not yet conducted any clinical trials.
If another drug with mifepristone as its active ingredient is approved in the EU for Cushing's syndrome before our drug, we will not receive the ten years of marketing exclusivity from the date of drug approval in the EU and other potential benefits. Any delay in our commercialization of Korlym may have a negative impact on the revenue that we might be able to realize from the exclusivity provided during the applicable periods.
If we cannot continue to obtain acceptable prices or adequate coverage and reimbursement for Korlym from third-party payors, we will be unable to generate significant revenues.
There is significant uncertainty related to the availability of third-party insurance coverage and reimbursement for newly approved medications. The commercial success of our medications in both domestic and international markets depends on whether third-party coverage and reimbursement is available for them. Government payors, including Medicare and Medicaid, health maintenance organizations and other third-party payors are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement of new medicines, and, as a result, they may not cover or provide adequate payment for our medications. Our near-term dependence on the commercial success of Korlym makes us particularly susceptible to any such cost containment or reduction efforts. Accordingly, even though Korlym has been approved for commercial sale, unless government and other third-party payors continue to provide adequate and timely coverage and reimbursement, physicians may not prescribe it and patients may not purchase it. In addition, meaningful delays in insurance coverage for individual patients may increase our costs and reduce our revenues. Further, we may need to obtain approvals from hospital formularies before Korlym can be covered for in-patient treatment. If we fail to obtain such approvals, this will reduce the level of revenues that we are able to attain.
In some foreign markets, pricing and profitability of prescription pharmaceuticals are subject to government control. In the United States, we expect that there will continue to be federal and state proposals for similar controls. Also, the trends toward managed health care in the United States and recent laws and legislation intended to reduce the cost of government insurance programs could significantly influence the purchase of health care services and products and may result in lower prices for our products or the exclusion of such products from reimbursement programs.
The PPACA, which was passed in 2010, included, among other things, the following measures:
| • | annual, non-deductible fees on any entity that manufactures or imports certain prescription drugs and biologics; |
| • | increases in Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program for both branded and generic drugs; |
| • | expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for certain individuals with income at or below 133% of the federal poverty level, thereby potentially increasing a manufacturer's Medicaid rebate liability; |
24
| • | expansion of access to commercial health insurance coverage through new state-based health insurance marketplaces, or exchanges; |
| • | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in and conduct comparative clinical research; |
| • | new requirements for manufacturers to discount drug prices to eligible patients by 50 percent at the pharmacy level and for mail order services in order for their outpatient drugs to be covered under Medicare Part D; |
| • | an increase in the number of entities eligible for discounts under the Public Health Service pharmaceutical pricing program; and |
| • | establishment of a licensure framework for follow-on biologic products. |
The PPACA provisions on comparative clinical effectiveness research extended the initiatives of the American Recovery and Reinvestment Act of 2009, also known as the stimulus package, which included $1.1 billion in funding to study the comparative effectiveness of health care treatments. This stimulus funding was designated for, among other things, conducting, supporting or synthesizing research that compares and evaluates the risks and benefits, clinical outcomes, effectiveness and appropriateness of products. The PPACA also appropriated additional funding to comparative clinical effectiveness research. Although Congress has indicated that this funding is intended to improve the quality of health care, it remains unclear how the research will impact current Medicare coverage and reimbursement or how new information will influence other third-party payor policies. It also is unclear what the full impact of PPACA's extension of coverage to previously uninsured individuals will be on the demand for our products.
In addition, other legislative changes have been proposed and adopted in the United States since the PPACA was enacted. In August 2011, the Budget Control Act of 2011 among other things, created the Joint Select Committee on Deficit Reduction to recommend proposals in spending reductions to Congress. The Joint Select Committee did not achieve its targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, triggering the legislation's automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of two percent per fiscal year, which went into effect on April 1, 2013. On January 2, 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, or the ATRA, which, among other things, further reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
These new laws and the regulations and policies implementing them, as well as other healthcare reform measures that may be adopted in the future, may have a material adverse effect on our industry generally and on our ability to successfully develop and commercialize our products.
We will need to continue to develop our medical education, sales and marketing capabilities to successfully commercialize Korlym and our other proprietary, selective GR-II antagonists.
To achieve commercial success for any approved product, we must either develop sales and marketing capabilities internally or enter into arrangements with third parties to market and sell our current and future products, and we may not be successful in doing so. We continue to hire experienced field and internal personnel to commercialize Korlym in the United States, which is expensive and time consuming. Any failure or delay in the development or failure to maintain effectively our internal capabilities for the marketing and sales of Korlym would adversely impact the commercialization of the product. If our efforts to develop an internal commercial marketing and sales team are not successful, cost-effective and timely, we may not achieve profitability.
We will need to increase the size of our organization, and we may experience difficulties in managing growth.
We expect that the further development of our commercial organization and the likely future expansion of our research and development efforts will strain our administrative, operational and management resources.
25
Future growth will impose significant added responsibilities on members of management, including the need to identify, recruit, maintain and integrate additional employees. To date, we have relied on a small management team, including a number of part-time contributors. Our future financial performance and our ability to compete effectively will depend, in part, on our ability to manage any future growth effectively.
To that end, we must be able to:
| • | integrate additional management, clinical development, administrative and sales and marketing personnel; |
| • | expand the size and composition of our management team; |
| • | develop our administrative, accounting and management information systems and controls; |
| • | hire and train additional qualified personnel; |
| • | manage our sales and marketing efforts effectively; |
| • | manage our supply chain effectively; |
| • | manage our clinical trials effectively; and |
| • | manage our research and development efforts effectively. |
We may not be able to accomplish these tasks, and our failure to accomplish any of them could harm our business.
Public perception of the active ingredient in Korlym, mifepristone (also known as "RU-486"), may limit our ability to market and sell Korlym.
The active ingredient in Korlym, mifepristone (RU-486), is approved by the FDA in another drug for the termination of early pregnancy. As a result, mifepristone has been and continues to be the subject of considerable ethical and political debate in the United States and elsewhere. Public perception of mifepristone may limit our ability to engage alternative manufacturers and may limit the commercial acceptance of Korlym by patients and physicians. Even though we have taken measures to minimize the likelihood of the prescribing of Korlym to a pregnant woman, physicians may choose not to prescribe Korlym to a woman simply to avoid any risk of unintentionally terminating a pregnancy. We have taken measures to control the distribution of Korlym to reduce the potential for diversion and this controlled distribution may negatively impact sales of Korlym.
We have no manufacturing capabilities and we currently depend on third parties to manufacture the active ingredient and the tablets for Korlym, both of which are single-source suppliers. If these suppliers are unable or unwilling to continue manufacturing Korlym and we are unable to contract quickly with alternative sources, or if these third-party manufacturers fail to comply with FDA regulations or otherwise fail to meet our requirements, our business will be harmed.
We currently have no experience in, and we do not own facilities for, nor do we plan to develop facilities for, manufacturing any products. We depend on a single-source, third-party contract manufacturer to supply the active pharmaceutical ingredient, or API, in Korlym. While we are in the process of negotiating a new agreement with this manufacturer, our existing agreement expires on March 31, 2014. We also depend on a single-source, third-party contract manufacturer to manufacture the Korlym tablet. In November 2012, the FDA approved AAI as a qualified site for the manufacture of Korlym tablets, subject to the successful manufacture of validation batches. We plan to undertake this validation process at AAI early in 2014 and are currently negotiating a long-term commercial supply agreement with AAI. If either of these manufacturers is unable or unwilling to meet our future demands in the quantities and time frame required, we may not be able to manufacture our product in a timely manner. Our current arrangements with these manufacturers are terminable by such manufacturers. If we are unable, for whatever reason, to obtain the API or Korlym tablets from our contract manufacturers, we may not be able to manufacture our required quantities or identify alternate manufacturers of mifepristone or Korlym tablets in a timely manner or on reasonable terms, if at all, which would harm our business. In addition, we expect to use third-party manufacturers and suppliers if and when our other product candidates are approved.
26
The facilities used by our contract manufacturers to manufacture our products must be approved by the FDA pursuant to inspections. We do not control the manufacturing processes of, and are completely dependent on, our contract manufacturing partners for compliance with the regulatory requirements, known as current good manufacturing practices, or cGMPs. If our contract manufacturers cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA or others, they will not be able to secure and/or maintain regulatory approval for their manufacturing facilities. In addition, we have no control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and qualified personnel. If the FDA or a comparable foreign regulatory authority does not approve these facilities for the manufacture of our products or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for or market our products. In addition, sanctions could be imposed on us, including fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing approval of our product candidates, delays, suspension or withdrawal of approvals, seizures or recalls of products, operating restrictions and criminal prosecutions, any of which could harm our business. If our suppliers fail to manufacture tablets on a timely basis in the quantities that we require, or fail to maintain manufacturing capabilities that meet FDA standards, we would likely experience a lengthy delay in our manufacturing processes.
If we or others identify previously unknown, serious side effects of mifepristone, we may be required to perform lengthy additional clinical trials, change the labeling of Korlym or withdraw it from the market, any of which would hinder or preclude our ability to generate revenues.
The FDA's approval of Korlym requires that we conduct a study of the interactions between Korlym and ketoconazole, an anti-fungal agent sometimes used to treat patients with Cushing's syndrome. It also requires us to study drug utilization to better characterize the reporting rates of adverse events associated with the long-term use of Korlym. If we or others identify previously unknown, serious side effects of mifepristone:
| • | regulatory authorities may withdraw their approvals; |
| • | we may be required to conduct additional clinical trials, make changes in labeling, implement changes to or obtain re-approvals of our manufacturing facilities; |
| • | we may experience a significant drop in the sales of Korlym; |
| • | our reputation in the marketplace may suffer; and |
| • | we may become the target of lawsuits, including class action lawsuits. |
Any of these events could harm or prevent sales of the affected products or could increase the costs and expenses of commercializing and marketing Korlym.
We may have substantial exposure to product liability claims and may not have adequate insurance to cover those claims.
We may be subject to product liability or other claims based on allegations that the use of our products has resulted in adverse effects or that our product candidates are not effective, whether by participants in our clinical trials for Korlym or other product candidates, or by patients using Korlym. A product liability claim may damage our reputation by raising questions about Korlym or any of our product candidates' safety or efficacy and could limit our ability to sell a product by preventing or interfering with product commercialization. In some cases, less common adverse effects of a pharmaceutical product are not known until long after the FDA approves the product for marketing. The active ingredient in Korlym is used to terminate pregnancy. Therefore, clinicians using the medicine in our clinical trials and physicians prescribing the medicine to women with childbearing potential must take necessary and strict precautions to ensure that the medicine is not administered to pregnant women. The failure to observe these precautions could result in significant product claims.
We have only limited product liability insurance coverage, with limits that we believe to be customary for a company beginning to commercialize its first pharmaceutical product. We intend to expand our product liability insurance coverage to any product candidates for which we obtain marketing approval. However, this insurance
27
may be prohibitively expensive or may not fully cover our potential liabilities. Our inability to obtain adequate insurance coverage at an acceptable cost could prevent or inhibit the commercialization of Korlym or any of our product candidates, or result in meaningful underinsured or uninsured liability. Defending a lawsuit could be costly and significantly divert management's attention from conducting our business. If a third party successfully sues us for any injury caused by our product candidates, our liability could exceed our total assets.
Even if we receive regulatory approval for our product candidates, we will be subject to ongoing and continued regulatory review, and if we are unable to maintain regulatory approval of Korlym, or if we fail to comply with regulatory requirements, we will be unable to generate revenue or may be subject to penalties and our business will be harmed.
Even after we obtain U.S. regulatory approval for a product, the FDA may still impose significant restrictions on the approved indicated uses for which the product may be marketed or on the conditions of approval. For example, a product's approval may contain requirements for potentially costly post-approval studies and surveillance, including Phase 4 clinical trials, to monitor the safety and efficacy of the product. The FDA's approval of Korlym was subject to limitations on the indicated uses for which the product may be marketed and requirements for post-marketing follow-up studies and information reporting. In addition, the FDA's approval of Korlym requires that we conduct a study of the interactions between Korlym and ketoconazole, an anti-fungal agent sometimes used to treat patients with Cushing's syndrome. It also requires us to conduct a drug utilization study to better characterize the reporting rates of adverse events associated with the long-term use of Korlym.
We are also subject to ongoing obligations and continued regulatory review by the FDA and other regulatory authorities in the United States and other countries with respect to the research, testing, manufacturing, labeling, distribution, adverse event reporting, storage, selling, advertising, promotion, recordkeeping and marketing of products. These requirements include submissions of safety and other post-marketing information and reports, annual updates on manufacturing activities and continued compliance with current good manufacturing practices, or cGMPs, and current good clinical practices, or cGCPs, for any clinical trials that we conduct post-approval. cGMPs and cGCPs are regulations and guidelines enforced by the FDA and comparable foreign regulatory authorities through periodic inspections of manufacturing sites, trial sponsors, clinical investigators and clinical sites. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with FDA regulations and other applicable foreign and U.S. regulatory requirements may result in, among other things, warning letters, civil and criminal penalties, injunctions, holds on clinical trials, product seizure or detention, refusal to permit the import or export of products, restrictions on product marketing, withdrawal of the product from the market, voluntary or mandatory product recalls, total or partial suspension of production, refusal to approve pending NDAs or supplements to approved NDAs, and suspension or revocation of product approvals.
The FDA's policies may change and additional governmental regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may place at risk the FDA marketing approval for Korlym and any other marketing approval that we may obtain, which would adversely affect our business, prospects and ability to achieve or sustain profitability.
The sale of our products is subject to regulatory approvals, and our business is subject to extensive regulatory requirements, and if we are unable to obtain regulatory approval for future product candidates, including mifepristone for the treatment of the psychotic features of psychotic depression, we will be limited in our ability to commercialize such product candidates and our business will be harmed.
We are not permitted to market or promote any of our product candidates before we receive regulatory approval from the FDA or comparable foreign regulatory authorities and, while we have received FDA
28
marketing approval for Korlym, we may be unable to maintain such approval and we may never receive such regulatory approval for any of our product candidates. Obtaining regulatory approval of a new drug is an uncertain, lengthy and expensive process, and success is never guaranteed. Despite the time, resources and effort expended, failure can occur at any stage. In order to receive approval from the FDA for each product candidate, we must demonstrate that the new drug product is safe and effective for its intended use and that our manufacturing processes for the product candidate comply with the FDA's cGMPs. cGMPs include requirements related to production processes, quality control and assurance, and recordkeeping. The FDA has substantial discretion in the approval process for human medicines. The FDA may require substantial additional clinical testing or find our drug products do not satisfy the standards for approval. Our inability or the inability of our suppliers to comply with applicable FDA and other regulatory requirements can result in, among other things, delays in or denials of new product approvals, warning letters, fines, consent decrees restricting or suspending manufacturing operations, injunctions, civil penalties, recall or seizure of products, total or partial suspension of sales, and/or criminal prosecution. Any of these or other regulatory actions could materially adversely affect our business and our financial condition.
Future governmental action or changes in FDA law, policy or personnel may also result in delays or rejection of an NDA in the United States. In addition, because the only other currently FDA-approved use of mifepristone is the termination of pregnancy, we expect that the label for mifepristone for any indication will include, as Korlym's does, some limitations, including a so-called "black-box" warning that it should not be used by pregnant women or women seeking to become pregnant.
If we receive regulatory approval for our future product candidates, including mifepristone for the treatment of psychotic depression and triple-negative breast cancer, we will be subject to ongoing FDA obligations and continued regulatory oversight and review, such as continued safety reporting requirements; and we may also be subject to additional FDA post-marketing restrictions and obligations. If we are not able to maintain regulatory compliance, we may not be permitted to market our product candidates and/or may be subject to product recalls or seizures.
Any regulatory approvals that we receive for our future product candidates may also be subject to limitations on the indicated uses for which the medicine may be marketed or contain requirements for potentially costly post-marketing follow-up studies. In addition, if the FDA approves any of our product candidates, we will be subject to ongoing and continuing regulatory requirements. See also the discussion above under "Even if we receive regulatory approval for our product candidates, we will be subject to ongoing and continued regulatory review, and if we are unable to maintain regulatory approval of Korlym, or if we fail to comply with regulatory requirements, we will be unable to generate revenue or may be subject to penalties and our business will be harmed."
If we market products in a manner that violates FDA regulations or health care fraud and abuse laws, we may be subject to civil or criminal penalties.
In the United States, we are subject to FDA regulations governing the promotion of health care products. Although physicians are permitted, based on their medical judgment, to prescribe drugs for indications other than those approved by the FDA, manufacturers are prohibited from promoting their products for such "off-label" uses. In the United States, we are marketing Korlym for treatment of hyperglycemia secondary to hypercortisolism in adult patients with endogenous Cushing's syndrome who have type 2 diabetes mellitus or glucose intolerance and have failed surgery or are not candidates for surgery and provide promotional materials and training programs to physicians regarding the use of Korlym for this indication. Although we believe our marketing materials and training programs for physicians do not constitute "off-label" promotion of Korlym, the FDA may disagree. If the FDA determines that our promotional materials, training or other activities by our employees or agents constitute "off-label" promotion of Korlym, it could request that we modify our training or promotional materials or other activities or subject us to regulatory enforcement actions, including the issuance of a warning letter, injunction, seizure, civil fine and criminal penalties. It is also possible that other federal or
29
state enforcement authorities might take action if they believe that the alleged improper promotion led to the submission and payment of claims for an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement. Even if it is later determined that we are not in violation of these laws, we may be faced with negative publicity, incur significant expenses defending our position and have to divert significant management resources from other matters.
In addition, there are health care fraud and abuse regulations and enforcement by both the federal government and the states in which we conduct our business. The laws that may affect our ability to operate include:
| • | the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made under federal health care programs such as the Medicare and Medicaid programs; |
| • | federal false claims laws, which prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to get a false claim paid. Pharmaceutical companies have been prosecuted under these laws for a variety of promotional and marketing activities, such as allegedly providing free product to or entering into "sham" consulting arrangements with customers to induce such customers to purchase, order or recommend the company's products in violation of the Anti-Kickback Statute and federal false claims laws and regulations; reporting to pricing services inflated average wholesale prices that were then used by certain governmental programs to set reimbursement rates; engaging in the promotion of "off-label" uses that caused customers to submit claims to and obtain reimbursement from governmental payors for non-covered "off-label" uses; and submitting inflated best price information to the Medicaid Drug Rebate Program; |
| • | the federal Health Insurance Portability and Accountability Act of 1996 (HIPAA), which created federal criminal laws that prohibit executing a scheme to defraud any health care benefit program or making false statements relating to health care matters; |
| • | federal "sunshine" laws that require transparency regarding financial arrangements with health care providers, such as the reporting and disclosure requirements imposed by the PPACA on drug manufacturers regarding any "transfer of value" made or distributed to prescribers and other health care providers, and ownership or investment interests held by physicians and their immediate family members. Manufacturers were required to begin data collection on August 1, 2013 and will be required to report such data to the Centers for Medicare & Medicaid Services (known as CMS) by March 31, 2014, and by the 90 th day of each calendar year thereafter; |
| • | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 (HITECH) and their respective implementing regulations, which impose obligations on covered healthcare providers, health plans, and healthcare clearinghouses, as well as their business associates that create, receive, maintain or transmit individually identifiable health information for or on behalf of a covered entity, with respect to safeguarding the privacy, security and transmission of individually identifiable health information; and |
| • | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry's voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or otherwise restrict payments that may be made to healthcare providers; state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
30
The risk of our being found in violation of these laws and regulations is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. Moreover, recent health care reform legislation has strengthened these laws. For example, the PPACA, among other things, amended the intent requirement of the federal anti-kickback and criminal health care fraud statutes; a person or entity no longer needs to have actual knowledge of these statutes or specific intent to violate them. In addition, the PPACA provided that the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the false claims statutes. If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from governmental health care programs, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our financial results.
Clinical drug development involves a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results.
Clinical development is a long, expensive and uncertain process, and data obtained from clinical trials and supportive studies are susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The results from early clinical trials may not be predictive of results eventually obtained in later clinical trials. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through preclinical studies and initial clinical trials. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profile of their medication candidate, despite promising results in earlier trials. Clinical trials may not demonstrate sufficient safety and efficacy to obtain regulatory approval.
To gain regulatory approval from the FDA to market mifepristone for the psychotic features of psychotic depression, our ongoing Phase 3 clinical trial (Study 14) must demonstrate the safety and efficacy of mifepristone for that indication, which it may fail to do. Even if the results of Study 14 are positive, the FDA may not accept a single positive trial with supportive data as sufficient to support the filing of our NDA and may require us to perform additional studies. If our ongoing Phase 3 clinical trial is not completed or conducted as planned or if mifepristone does not prove to be safe and effective or does not receive required regulatory approvals, the commercialization of mifepristone for the psychotic features of psychotic depression would be delayed or prevented, and our ability to generate revenues would be impaired.
Our ongoing Phase 1 study of mifepristone in combination with chemotherapy to treat triple-negative breast cancer is too small to demonstrate definitively the safety or efficacy of mifepristone for that indication. Even if the trial generates positive results, those results would have to be confirmed in at least one substantially larger, more expensive, and lengthier trial if we are to have sufficient basis for seeking regulatory approval.
Moreover, the commencement and completion of clinical trials may be delayed by many factors that are beyond our control, including:
| • | delays obtaining regulatory approval to commence a trial; |
| • | reaching agreement on acceptable terms with contract research organizations, or CROs, and clinical trial sites; |
| • | obtaining institutional review board, or IRB, approval at each site; |
| • | slower than anticipated patient enrollment; |
| • | scheduling conflicts with participating clinicians and clinical institutions; |
| • | lack of funding; |
| • | negative or inconclusive results; |
| • | patient noncompliance with the protocol; |
31
| • | adverse medical events or side effects among patients during the clinical trials; |
| • | negative or problematic FDA inspections of our clinical operations or manufacturing operations; and |
| • | real or perceived lack of effectiveness or safety of mifepristone. |
We could encounter delays if a clinical trial is suspended or terminated by us, the IRBs of the clinical trial sites in which such trials are being conducted, by the Data Safety Monitoring Board for such trial or by the FDA or other regulatory authorities. Such authorities may impose such a suspension or termination due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA or other regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a drug, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial.
We may decide, or the FDA or other regulatory authorities may require us, to pursue additional clinical or preclinical studies on mifepristone for the treatment of the psychotic features of psychotic depression. Additional trials or studies may require additional funding, the availability of which is not assured. Also, it is possible that additional trials or studies that we decide are necessary or desirable will delay or prevent the completion of our development programs. Even if we are able to conduct all of the clinical trials and supportive studies that we consider appropriate, we may never receive regulatory approval to market mifepristone for psychotic depression or triple-negative breast cancer.
Our use of MedAvante to provide centralized psychiatric rating services in Study 14, our ongoing clinical trial evaluating mifepristone for the psychotic features of psychotic depression, may not result in any improvement in the accuracy and consistency of the study's psychiatric assessments.
In connection with our ongoing Phase 3 trial evaluating mifepristone for the psychotic features of psychotic depression, Study 14, we engaged MedAvante to provide centralized psychiatric rating services. MedAvante is providing centralized psychometric assessments via high resolution video-conferencing. The use of MedAvante's centralized rating services is intended to increase the accuracy and consistency of the psychiatric assessments.
MedAvante has provided similar centralized rating services to companies conducting clinical studies in various psychiatric disorders. However, they have not previously provided centralized rating services to any study in patients with psychotic depression. Although we and MedAvante conducted a small pilot evaluation in patients with psychotic depression to assess patient receptivity, we cannot be certain that centralized rating will be successful with the patients enrolled in our study.
If patients are uncomfortable or unwilling to participate in the centralized rating process or if MedAvante is unable to provide services in a satisfactory manner over the course of the trial, we may not see any improvement in the accuracy or reliability of the psychiatric assessments. Such a result might diminish the likelihood of a successful trial or a definitive demonstration of the efficacy of mifepristone in treating the psychotic features of psychotic depression.
During screening for Study 14, there has been a higher than anticipated incidence of potential patients who do not meet appropriate criteria for entrance into the trial for diagnostic and other clinical reasons. Although we believe that this is the result of improved accuracy in the screening process resulting from the use of the MedAvante centralized rating services as an additional step in the selection of patients appropriate for inclusion in the study, MedAvante's diagnostic screening may not actually improve trial performance.
32
We will perform an interim analysis of the data from Study 14, however, the results may not establish the efficacy of mifepristone for the treatment of psychotic depression. If the results are inconclusive and we continue the study, conducting an interim analysis will make achieving a final, statistically significant result more difficult.
If we choose to perform an interim analysis of the data from Study 14, its results may be negative or inconclusive. If they are negative, then we will terminate the trial and either incur the substantial additional expense and delay of undertaking a new trial, or discontinue the study of mifepristone for the psychotic features of psychotic depression, which may reduce our future revenue. If the results are inconclusive, and we choose to continue the trial, we will incur additional expense and delay the possibility of our obtaining regulatory approval of a treatment for this disease.
In addition, performing an interim analysis makes the measure of statistical significance in any continued trial more rigorous and more difficult to meet. Our interim analysis will examine results from the first 226 patients enrolled in the study. To demonstrate efficacy with statistical significance, the results must have a "p value" (a measure of statistical significance) of £ 0.02967, which is a more difficult standard to meet than the 0.05 that would apply if we had deferred our analysis until we reached the trial's originally specified enrollment of 450 patients. If the results of our analysis are inconclusive and we continue our trial to full enrollment, then the results generated at that time must also meet a "p value" of £ 0.02967. Therefore, a continued trial following an interim analysis is less likely to achieve a statistically meaningful positive outcome.
We depend on third parties to conduct and manage many of our clinical trials and to perform related data collection and analysis and, if these third parties do not successfully carry out their contractual duties or meet expected timelines, we may face costs and delays that may prevent or delay us from obtaining regulatory approval for or commercializing our product candidates, which could substantially harm our business.
We rely on clinical investigators and clinical sites to enroll patients and other third parties such as clinical research organizations (CROs) to manage many of our trials and to perform related data collection and analysis. We control only certain aspects of these third parties' activities. Nevertheless, we are responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards, and our reliance on third parties does not relieve us of our regulatory responsibilities. We and these third parties are required to comply with cGCPs. If we or any of the third parties working on or conducting our trials fail to comply with applicable cGCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approval of our marketing applications, if at all. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials complies with cGCP requirements. In addition, our clinical trials must be conducted with product produced under cGMP regulations. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process. Moreover, we may not be able to control the timing of identification and selection of appropriate sites for our planned trials and the amount and timing of resources that the clinical sites that conduct the clinical testing may devote to our clinical trials. If our clinical investigators and clinical sites fail to enroll a sufficient number of patients in our clinical trials or fail to enroll them on our planned schedules, we will be unable to complete our trials or to complete them as planned, which could delay or prevent us from completing the clinical development of mifepristone for the psychotic features of psychotic depression, triple-negative breast cancer or other development programs.
We have agreements with CROs that are conducting our ongoing Phase 3 trial evaluating mifepristone for the treatment of the psychotic features of psychotic depression (Study 14) and our Phase 1 trial of mifepristone for the treatment of triple-negative breast cancer to supervise and monitor clinical site performance and to perform investigator supervision, data collection and analysis for this trial. The conduct of future clinical trials may also be conducted through the use of CROs and third party clinical sites. We may not be able to maintain
33
relationships with this or other CROs or with the clinical investigators and the clinical sites through the completion of all trial activities without delays in anticipated timing of trial activities or excessive expenditures. If any of our relationships with CROs or other third parties terminates, we may not be able to enter into arrangements with alternative CROs or third parties on commercially reasonable terms, or at all. If these CROs, clinical investigators, clinical sites or other third parties do not carry out their contractual duties or obligations or fail to meet expected deadlines, or if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to their failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our clinical trials may be extended, delayed or terminated and we may be unable to obtain regulatory approval for, or successfully commercialize, mifepristone for the psychotic features of psychotic depression or triple-negative breast cancer.
Failure to obtain regulatory approval in foreign jurisdictions will prevent us from commercializing Korlym and our other product candidates abroad.
We may seek to commercialize our products and product candidates in international markets with the help of one or more partners or on our own. Outside the United States, we may commercialize a product only if we receive a marketing authorization and, in many cases, pricing approval, from the appropriate regulatory authorities, whose approval processes include all of the risks associated with the FDA approval process, and, in some cases, additional risks. The approval procedure varies among countries and can involve additional testing, and the time required to obtain approval may differ from that required to obtain FDA approval. Other than seeking and receiving Orphan Drug Designation in the EU and the submission of our MAA to the EMA in October 2013, we have not taken any actions to obtain foreign approvals. We may not develop our product candidates in the clinic in order to obtain foreign regulatory approvals on a timely basis, if at all.
Approval by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA, but a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in others. We may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our product candidates in any foreign market.
The "fast track" designation for the development program of mifepristone for the treatment of the psychotic features of psychotic depression may not lead to a faster development or regulatory review or approval process.
If a human medicine is intended for the treatment of a serious or life-threatening disease or condition and the medicine demonstrates the potential to address unmet medical needs for this disease or condition, the sponsor of an IND may apply for FDA "fast track" designation for a particular indication. Marketing applications submitted by sponsors of product candidates in fast track development may qualify for expedited FDA review under the policies and procedures offered by the FDA, but the fast track designation does not assure any such qualification. Although we have obtained a fast track designation from the FDA for mifepristone for the treatment of the psychotic features of psychotic depression, we may not experience a faster development process, review or approval compared to applications considered for approval under conventional FDA procedures. In addition, the FDA may withdraw our fast track designation at any time. If we lose our fast track designation, the approval process may be delayed. In addition, our fast track designation does not guarantee that we will qualify for or be able to take advantage of the expedited review procedures and does not increase the likelihood that mifepristone will receive regulatory approval for the treatment of the psychotic features of psychotic depression.
34
We face competition from companies with substantial financial, technical and marketing resources, which could limit our future revenues from the commercialization of mifepristone for the treatment of the psychotic features of psychotic depression or for other indications.
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. If approved for commercial use as a treatment for the psychotic features of psychotic depression, mifepristone will compete with established treatments, including electroconvulsive therapy (ECT) and combination medicinal therapy.
Combination medicinal therapy consists of the use of antipsychotic and antidepressant medicines not currently approved for the treatment of psychotic depression. The antipsychotics are prescribed by physicians for off-label use to treat the psychotic features of psychotic depression, which is the clinical target of mifepristone. Antipsychotics include Abilify ® (Bristol-Myers Squibb), Clozaril ® (Novartis), Geodon ® and Navane ® (Pfizer), Haldol ® (Ortho-McNeil), Mellaril ® (Mylan), Risperdal ® (Janssen Pharmaceuticals), Seroquel ® (AstraZeneca), Stelazine ® and Thorazine ® (GlaxoSmithKline) and Zyprexa ® (Eli Lilly). Mifepristone may not compete effectively with these established treatments. In addition, we are aware of one clinical trial conducted by Organon, now a division of Merck & Co., for a new chemical entity for the treatment of psychotic depression. Organon's new chemical entity is a GR-II antagonist; we believe that its commercial use would be covered by our patent.
Our present and potential competitors include major pharmaceutical companies such as the makers of the drugs identified above, as well as specialized pharmaceutical firms, universities and public and private research institutions. Moreover, we expect competition to intensify as technical advances are made. These competitors, either alone or with collaborative parties, may succeed with the development and commercialization of medicinal products that are superior to and more cost-effective than mifepristone. Many of our competitors and related private and public research and academic institutions have greater experience, more financial and marketing resources and larger research and development staffs than we do. In addition, many of these competitors, either alone or together with their collaborative partners, have significantly greater experience than we do in developing human medicines, obtaining regulatory approvals, manufacturing and commercializing products.
Accordingly, mifepristone may not be an effective competitor against established treatments and our present or potential competitors may succeed in developing medicinal products that are superior to mifepristone or render mifepristone obsolete or non-competitive. If we are unable to establish mifepristone as a superior and cost-effective treatment for the psychotic features of psychotic depression, or any future use, we may be unable to generate the revenues necessary to support our business.
Our efforts to discover, develop and commercialize new product candidates beyond mifepristone are at a very early stage. If we fail to identify and develop additional uses for GR-II antagonists, we may be unable to market additional products.
To develop additional potential sources of revenue, we believe that we must identify and develop additional product candidates or new therapeutic uses for mifepristone. We own or have exclusively licensed issued U.S. patents covering the use of GR-II antagonists to treat psychotic depression, triple-negative breast cancer, mental disorders by optimizing mifepristone levels in plasma serum, mild cognitive impairment, weight gain due to treatment with antipsychotic medication, stress disorders, early dementia, delirium, gastroesophageal reflux disease, Down's Syndrome, catatonia, psychosis associated with cocaine addiction, psychosis associated with Interferon-alpha therapy, migraine headaches, and to increase the therapeutic response to ECT. In addition, we have six U.S. method of use patent applications covering GR-II antagonists for the treatment of a number of other metabolic and psychiatric disorders, six U.S. composition of matter patents covering specific GR-II antagonists, and one additional U.S. composition of matter patent application is pending. We have also filed patent applications in the major international markets.
35
The use of GR-II antagonists may not be effective to treat these conditions or any other indications. Moreover, we could discover that the use of GR-II antagonists in these patient populations has unacceptable side effects or is otherwise not safe. Due to the risks of efficacy and side effects inherent in developing novel compounds, we are likely to enter multiple compounds into development, which would increase our rate of spending with no assurance that we will be successful in developing new drugs that are safe and effective.
In addition, we may not develop or continue to develop product candidates for any of the indications or compounds covered by our patents and patent applications. Typically, there is a high rate of attrition for product candidates in preclinical and clinical trials, and our product development efforts may not lead to commercially viable products. For example, although we plan to advance one or more of the new compounds to the clinic in 2014, we may fail to do so.
We may elect to enter into collaboration arrangements with respect to one or more of our product candidates. If we do enter into such an arrangement, we would be dependent on a collaborative partner for the success of the product candidates developed under the arrangement. Any future collaborative partner may fail to successfully develop or commercialize a product candidate under a collaborative arrangement.
We only have significant clinical experience with mifepristone and we may determine that mifepristone is not desirable for uses other than for the treatment of hyperglycemia secondary to hypercortisolism in adult patients with endogenous Cushing's syndrome who have type 2 diabetes mellitus or glucose intolerance and have failed surgery or are not candidates for surgery and, potentially, for the psychotic features of psychotic depression and triple-negative breast cancer. For example, we do not intend to develop mifepristone for mitigation of the weight gain associated with the use of Zyprexa, Risperdal or other atypical antipsychotics, even though we have reported positive results in the proof of concept studies described above in Part I, Item 1, Business – Overview – Mifepristone Proof-of-Concept Studies. We may pursue other GR-II antagonists for this use. The compounds developed pursuant to our early clinical, preclinical and discovery research programs may fail to become viable product candidates regardless of the resources we may dedicate to the program. Even if product candidates are identified, we may abandon further development efforts before we reach clinical trials or after expending significant expense and time conducting clinical trials due to financial constraints, concerns over the safety or efficacy of the product candidates, manufacturing difficulties or other reasons. Moreover, governmental authorities may enact new legislation or regulations that could limit or restrict our development efforts. If we are unable to successfully discover and commercialize new uses for GR-II antagonists, we may be unable to generate sufficient revenue to support our operations.
If we lose our key personnel or are unable to attract and retain additional skilled personnel, we may be unable to pursue our product development and commercialization efforts.
Our ability to operate successfully and manage our potential future growth depends significantly upon retaining key research, technical, sales, marketing, managerial and financial personnel, and attracting and retaining additional highly qualified personnel in these areas. For example, we do not currently employ a Chief Medical Officer to manage our clinical development efforts, although our efforts to hire such an executive are ongoing. We depend substantially on the principal members of our management and scientific staff. We do not have agreements with any of our executive officers that provide for their continued employment with us or employment insurance covering any of our key personnel. Any officer or employee can terminate his or her relationship with us at any time and work for one of our competitors. The loss of these key individuals could result in competitive harm because we could experience delays in our product research, development and commercialization efforts without their expertise.
We face intense competition for qualified personnel from numerous companies, as well as universities and nonprofit research organizations in the highly competitive San Francisco Bay Area. Although we believe that we have been successful in attracting and retaining qualified personnel to date, we may not be able to attract and retain sufficient qualified personnel in the future. The inability to attract and retain these personnel could result in delays in the research, development and commercialization of our potential products.
36
Rapid technological change could make our product and product candidates obsolete.
Pharmaceutical technologies have undergone rapid and significant change and we expect that they will continue to do so. Our future will depend in large part on our ability to maintain a competitive position with respect to these technologies. Korlym and any products and processes that we develop may become obsolete or uneconomical before we recover any or all expenses incurred in connection with their development. Rapid technological change could make Korlym and our product candidates obsolete or uneconomical, which could materially adversely affect our business, financial condition and results of operations.
The occurrence of a catastrophic disaster or other similar events could cause damage to our own or our manufacturers' facilities and equipment, which could require us to cease or curtail operations.
Because our executive offices are located in the San Francisco Bay Area and some of our current manufacturers are also located in earthquake-prone areas, our business is vulnerable to damage from various types of disasters or other similarly disruptive events, including earthquake, fire, flood, power loss and communications failures. In addition, political considerations relating to mifepristone may put us and our manufacturers at increased risk for terrorist attacks, protests or other disruptive events. If any disaster or other similar event were to occur, we may not be able to operate our business and our manufacturers may not be able to produce Korlym or our product candidates. Our insurance may not be adequate to cover, and our insurance policies may exclude coverage for, our losses resulting from disasters or other business interruptions.
We rely significantly on information technology and any failure, inadequacy, interruption or security lapse of that technology, including any cyber security incidents, could harm our ability to operate our business effectively.
Despite the implementation of security measures, our internal computer systems and those of third parties with which we contract are vulnerable to damage from cyber-attacks, computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. System failures, accidents or security breaches could cause interruptions in our operations, and could result in a material disruption of our clinical and commercialization activities and business operations, in addition to possibly requiring substantial expenditures of resources to remedy. The loss of clinical trial data could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and our product research, development and commercialization efforts could be delayed.
Risks Related to Our Capital Needs and Financial Results
We may need additional capital in order to complete the development and commercialization of mifepristone for psychotic depression, triple-negative breast cancer or other indications or for the development and commercialization of our proprietary, selective GR-II antagonists. Additional capital may not be available to us at all or on favorable terms, which could adversely affect our business.
We may have to perform more clinical trials, in addition to our ongoing Phase 3 trial, prior to the filing by the FDA of our NDA for mifepristone for the treatment of the psychotic features of psychotic depression. If so, we may need to raise additional funds to complete the development of mifepristone for that indication. In addition, we may need to raise additional funds to continue and expand the development of mifepristone for the treatment of triple-negative breast cancer and of our proprietary, selective GR-II antagonists in various indications. We may also raise additional funds for other research and development activities, including clinical trials, and working capital and for other general corporate purposes, or to acquire or invest in businesses, products and technologies that are complementary to our own.
37
Factors impacting our cash position and future prospects of liquidity include the following:
| • | the amount and timing of revenues from the commercialization of Korlym; |
| • | the pace at which physicians adopt Korlym as a treatment; |
| • | the willingness of insurance companies, the government and other third-party payors to provide coverage for Korlym at reasonable rates; |
| • | changes in the coverage and reimbursement policies of third-party insurance companies or government agencies; |
| • | the costs, timing of site selection and enrollment of our clinical trials; |
| • | the results of our research efforts and clinical trials; |
| • | the need to perform additional clinical trials and other supportive studies; |
| • | the results of our interim analysis of Study 14 and the timing of the submission of an NDA to the FDA, the acceptance of the NDA submission, and the outcome of the FDA approval process for the marketing of mifepristone for the treatment of the psychotic features of psychotic depression; |
| • | the timing of commercialization of mifepristone for the treatment of psychotic depression; |
| • | the timing and outcome of our Phase 1 study of mifepristone for the treatment of triple-negative breast cancer; |
| • | developments or disputes concerning patents or proprietary rights, including announcements of claims of infringement, interference or litigation against us or our licensors; |
| • | actual or anticipated fluctuations in our operating results; |
| • | changes in our growth rates; and |
| • | changes in our research and development plans for our proprietary, selective GR-II antagonists. |
Consequently, we may need additional funding sooner than anticipated. In addition, we may choose to raise additional capital due to market conditions or strategic considerations even if we believe we have sufficient funds for our current and future operating plans.
We cannot be certain that additional funding will be available on acceptable terms or at all. Even though we have raised funds a number of times in the past, market and economic conditions may make it difficult for us to raise any or sufficient additional capital. Our sales of common stock and warrants and the exercises of warrants have been dilutive to stockholders and any exercise of outstanding warrants and additional equity financing could cause further dilution to stockholders. Debt financing, if available, may involve restrictive covenants. If we obtain funds through collaborations with others, these arrangements may be on unfavorable terms or may require us to relinquish certain rights to Korlym, our technologies or product candidates, which we would otherwise seek to develop on our own. If adequate funds are not available, we may be required to delay, reduce the scope of or eliminate one or more of our research or development programs or we may be required to discontinue operations.
We have incurred losses since inception and anticipate that we will incur continued losses for at least the next year.
We have a limited history of operations and have focused primarily on clinical trials. We have begun to commercialize Korlym and, if the outcome of our clinical trials supports it, we plan to seek FDA regulatory clearance to market mifepristone for the treatment of the psychotic features of psychotic depression. Historically, we have funded our operations primarily from the sale of our equity securities. We have incurred losses in each year since our inception in 1998. As of December 31, 2013, we had an accumulated deficit of $292.6 million. We began to sell our first commercial product, Korlym, in the United States in April 2012. Based on this limited experience marketing Korlym, it is difficult for us to predict the magnitude or timing of future product sales. We expect our research and development expenses to increase in connection with the clinical trials and other development activities for mifepristone and for other product candidates. We expect to incur significant expenses related to commercializing Korlym. We are unable to predict the extent of any future losses or whether or when we will become profitable.
38
We may not be able to pursue all of our product research and development opportunities if we are unable to generate sufficient revenue or secure adequate funding for these programs.
The costs required to start or continue many of the programs that our intellectual property allows us to consider for further development are collectively greater than the funds currently available to us. For example, we have successfully discovered three series of compounds that are selective GR-II antagonists but do not appear to block the progesterone receptor. Further development of these proprietary compounds or any further development stemming from our method of use patents may be delayed or cancelled if we determine that such development may jeopardize our ability to complete the clinical development of mifepristone for the treatment of psychotic depression.
Global economic conditions could adversely affect our liquidity and financial condition.
In the United States and globally, market and economic conditions have been volatile over the past few years, with significantly tighter credit conditions in the markets in which we conduct our operations. Renewed concerns about the recent recession and the systemic impact of adverse economic conditions, such as unstable global financial markets, adverse effects on the cost and availability of capital, high corporate, consumer and governmental debt levels and high unemployment may cause lenders and institutional investors to reduce, and in some cases, cease, to provide credit to businesses. Renewed or increased turbulence in the global markets and economies may adversely affect our liquidity and financial condition.
If we do not have sufficient cash flow to continue operating our business and are unable to borrow funds or raise equity or debt capital, we may need to find alternative ways to increase our liquidity. Such alternatives may include, without limitation, curtailing clinical or drug development activity, or limiting our commercial efforts, product manufacturing or sales and marketing support, which would have an adverse effect on our business, results of operations, cash flows and financial condition.
If we acquire other selective GR-II antagonists or other technologies or potential products, we will incur a variety of costs and may never realize the anticipated benefits of the acquisition.
If appropriate opportunities become available, we may attempt to acquire other GR-II antagonists, particularly GR-II antagonists that do not terminate pregnancy. We may also be able to acquire other technologies or potential products that are complementary to our operating plan. We currently have no commitments, agreements or plans for any acquisitions. The process of acquiring rights to another GR-II antagonist or any other potential product or technology may result in unforeseen difficulties and expenditures and may absorb significant management attention that would otherwise be available for ongoing development of our business. In addition, we may fail to realize the anticipated benefits of any acquired potential product or technology. Future acquisitions could dilute our stockholders' ownership interest in us and could cause us to incur debt, expose us to future liabilities and result in amortization or other expenses related to goodwill and other intangible assets.
Failure to meet our obligations under our Financing Agreement with Biopharma Secured Debt Fund II Sub, S.àr.l (Biopharma), could adversely affect our financial results and liquidity.
Pursuant to our Financing Agreement with Biopharma entered into in August 2012, we are obligated to make payments to Biopharma equal to 20 percent of our net product sales of Korlym, any future mifepristone-based products and our next-generation selective GR-II antagonists (Covered Products), subject to certain quarterly caps, as well as an un-capped 20 percent of any upfront, milestone or other contingent payments we receive with respect to Covered Products, until such payments to Biopharma total $45.0 million.
Pursuant to this agreement, we may not: (i) incur indebtedness greater than the sum of earnings before interest, taxes, depreciation and amortization, including such items as non-cash stock-based compensation, for
39
the four calendar quarters preceding such incurrence, which we refer to as the Indebtedness Covenant; (ii) pay a dividend or other cash distribution, unless we have cash and cash equivalents in excess of $50.0 million after such payment; (iii) amend or restate our certificate of incorporation or bylaws unless such amendments or restatements do not affect Biopharma's interests under the transaction; and (iv) encumber any of the collateral securing our performance under the agreement.
The percentage used to calculate our payments to Biopharma would increase to 50 percent and any applicable payment caps would lapse if we (i) fail to provide Biopharma with certain information regarding our promotion and sales of Covered Products, (ii) do not devote a commercially reasonable amount of resources to the promotion and marketing of the Covered Products or (iii) violate the Indebtedness Covenant and, in each case, fail to cure within the applicable cure period.
Upon a Corcept change of control transaction, as defined in the agreement, Biopharma will be automatically entitled to receive any amounts not previously paid, up to our maximum repayment obligation of $45.0 million. As defined in the agreement, "Change of Control" includes, among other things, (i) a greater than 50 percent change in the ownership of Corcept, (ii) certain changes in Board composition of Corcept and (iii) the licensing of Korlym to a third party for sale in the United States.
To secure our obligations under the agreement, we granted Biopharma a security interest in our rights in patents, trademarks, trade names, domain names, copyrights, know-how and regulatory approvals related to the Covered Products, all books and records relating to the foregoing and all proceeds of the foregoing, which we refer to as the Collateral. If we (i) fail to deliver a royalty payment when due and do not remedy that failure within 30 days, (ii) fail to maintain a first-priority perfected security interest in the Collateral in the United States and do not remedy that failure within five business days of receiving notice of such failure or (iii) become subject to an event of bankruptcy, then Biopharma may attempt to recover up to $45.0 million (after deducting any payments we have already made).
We cannot assure that we will not breach the covenants or other terms of, or that an event of default will not occur under this agreement and, if a breach or event of default occurs, we cannot assure that we will be able to cure the event within the time permitted. Any failure to pay our obligations when due, any breach or default of our covenants or other obligations, or any other event that causes an acceleration of payment at a time when we do not have sufficient resources to meet these obligations, could have a material adverse effect on our business, results of operations, financial condition and future viability.
The acceleration of the payment obligation in the event of a change of control transaction may make us less attractive to potential acquirers, and the payment of such funds out of our available cash or acquisition proceeds would reduce acquisition proceeds for our stockholders.
Risks Relating to Our Intellectual Property
If Korlym or future product candidates conflict with the patents of others or if we become involved in other intellectual property disputes, we may have to engage in costly litigation or obtain a license and we may be unable to commercialize our product candidates.
Our success depends in part on our ability to obtain and maintain adequate patent protection for the use of mifepristone for the treatment of the psychotic features of psychotic depression and other potential uses of GR-II antagonists. If we do not adequately protect our intellectual property, competitors may be able to use our intellectual property and erode our competitive advantage.
To date, we own fifteen issued U.S. method of use patents and have exclusively licensed three issued U.S. method of use patents. We have six U.S. method of use patent applications pending for GR-II antagonists. We own six composition of matter patents and have one composition of matter patent application pending. We have
40
applied, and will continue to apply, for patents covering our product candidates as we deem appropriate. We have also filed, where we deemed appropriate, foreign patent applications corresponding to our U.S. patents and applications.
We have exclusively licensed three issued U.S. patents from Stanford University for the use of GR-II antagonists in the treatment of psychotic major depression, which is commonly referred to as psychotic depression, cocaine-induced psychosis and early dementia, including early Alzheimer's disease. We have also exclusively licensed from the University of Chicago allowed U.S. patent claims for the use of mifepristone in the treatment of triple-negative breast cancer, which claims are covered in U.S. Patent Application No. 13/071,363 "Methods and Compositions Related to Glucocorticoid Receptor Antagonists and Breast Cancer." On August 8, 2013 the U.S. Patent and Trademark Office notified the University of Chicago that certain claims in the application had been allowed, although the patent has not yet issued.
We bear the costs of prosecuting, protecting and defending the rights to these patents. In order to maintain the exclusive license to these patents until their expiration, we are obligated to make milestone and royalty payments to both universities. If we become noncompliant with our obligations under our agreements, we may lose the right to commercialize mifepristone for the treatment of psychotic depression, cocaine-induced psychosis, early dementia and triple-negative breast cancer and our business would be materially harmed. In addition, if Stanford University were to terminate our mifepristone license due to breach of the license on our part, we would not be able to commercialize mifepristone for the treatment of the psychotic features of psychotic depression, cocaine-induced psychosis or early dementia. If the University of Chicago were to terminate our license, we would not be able to commercialize mifepristone for the treatment of triple-negative breast cancer.
Our patent applications and patents licensed or issued to us may be challenged by third parties and our patent applications may not result in issued patents. For example, in 2004, Akzo Nobel (now a division of Merck & Co.) filed an observation challenging the claims of our exclusively licensed European patent application with claims directed to psychotic depression. In this instance, the patent later issued and, in 2007, we received notice from the European Patent Office that there will be no opposition proceedings in Europe in regard to this patent.
Our presently pending and future patent applications may not issue as patents, and any patent issued to us may be challenged, invalidated, held unenforceable or circumvented. For example, the arguments presented by Akzo Nobel could be raised in the United States either before the U.S. Patent and Trademark Office or in a court of law. Furthermore, the claims in patents which have been issued to us, or which may be issued to us in the future, may not be sufficiently broad to prevent third parties from producing competing products. In addition, the laws of various foreign countries in which we compete may not protect our intellectual property to the same extent as do the laws of the United States. If we fail to obtain adequate patent protection for our proprietary technology, our competitors may produce competing products based on our technology, which would impair our ability to compete.
If a third party were successful in asserting an infringement claim against us, we could be forced to pay damages and prevented from developing, manufacturing or marketing our potential products. We do not have liability insurance for patent infringements. A third party could require us to obtain a license to continue to use their intellectual property, and we may not be able to do so on commercially acceptable terms, or at all. We believe that significant litigation will continue in our industry regarding patent and other intellectual property rights. If we become involved in litigation, it could consume a substantial portion of our resources. Regardless of the merit of any particular claim, defending a lawsuit takes significant time, is expensive and diverts management's attention from other business.
If we are unable to protect our trade secrets and proprietary information, our ability to compete in the market could be diminished.
In addition to patents, we rely on a combination of confidentiality, nondisclosure and other contractual provisions, laws protecting trade secrets and security measures to protect our trade secrets and proprietary
41
information. Nevertheless, these measures may not adequately protect our trade secrets or other proprietary information. If they do not adequately protect our rights, third parties could use our proprietary information, which could diminish our ability to compete in the market. In addition, employees, consultants and others who participate in the development of our product candidates may breach their agreements with us regarding our trade secrets and other proprietary information, and we may not have adequate remedies for the breach. We also realize that our trade secrets may become known through means not currently foreseen. Notwithstanding our efforts to protect our trade secrets and proprietary information, our competitors may independently develop similar or alternative products that are equal or superior to our product candidates without infringing on any of our proprietary information or trade secrets.
Our licensed patent covering the use of mifepristone to treat psychotic depression and our licensed patent rights covering the use of mifepristone to treat triple-negative breast cancer cover only mifepristone's method of use and not its composition of matter, which may make it more difficult for us to prove patent infringement if physicians prescribe another manufacturer's mifepristone for the treatment of Cushing's syndrome, psychotic depression or triple-negative breast cancer or if patients acquire mifepristone from other sources, such as the internet or underground market.
We have an exclusive license from Stanford University to a patent covering the use of GR-II antagonists, including mifepristone, for the treatment of psychotic depression and an exclusive license from the University of Chicago to certain allowed patent claims covering the use of mifepristone to treat triple-negative breast cancer. A method of use patent covers only a specified use of a particular compound, not a particular composition of matter. Because none of the patents we have licensed from Stanford University and none of the allowed patent rights we have licensed from the University of Chicago cover the composition of mifepristone, we cannot prevent others from commercializing mifepristone in indications not covered by these or our other method of use patents. Although any such "off-label" use would violate our patents, effectively monitoring compliance with our patents may be difficult and costly.
In addition, we cannot be assured that patients will not obtain mifepristone from other sources. As with other pharmaceutical products, patients may be able to purchase mifepristone through the internet or underground market. Mifepristone is also sold in the United States by Danco Laboratories for the termination of early pregnancy. While distribution is limited to a single dose provided in the physician's office and covered by other restrictions, we cannot be certain that Cushing's syndrome patients will not be able to obtain mifepristone from this source or others, should another company receive approval to market mifepristone for another indication.
Risks Related to Our Stock
The market price of our common stock has been and is likely to continue to be highly volatile due to the limited number of shares of our common stock held by non-affiliates or factors influencing the stock market and opportunities for sale at any given time may be limited.
We cannot assure you that an active trading market for our common stock will exist at any time. Holders of our common stock may not be able to sell shares quickly or at the market price if trading in our common stock is not active. During the 52-week period ended March 3, 2014, our average daily trading volume was approximately 231,000 shares and the intra-day sales prices per share of our common stock on The NASDAQ Stock Market ranged from $1.47 to $3.98. As of March 3, 2014, our officers, directors and principal stockholders controlled 36 percent of our common stock. The trading price of our common stock has been and is likely to continue to be highly volatile and could be subject to wide fluctuations in price in response to various factors, many of which are beyond our control, including:
| • | the pace of market acceptance of Korlym or the timing and level of coverage and reimbursement attained; |
| • | our cash and short-term investment position; |
| • | actual or anticipated timing and results of our clinical trials; |
42
| • | new products or services introduced or announced by us or our competitors; |
| • | actual or anticipated regulatory approvals of our product candidates or of competing products; |
| • | changes in laws or regulations applicable to our product candidates or our competitors' products; |
| • | changes in the expected or actual timing of our development programs or our competitors' potential development programs; |
| • | actual or anticipated variations in quarterly operating results, including potential product returns and timing of revenue recognition; |
| • | announcements of technological innovations by us, our collaborators or our competitors; |
| • | general market and economic conditions; |
| • | changes in financial estimates or recommendations by securities analysts; |
| • | conditions or trends in the biotechnology and pharmaceutical industries; |
| • | changes in the market valuations of similar companies; |
| • | announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures or capital commitments; |
| • | additions or departures of key personnel; |
| • | disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
| • | developments concerning collaborations; |
| • | trading volume of our common stock; |
| • | limited number of shares of our common stock held by our non-affiliates; |
| • | maintaining compliance with the listing requirements of the stock exchange on which we are listed; |
| • | success of additional financing efforts; and |
| • | purchases or sales of our common stock by us, our officers, directors or our stockholders. |
In addition, the stock market in general, The NASDAQ Stock Market and the market for biotechnology and life sciences companies in particular have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies. These broad market and industry factors may seriously harm the market price of our common stock, regardless of our operating performance. In the past, following periods of volatility in the market, securities class-action litigation has often been instituted against companies. Such litigation, if instituted against us, could result in substantial costs and diversion of management's attention and resources.
If our operating and financial performance in any given period does not meet the guidance that we provide to the public, estimates published by research analysts or other investor expectations, our stock price may decline.
We have provided guidance as to our expected 2014 net revenue. Our guidance is only an estimate of what management believes is realizable as of the date of the release of such guidance. Our actual results may vary from our guidance and the variations may be material.
There are a number of reasons why we might fail to meet our financial guidance or other expectations about our business, including, but not limited to, the risks and uncertainties described in this report and in our other public filings and public statements. In particular, there are inherent difficulties in predicting the amount of Korlym that will be sold. For example, the rate of physician adoption of Korlym is uncertain. Research analysts who cover our business have put forth a wide range of revenue estimates, based on their own analyses. We believe research analysts will consider the guidance we have provided as one factor in determining their own annual revenue estimates. Estimating our net revenue for future periods is difficult and you should rely on our guidance and the estimates of research analysts at your own discretion. If, in the future, our operating or financial results for a particular period do not meet our guidance, analyst estimates or the expectations of investors, or if we reduce our guidance for future periods, our stock price may decline.
43
Research analysts may not continue to provide or initiate coverage of our common stock or may issue negative reports, which may have a negative impact on our common stock's market price.
Securities analysts currently covering our common stock may discontinue research coverage. Additional securities analysts may elect not to provide research coverage of our common stock. A lack of research coverage may adversely affect our common stock's market price. The trading market for our common stock may be affected in part by the research and reports that industry or financial analysts publish about us or our business. If one or more of the analysts who elects to cover us downgrades our stock, our stock price would likely decline rapidly and significantly. If one or more of these analysts ceases coverage of our company, we could lose visibility in the market, which in turn could cause our stock price to decline. In addition, rules mandated by the Sarbanes-Oxley Act of 2002, and a global settlement reached in 2003 between the SEC, other regulatory analysts and a number of investment banks have led to a number of fundamental changes in how analysts are reviewed and compensated. In particular, many investment banking firms are required to contract with independent financial analysts for their stock research. It may be difficult for companies such as ours with smaller market capitalizations to attract independent financial analysts that will cover our common stock. This could have a negative effect on our market price.
Sale of a substantial number of shares of our common stock may cause the price of our common stock to decline.
Sales of a substantial number of shares of our common stock in the public market could harm the market price of our common stock. As additional shares of our common stock become available for resale in the public market, whether as a result of equity financings by us or due to the release of trading restrictions, the supply of our common stock will increase, which could decrease the price. Substantially all of the shares of our common stock are eligible for sale, subject to applicable volume and other resale restrictions.
We may be required to pay significant penalties if we are not able to meet our obligations under our outstanding registration rights agreements.
We have entered into registration rights agreements in connection with certain of our securities offerings. We may be obligated to pay liquidated damages if we do not meet our obligations under those agreements.
If we are required to pay significant amounts, such as the liquidated damages described above, under these or future registration rights agreements, it could have a material adverse effect on our financial condition and ability to finance our operations.
Our officers, directors and principal stockholders, acting as a group, will be able to significantly influence corporate actions.
As of March 3, 2014, our officers, directors and principal stockholders control 36 percent of our common stock. As a result, these stockholders, acting together, will be able to significantly influence all matters requiring approval by our stockholders, including the election of directors and the approval of mergers or other business combination transactions. The interests of this group of stockholders may not always coincide with our interests or the interests of other stockholders and may prevent or delay a change in control. This significant concentration of share ownership may adversely affect the trading price of our common stock because investors often perceive disadvantages to owning stock in companies with controlling stockholders.
Changes in laws and regulations may result in increased costs to us, which may harm our financial results.
New laws and regulations, as well as changes to existing laws and regulations, affecting our company, including the provisions of the PPACA requiring the reporting of aggregate spending related to health care professionals, the provisions of the Sarbanes-Oxley Act of 2002 and rules adopted by the SEC and by The
44
NASDAQ Stock Market have and will likely continue to result in increased costs to us as we respond to their requirements. We are investing resources to comply with evolving laws and regulations, and this investment may result in increased selling, general and administrative expenses and a diversion of management's time and attention from revenue-generating activities to compliance activities.
In addition, new rules and regulations could make it more difficult or costly for us to obtain certain types of insurance, including director and officer liability insurance, and we may be forced to accept reduced policy limits and coverage or incur higher costs to obtain the same or similar coverage. The impact of these events could also make it more difficult for us to attract and retain qualified persons to serve on our Board of Directors, or our board committees, or as executive officers. At present, we cannot predict or estimate the amount of the additional costs related to new rules and regulations or the timing of such costs.
Compliance with public company obligations, including the securities laws and regulations, is costly and requires significant management resources, and we may fail to comply.
We are a small company with limited resources.
The federal securities laws and regulations, including the corporate governance and other requirements of the Sarbanes-Oxley Act of 2002, impose complex and continually changing regulatory requirements on our operations and reporting. These requirements have increased and will continue to increase our legal compliance costs.
Section 404 of the Sarbanes-Oxley Act of 2002 requires that we evaluate and determine the effectiveness of our internal controls over financial reporting and provide a management report on the internal control over financial reporting. This same legislation also requires that the independent registered public accounting firm auditing our financial statements must attest to and report on the effectiveness of our internal controls over financial reporting. If we are unable to complete the required assessment as to the adequacy of our internal control over financial reporting in future years or if our independent registered public accounting firm is unable to provide us with an unqualified report as to the effectiveness of our internal control over financial reporting as of future year ends, investors could lose confidence in the reliability of our financial reporting.
Changes in or interpretations of accounting rules and regulations could result in unfavorable accounting charges or require us to change our accounting policies or operating practices.
Accounting methods and policies for business and marketing practices of pharmaceutical companies are subject to continual review, interpretation and guidance from relevant accounting authorities, including the SEC. Although we believe that our accounting practices are consistent with current accounting pronouncements, changes to or interpretations of accounting methods or policies in the future may require us to reclassify, restate or otherwise change or revise our financial statements. Any such changes could result in corresponding changes to the amounts of assets, liabilities, revenues, expenses and income. Any such changes could have a material adverse effect on our business, financial position and results of operations and could cause the market value of our common stock to decline.
If we fail to continue to meet all applicable NASDAQ Stock Market requirements, our stock could be delisted by The NASDAQ Stock Market. If delisting occurs, it would adversely affect the market liquidity of our common stock and harm our business.
If we are unable to meet any of The NASDAQ listing requirements in the future, including, for example, if the closing bid price for our common stock is below $1 per share for 30 consecutive trading days, The NASDAQ Stock Market could determine to delist our common stock, the delisting could adversely affect the market liquidity of our common stock and the market price of our common stock could decrease. During the 52-week period ended March 3. 2014, the intra-day sales prices per share of our common stock on The NASDAQ Stock
45
Market ranged from $1.47 to $3.98. Such delisting could also adversely affect our ability to obtain financing for the continuation of our operations and could result in the loss of confidence by investors, suppliers and employees.
Anti-takeover provisions in our charter and bylaws and under Delaware law and payment acceleration provisions under the Biopharma Financing Agreement may make an acquisition of us or a change in our management more expensive or difficult, even if an acquisition or a management change would be beneficial to our stockholders.
Provisions in our charter and bylaws may delay or prevent an acquisition of us or a change in our management. Some of these provisions allow us to issue preferred stock without any vote or further action by the stockholders, require advance notification of stockholder proposals and nominations of candidates for election as directors and prohibit stockholders from acting by written consent. In addition, a supermajority vote of stockholders is required to amend our bylaws. Our bylaws provide that special meetings of the stockholders may be called only by our Chairman, President or the Board of Directors and that the authorized number of directors may be changed only by resolution of the Board of Directors. These provisions may prevent or delay a change in our Board of Directors or our management, which is appointed by our Board of Directors. In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law. Section 203 may prohibit large stockholders, in particular those owning 15 percent or more of our outstanding voting stock, from merging or combining with us. In addition, our payment obligations to Biopharma accelerate in the event of a change of control transaction. See "Risk Factors – Failure to meet our obligations under our Financing Agreement with Biopharma Secured Debt Fund II Sub, S.à r.l, could adversely affect our financial results and liquidity." These provisions in our charter and bylaws and under Delaware law and the Financing Agreement could reduce the price that investors might be willing to pay for shares of our common stock in the future and result in the market price being lower than it would be without these provisions.
| ITEM 1B. | UNRESOLVED STAFF COMMENTS |
None.
| ITEM 2. | PROPERTIES |
We lease 13,225 square feet of office space in Menlo Park, California for our corporate facilities. Our current lease extended our occupancy through December 2014. We expect that these facilities will accommodate our operations for the next year.
| ITEM 3. | LEGAL PROCEEDINGS |
We are not currently involved in any material legal proceedings.
| ITEM 4. | MINE SAFETY DISCLOSURES |
Not applicable.
46
PART II
ITEM 5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock is traded on The NASDAQ Capital Market under the symbol "CORT". The following table sets forth the high and low intra-day sale prices per share of our common stock on The NASDAQ Capital Market for the periods indicated. These prices represent quotations among dealers without adjustments for retail mark-ups, markdowns or commissions, and may not represent prices of actual transactions.
| 2013 | High | Low | ||||||
First Quarter | $ | 2.23 | $ | 1.43 | ||||
Second Quarter | $ | 2.01 | $ | 1.60 | ||||
Third Quarter | $ | 2.20 | $ | 1.47 | ||||
Fourth Quarter | $ | 3.24 | $ | 1.57 | ||||
2012 | ||||||||
First Quarter | $ | 4.90 | $ | 2.50 | ||||
Second Quarter | $ | 4.55 | $ | 3.49 | ||||
Third Quarter | $ | 4.51 | $ | 2.48 | ||||
Fourth Quarter | $ | 2.84 | $ | 1.27 | ||||
Stockholders of Record and Dividends
As of March 3, 2014, we had 100,579,438 shares of common stock outstanding held by 105 stockholders of record. We have never declared or paid cash dividends on our capital stock. We currently intend to retain any future earnings to finance the growth and development of our business and therefore, do not anticipate paying any cash dividends in the foreseeable future. In addition, the Biopharma Financing Agreement prohibits payment of dividends unless we have cash and cash equivalents in excess of $50 million after such payment.
Sale of Unregistered Securities
None.
Repurchases of Securities
None.
Market Performance Graph
The graph and the accompanying text below is not "soliciting material," is not deemed filed with the SEC and is not to be incorporated by reference in any filings by us under the Securities Act or the Exchange Act, whether made before or after the date hereof and irrespective of any general incorporation language in such filing.
The rules of the SEC require that we include a line-graph comparing cumulative stockholder returns on our common stock with the NASDAQ Composite Index (which tracks the aggregate price performance of equity securities of companies traded on NASDAQ) and either a published industry or line-of-business standard index or an index of peer companies selected by us. We have elected to use the NASDAQ Biotechnology Index (consisting of a group of 120 companies in the biotechnology sector, including us) for purposes of the performance comparison that appears below.
47
The graph shows the cumulative total stockholder return assuming the investment of $100.00 and the reinvestment of dividends and is based on the returns of the component companies weighted according to their market capitalizations as of the end of the period for which returns are indicated. No dividends have been declared on our common stock.
The stockholder return shown on the graph below is not necessarily indicative of future performance, and we do not make or endorse any predictions as to future stockholder returns.
COMPARISON OF 5 YEAR CUMULATIVE TOTAL RETURN* AMONG
CORCEPT THERAPEUTICS, THE NASDAQ STOCK MARKET (U.S.) INDEX
THE NASDAQ US BENCHMARK TOTAL RETURN (TR) INDEX
AND THE NASDAQ BIOTECHNOLOGY INDEX
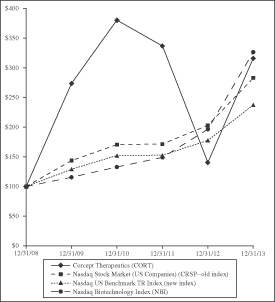
* $100 invested on December 31, 2008 including reinvestment of dividends. Fiscal year ended December 31.
As a result of a change in the total return data made available to us through our vendor provider, our performance graphs going forward will be using a comparable index provided by NASDAQ OMX Global Indexes. Please note, information for the NASDAQ US (CRSP) index will not be provided in the future, as December 31, 2013 is last day data is available.
48
ITEM 6. SELECTED FINANCIAL DATA
SELECTED FINANCIAL DATA
(in thousands, except per share data)
The selected financial data set forth below are derived from our financial statements. The statement of operations data for the years ended December 31, 2013, 2012, and 2011 and the balance sheet data as of December 31, 2013 and 2012 are derived from our audited financial statements included in this Annual Report on Form 10-K (Form 10-K). The statements of operations data for the years ended December 31, 2010 and 2009, and the balance sheet data as of December 31, 2011, 2010 and 2009 have been derived from our audited financial statements, which are not included in this Form 10-K. Our historical results are not necessarily indicative of our results to be expected for 2014 or for any future period The selected financial data set forth below should be read in conjunction with our financial statements, the related notes and "Management's Discussion and Analysis of Financial Condition and Results of Operations" included elsewhere in this Form 10-K.
| Year Ended December 31, | ||||||||||||||||||||
| 2013 | 2012 | 2011 | 2010 | 2009 | ||||||||||||||||
| (In thousands, except per share data) | ||||||||||||||||||||
Statement of Operations Data: | ||||||||||||||||||||
Revenues: | ||||||||||||||||||||
Product sales, net | $ | 10,357 | $ | 3,307 | $ | - | $ | - | $ | - | ||||||||||
Collaboration revenue | - | - | - | - | 29 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Total revenues | 10,357 | 3,307 | - | - | 29 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Operating expenses: | ||||||||||||||||||||
Cost of sales | 143 | 91 | - | - | - | |||||||||||||||
Research and development* | 20,470 | 14,074 | 21,001 | 18,949 | 14,402 | |||||||||||||||
Selling, general and administrative* | 31,240 | 25,414 | 11,331 | 8,488 | 5,877 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Total operating expenses | 51,853 | 39,579 | 32,332 | 27,437 | 20,279 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Loss from operations | (41,496) | (36,272) | (32,332) | (27,437) | (20,250) | |||||||||||||||
Non-operating income (expense), net* | (4,515) | (1,776) | (22) | 1,471 | 84 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Net loss | $ | (46,011) | $ | (38,048) | $ | (32,354) | $ | (25,966) | $ | (20,166) | ||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Net loss per share: | ||||||||||||||||||||
Basic and diluted | $ | (0.46) | $ | (0.41) | $ | (0.39) | $ | (0.38) | $ | (0.38) | ||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Weighted average shares – basic and diluted | 99,819 | 93,015 | 83,309 | 68,336 | 52,443 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
* Includes significant non-cash expenses, of the following: | ||||||||||||||||||||
Stock-based compensation | ||||||||||||||||||||
Research and development | $ | 618 | $ | 546 | $ | 547 | $ | 220 | $ | 263 | ||||||||||
Selling, general and administrative | 4,578 | 4,764 | 2,888 | 1,896 | 1,552 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Total stock-based compensation | 5,196 | 5,310 | 3,435 | 2,116 | 1,815 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Non-operating expense related to accretion of interest on long-term obligation | 4,410 | 1,680 | - | - | - | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Total significant non-cash expenses | $ | 9,606 | $ | 6,990 | $ | 3,435 | $ | 2,116 | $ | 1,815 | ||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
| As of December 31, | ||||||||||||||||||||
| 2013 | 2012 | 2011 | 2010 | 2009 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
Balance Sheet Data: | ||||||||||||||||||||
Cash, cash equivalents and investments | $ | 54,877 | $ | 93,032 | $ | 39,635 | $ | 24,578 | $ | 23,867 | ||||||||||
Working capital | 45,573 | 86,703 | 34,749 | 21,136 | 22,001 | |||||||||||||||
Total assets | 63,077 | 99,166 | 39,833 | 25,104 | 24,511 | |||||||||||||||
Long-term obligation – current portion | 5,743 | 2,650 | - | - | - | |||||||||||||||
Long-term obligation, net of current portion | 29,322 | 29,030 | - | - | - | |||||||||||||||
Total stockholders' equity | 21,017 | 61,777 | 34,807 | 21,244 | 22,092 | |||||||||||||||
49
ITEM 7. | MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
Forward-Looking Statements
This Management Discussion should be read in conjunction with the financial statements and notes thereto appearing elsewhere in this report. We make statements in this section that are forward-looking statements within the meaning of the federal securities laws. For a complete discussion of such forward-looking statements and the potential risks and uncertainties that may impact upon their accuracy, see "Forward-Looking Statements" included in "Risk Factors" in Part I, Item 1A of this Form 10-K and the "Overview" and "Liquidity and Capital Resources" sections of this Management's Discussion and Analysis of Financial Condition and Results of Operations.
Overview
We are a pharmaceutical company engaged in the discovery, development and commercialization of drugs for the treatment of severe metabolic, psychiatric and oncologic disorders. Our focus is on disorders associated with the steroid hormone cortisol. Elevated levels and abnormal release patterns of cortisol have been implicated in a broad range of human disorders.
Since our inception in 1998, we have been developing mifepristone, a potent, competitive glucocorticoid receptor II (GR-II) antagonist. In February 2012, the FDA approved Korlym ® (mifepristone) 300 mg Tablets in the United States as a once-daily oral medication for treatment of hyperglycemia secondary to hypercortisolism in adult patients with endogenous Cushing's syndrome who have type 2 diabetes mellitus or glucose intolerance and have failed surgery or are not candidates for surgery. We first made the drug available to patients in the United States in April 2012. We have an ongoing Phase 3 study of mifepristone, the active ingredient in Korlym, for treatment of the psychotic features of psychotic depression. We have also begun a Phase 1 safety and efficacy study of mifepristone in combination with chemotherapy in the treatment of triple-negative breast cancer – a form of cancer with a particularly poor prognosis. We have discovered and patented three series of selective GR-II antagonists that, like Korlym, competitively block GR-II but do not bind to the progesterone receptor and thus do not interfere with pregnancy.
Unless otherwise stated, all references in this document to "we," "us," "our," "Corcept," the "Company," "our company" and similar designations refer to Corcept Therapeutics Incorporated.
Cushing's Syndrome. Cushing's syndrome is a disorder caused by prolonged exposure of the body's tissues to high levels of the hormone cortisol. Sometimes called "hypercortisolism," it is uncommon and most often affects adults aged 20 to 50. An estimated 10 to 15 of every one million people are newly diagnosed with this syndrome each year, resulting in approximately 3,000 new patients and an estimated prevalence of 20,000 patients with Cushing's syndrome in the United States.
The FDA approval of Korlym allows us to market Korlym in the United States for its approved indication. Since Korlym's approval in February 2012, we have been carrying out our commercialization plans, including deploying medical science liaisons (MSLs) and sales representatives. We have also developed digital marketing capabilities and patient assistance programs to support physicians and patients. Korlym first became available to patients in April 2012.
We have Orphan Drug Designations for Korlym from the FDA for the approved indication and from the European Commission for the treatment of endogenous Cushing's syndrome. Orphan Drug Designation in the United States is a special status granted by the FDA to encourage the development of treatments for diseases or conditions that affect fewer than 200,000 patients in the United States. Drugs that receive Orphan Drug Designation obtain seven years of marketing exclusivity for the approved indication from the date of drug approval, as well as tax credits for clinical trial costs, marketing application filing fee waivers and assistance
50
from the FDA in the drug development process. Benefits of Orphan Drug Designation in the EU are similar to those in the United States, but include ten years of marketing exclusivity for the approved indication in all 28 member states, free scientific advice during drug development, access to a centralized review process and a reduction or complete waiver of fees levied by the European Medicines Agency (EMA). The EMA has accepted our plan to study the use of Korlym in children with Cushing's syndrome. We expect that the completion of this study will extend our period of marketing exclusivity by two years in the EU, provided our orphan protection is still in place at that time. We submitted our Marketing Authorization Application request to the EMA in October 2013.
In May 2013, we entered into an agreement with IDIS Limited (IDIS) to distribute Korlym on a named-patient basis in all countries outside of the United States. A named-patient program provides access to drugs for a single patient or group of patients in countries where they are not commercially available. Products offered through such a program can be investigational or can be approved in one country but not the patient's home country. Regulations covering named-patient programs vary by country. IDIS's right to distribute Korlym in a particular country will terminate automatically upon Korlym's approval by regulatory authorities in that country and its availability there on a commercial basis. IDIS received approval from the Medicines Healthcare Products Regulatory Agency to distribute Korlym on a named-patient basis in the third quarter of 2013.
Psychotic Depression. We are also developing mifepristone, the active ingredient in Korlym, for the treatment of the psychotic features of psychotic depression under an exclusive patent license from Stanford University. The FDA has granted "fast track" status to evaluate the safety and efficacy of mifepristone for the treatment of the psychotic features of psychotic depression.
In March 2008, we began enrollment in Study 14, our ongoing Phase 3 trial in psychotic depression. The protocol for this trial incorporates what we have learned from our three previously completed Phase 3 trials. It attempts to address the established relationship between increased drug plasma levels and clinical response and attempts to decrease the random variability observed in the results of the psychometric instruments used to measure efficacy. In one of the previously completed Phase 3 trials, Study 06, we prospectively tested and confirmed that patients whose plasma levels rose above a predetermined threshold statistically separated from both those patients whose plasma levels were below the threshold and those patients who received placebo; this threshold was established from data produced in earlier studies.
As expected, the group of patients who took 1200 milligrams (mg) of mifepristone in Study 06 developed higher average drug plasma levels than did the groups of patients who received lower doses. Further, there was no discernible difference in the incidence of adverse events between patients who received placebo in Study 06 and those who received 300 mg, 600 mg or 1200 mg of mifepristone in that study. In August 2011, we published our analysis of these data in The Journal of Clinical Psychopharmacology . Based on this information, we are testing a mifepristone dose of 1200 mg once per day for seven days in Study 14.
In addition, we are using a third-party centralized rating service to independently evaluate the patients for entry into the study as well as to evaluate their level of response throughout their participation in the study. We believe the centralization of this process will improve the consistency of rating across clinical trial sites and reduce the background statistical noise that was observed in earlier studies and is endemic to psychopharmacologic studies. We believe that the change in dose, as well as the other modifications to the protocol described above, should allow us to demonstrate the efficacy of mifepristone in the treatment of the psychotic symptoms of psychotic depression.
We will perform an interim analysis of the first 226 patients and expect to have the results of that analysis in the second quarter of 2014. If the outcome is positive, we plan to stop the trial and submit an NDA by the end of 2014.
Triple-Negative Breast Cancer . In January 2014 we began a Phase 1 study of mifepristone in combination with the chemotherapy drug eribulin in the treatment of triple-negative breast cancer.
51
We plan to conduct our study in two phases. First, the recommended dose for the second phase of the study will be determined in up to 20 patients with metastatic breast cancer. In the subsequent expansion phase, twenty patients with GR-II-positive triple-negative breast cancer will be dosed to determine a preliminary estimate of efficacy. Mifepristone will be administered orally with food once daily and eribulin will be administered intravenously. We began enrolling patients in this study in February 2014 and expect to have initial results in the first half of 2015.
Antipsychotic-induced Weight Gain Mitigation. In 2005, we announced the results of studies in rats that demonstrated that mifepristone both reversed the weight gain associated with the ongoing use of olanzapine and mitigated the weight gain associated with the initiation of treatment with olanzapine (the active ingredient in Zyprexa ® ). The results from this study were published in the journal Brain Behavioral Research in early 2006. This study was paid for by Eli Lilly and Company (Eli Lilly).
During 2007, we announced positive results from our clinical proof-of-concept study in lean healthy male volunteers evaluating the ability of mifepristone to mitigate weight gain associated with the use of Zyprexa. The results showed a statistically significant reduction in weight gain in those subjects who took Zyprexa plus mifepristone compared to those who took Zyprexa plus placebo. Also, the addition of mifepristone to treatment with Zyprexa had a beneficial impact on secondary metabolic measures such as fasting insulin, triglycerides and abdominal fat, as indicated by waist circumference. Eli Lilly provided Zyprexa and financial support for this study, the results of which were published in the journal Advances in Therapy in 2009. In January 2009, we announced positive results from a similar proof-of-concept study evaluating the ability of mifepristone to mitigate weight gain associated with the use of Johnson & Johnson's Risperdal ® . This study confirmed and extended the earlier results seen with mifepristone and Zyprexa, demonstrating a statistically significant reduction in weight gain and in the secondary metabolic endpoints of fasting insulin, triglycerides and abdominal fat, as indicated by waist circumference. The results from the study of mifepristone and Risperdal were presented at several scientific conferences, including the American Diabetes Association meeting in June 2009, and were published in the journal Obesity in 2010.
The combination of Zyprexa or Risperdal and mifepristone is not approved for any indication. The purpose of these studies was to explore the hypothesis that GR-II antagonists, such as mifepristone and our next generation of selective GR-II antagonists, would mitigate weight gain associated with antipsychotic medications. The group of medications known as second generation antipsychotic medication, including Zyprexa, Risperdal, Clozaril ® and Seroquel ® , are widely used to treat schizophrenia and bipolar disorder. All medications in this group are associated with treatment-emergent weight gain of varying degrees and carry a warning in their labels relating to treatment-emergent hyperglycemia and diabetes mellitus.
Selective GR-II Receptor Antagonists. In 2003, we initiated a discovery research program to identify and patent selective GR-II antagonists with the intent of developing a pipeline of products for proprietary use. Three distinct series of GR-II antagonists were identified. These compounds, like mifepristone, competitively antagonize the cortisol receptor (GR-II) but do not block the PR (progesterone), ER (estrogen), AR (androgen) or GR-I (mineralocorticoid) receptors. Both the United States Patent & Trademark Office (USPTO) and the European Patent Office (EPO) have issued composition of matter patents to us in each of the three series. One additional composition of matter patent application is pending.
Several of our new compounds have demonstrated positive results in animal or in vitro models for the prevention and reversal of alcohol dependence; amyotrophic lateral sclerosis (Lou Gehrig's disease); Alzheimer's disease; anti-psychotic-induced weight gain; breast, ovarian and prostate cancer in combination with a chemotherapeutic agent; electroconvulsive shock-induced retrograde amnesia; the metabolic syndrome; muscular dystrophy; obesity; ovarian and prostate cancer; prevention of glucocorticoid-induced neurological damage in premature infants; and stress disorders. We intend to continue our discovery research program with the goal of identifying new selective GR-II antagonists, to manufacture and conduct pre-clinical development of one or more of these compounds and to study the most promising of them in humans. We plan to advance one or more compounds to the clinic in 2014.
52
General
Our activities to date have included:
| • | product development, including drug formulation and manufacturing, as well as designing, funding and overseeing clinical trials and conducting non-human clinical investigatory activities, such as toxicological testing; |
| • | commercialization of Korlym, including hiring and training medical science liaisons and sales representatives, retention and management of third-party distribution partners, establishment of third-party coverage and reimbursement and patient assistance programs and marketing activities; |
| • | regulatory affairs; |
| • | discovery research; and |
| • | intellectual property prosecution and expansion. |
Historically, we have financed our operations and internal growth primarily through private placements of our preferred and common stock, the public sale of common stock and through our Financing Agreement with Biopharma, rather than through collaborative or partnership agreements.
As of December 31, 2013, we had an accumulated deficit of $292.6 million. Our historical operating losses have resulted principally from our research and development activities, including clinical trial activities for mifepristone, discovery research, non-clinical activities such as toxicology and carcinogenicity studies, manufacturing and regulatory activities, as well as selling, general and administrative expenses, including expenses related to the commercial launch of Korlym. We may continue to incur net losses over at least the next few years as we continue our mifepristone and selective GR-II antagonist discovery and clinical development programs, apply for regulatory approvals, acquire and / or develop treatments in other therapeutic areas, establish sales and marketing capabilities and expand our operations.
Our business is subject to significant risks, including the risks inherent in our research and development efforts, the results of our mifepristone and other clinical trials, uncertainties associated with securing financing, uncertainties associated with obtaining and enforcing patents, our investment in manufacturing set-up, the management of our supply chain, the lengthy and expensive regulatory approval process and competition from other products. Our ability to successfully generate revenues in the foreseeable future is dependent upon our ability, alone or with others, to finance our operations and develop, obtain regulatory approval for, manufacture and market our products.
Results of Operations
Net Product Sales – Net product sales includes product revenue resulting from sales to our customers, reduced by (1) trade allowances, such as discounts for prompt payment and distributor fees, (2) estimated government rebates and chargebacks, (3) reserves for expected product returns and 4) estimated costs of our patient assistance program.
In April 2012, we made Korlym commercially available in the United States through a specialty pharmacy that sells to individual patients and a specialty distributor that sells to hospital pharmacies. For the year ended December 31, 2013, we recorded $10.4 million in net product sales, as compared to $3.3 million for the year ended December 31, 2012. To calculate net product sales, we deducted from gross sales estimates of prompt-pay discounts (which we ceased to incur with respect to our specialty pharmacy customer beginning in the third quarter of 2013), distribution service fees, rebates and chargebacks owed to government payors and patient assistance program costs, which amounts are not material for any of the periods presented.
We provide cash donations to a non-profit third party organization that supports patients who meet certain eligibility requirements with financial assistance for the treatment of Cushing's syndrome, which treatment may include Korlym. We do not include as net product revenues sales of Korlym tablets funded through this source.
53
Cost of sales - Cost of sales includes the cost to manufacture Korlym (which includes material, third-party manufacturing costs and indirect personnel and other overhead costs) based on units sold in the current period, as well as the cost of stability testing and distribution. We began capitalizing Korlym production costs as inventory following approval by the FDA to market Korlym in February 2012. Prior to Korlym's approval, we expensed all costs related to the manufacturing of product (including stability costs and manufacturing overhead) as incurred, classifying these costs as research and development expense. A portion of the product manufactured prior to FDA approval is available for us to use commercially.
Cost of sales was $143,000 for the year ended December 31, 2013, as compared to $91,000 for the year ended December 31, 2012, which equals 1.4 percent and 2.8 percent of net product sales for the respective periods. The majority of these costs related to stability testing and distribution costs. The amount and timing of stability testing varies from period to period as determined by FDA regulations and our production schedule and is not a fixed percentage of our sales volumes. In addition, the cost of manufacturing Korlym reflected in our cost of sales through December 31, 2013, and for some period thereafter, will not reflect the full cost of production because we have previously expensed the majority of the raw materials, labor and overhead costs incurred to produce the product sold during these periods. We expect that our cost of sales of Korlym as a percentage of net product sales will fluctuate from period to period during 2014 as product manufactured prior to FDA approval is consumed.
Research and development expenses – Research and development expenses include (1) personnel costs related to our development activities, including facilities costs and non-cash stock-based compensation, (2) costs of discovery research, (3) costs associated with IND-enabling activities and pre-clinical studies, (4) costs of clinical trials, including trial preparation, enrollment, site monitoring and data management and analysis expenses, (5) regulatory costs, (6) costs of manufacturing development, including the development and activities to qualify a tablet manufacturing site, (7) costs of manufacture and / or acquisition of clinical trial materials and material used in registration and validation batches included in regulatory submissions and (8) other costs associated with the preparation and prosecution of the regulatory submissions related to Korlym or other product candidates.
Research and development expenses increased 45.4 percent to $20.5 million for the year ended December 31, 2013 from $14.1 million in 2012.
During the year ended December 31, 2013, as compared to 2012, there was a net increase of $983,000 in staffing and consultancy costs. The year ended December 31, 2012 included cash bonuses awarded on FDA approval of Korlym to employees working in research and development in the amount of $474,000. After adjusting for the effect of these bonuses, there was a net increase of $1.5 million in staffing and consulting costs between the year ended December 31, 2013 and 2012, $73,000 of which represented increases in non-cash stock-based compensation costs. Increases in staffing and consultancy costs between periods was due to the increased psychotic depression study activities, preparations for a submission to the EMA for approval of mifepristone for Cushing's syndrome in Europe and other research and development activities.
Clinical trial costs related to our Phase 3 product candidate reflected net increases of $4.6 million during the year ended December 31, as compared to 2012. During the year ended December 31, 2013 as compared to 2012, there were increases of $6.2 million related to our Phase 3 study with mifepristone for the treatment of psychotic depression, which were partially offset by decreases of $182,000 related to the clinical trials with Korlym in the treatment of Cushing's syndrome and $1.4 million related to drug-drug interaction and other NDA-supportive studies with Korlym that occurred in the prior year.
During the year ended December 31, 2013, as compared to 2012, there were also net increases of $701,000 related to research and development of our new GR-II antagonists, $220,000 in academic research grants to further basic scientific research regarding GR-II antagonism and $413,000 related to research regarding other products.
54
During the year ended December 31, 2013 as compared to 2012, there was a decrease of $784,000 related to our manufacturing costs of Korlym due to the completion of certain manufacturing process development efforts.
Research and development expenses decreased 33 percent to $14.1 million for the year ended December 31, 2012 from $21.0 million for 2011.
During the year ended December 31, 2012 as compared to 2011, there was an increase of $991,000 in staffing costs, which includes bonuses paid on FDA approval of Korlym in the amount of $474,000, and an increase of $164,000 for stock-based compensation expenses related to employees working in research and development functions. During the year ended December 31, 2012, as compared to 2011, there were decreases in consultancy costs of $2.8 million due primarily to the additional resources required during 2011 for the preparation, submission and prosecution of the NDA for Korlym in the treatment of Cushing's syndrome, which was submitted in April 2011 and filed by the FDA in June 2011. For the year ended December 31, 2012, non-cash stock-based compensation expense related to consultant options decreased $165,000 as compared to 2011 due primarily to the inclusion in 2011 of costs related to a stock option award to a consultant that vested in its entirety on the filing of the NDA by the FDA in June 2011.
Korlym manufacturing costs categorized as research and development expense decreased $3.8 million during the year ended December 31, 2012 as compared to 2011, due primarily to capitalizing to inventory the costs of Korlym's active pharmaceutical ingredient and the manufacture of Korlym tablets for commercial sale following the date of FDA approval.
Clinical trial costs reflected a net decrease of $1.6 million during the year ended December 31, 2012, as compared to 2011. During the year ended December 31, 2012, as compared to 2011, there were decreases of $1.2 million related to clinical studies with CORT 108297 and $917,000 related to the clinical trials with Korlym in the treatment of Cushing's syndrome. These decreases were partially offset by increases during year ended December 31, 2012, as compared to 2011, of $487,000 related to the psychotic depression study.
In addition, costs related to financial support for medical conferences and seminars in support of our Cushing's syndrome program decreased $337,000 for the year ended December 31, 2012, as compared to 2011, because, subsequent to product approval, the nature of our activities at medical meetings has changed, and now such costs relate to marketing activities that are classified as a component of selling, general and administrative expenses. Costs relating to IND-enabling activities and research efforts regarding our new GR-II antagonists increased $363,000 during the year ended December 31, 2012, as compared to 2011.
Below is a summary of our research and development expenses by major project:
| Year Ended | ||||||||||||
| December 31, | ||||||||||||
Project | 2013 | 2012 | 2011 | |||||||||
| (in thousands) | ||||||||||||
Development programs: | ||||||||||||
Psychotic depression | $ | 9,755 | $ | 2,613 | $ | 1,779 | ||||||
Cushing's syndrome | 2,740 | 4,093 | 10,925 | |||||||||
Cancer | 301 | - | - | |||||||||
Selective GR-II antagonists | 5,250 | 4,249 | 4,546 | |||||||||
Unallocated activities, including NDA supportive studies and manufacturing, regulatory and pre-clinical activities | 1,806 | 2,573 | 3,204 | |||||||||
Stock-based compensation | 618 | 546 | 547 | |||||||||
|
|
|
|
|
| |||||||
Total research and development expense | $ | 20,470 | $ | 14,074 | $ | 21,001 | ||||||
|
|
|
|
|
| |||||||
We expect research and development expenditures in 2014 to be approximately the same as they were in 2013. Research and development expenses in 2015 and beyond will depend on our strategic priorities. See also, "Liquidity and Capital Resources".
55
Many factors can affect the cost and timing of our trials including inconclusive results requiring more clinical trials, slow patient enrollment, adverse side effects in study patients, insufficient supplies of medicine for our clinical trials and real or perceived lack of effectiveness or safety of the drug in our trials. The cost and timing of development of our selective GR-II antagonists will depend on the success of our efforts and any difficulties that we may encounter. In addition, the development of all of our product candidates will be subject to extensive governmental regulation. These factors make it difficult for us to predict the timing and costs of the further development and approval of our product candidates.
Selling, general and administrative expenses – Selling, general and administrative expenses include (1) internal personnel, a contracted sales force and other consultancy costs related to administrative and commercialization activities, including facilities costs and non-cash stock-based compensation, (2) expenses of third-party vendors that we engage to execute our commercial plans related to Korlym, including marketing and promotion, strategy development, market research and analytics, reimbursement support services, pharmacovigilance, distribution of marketing materials and other logistical needs, (3) medical educational grants and donations and (4) legal, accounting and other professional fees.
For the year ended December 31, 2013, selling, general and administrative expenses increased 22.9 percent to $31.2 million from $25.4 million for 2012.
During the year ended December 31, 2013, as compared to 2012, staffing and consultancy costs reflected a net decrease of $312,000. The year ended December 31, 2012 included $1.6 million related to cash bonuses awarded to employees and officers working in selling, general and administrative functions and $1.3 million of non-cash stock-based compensation related to awards that vested in February 2012 upon the FDA approval of Korlym. After adjusting for these items, there was a $2.6 million increase in staffing and consultancy costs during 2013 as compared to 2012, due primarily to additional resources necessary to commercialize Korlym, of which $1.1 million represented increases in non-cash stock-based compensation costs.
During the year ended December 31, 2013, as compared to 2012, there were increases in other professional services costs related to commercialization activities of $3.6 million, which included $2.2 million related to our contracted sales force and $944,000 related to market research and marketing materials. In addition, there were increases of $2.6 million between the respective years in other commercial and non-commercial support costs, such as education, training and conference costs, medical education grants and donations, facilities and technology costs, travel and fleet vehicle costs, legal, insurance and other service fees.
For the year ended December 31, 2012, selling, general and administrative expenses increased to $25.4 million from $11.3 million for 2011.
During the year ended December 31, 2012 as compared to 2011, staffing and consultancy costs increased $6.9 million due primarily to additional resources necessary to commercialize Korlym. The increase for the year ended December 31, 2012 included the $1.6 million in cash bonuses awarded in the first quarter of 2012 to employees working in selling, general and administrative functions in recognition of the FDA's approval of Korlym, $1.3 million of non-cash stock-based compensation costs related to performance-based stock option awards to officers that vested in February 2012 upon the FDA approval of Korlym and $600,000 of increases related to other stock options to directors, officers and employees working in selling, general and administrative functions.
In addition, other professional services costs related to commercialization activities and other corporate matters increased $5.8 million during the year ended December 31, 2012 as compared to 2011. These costs reflect increased vendor activities after FDA approval, pricing strategy and market analysis, patient registry and reimbursement programs, focus groups, internet marketing and communications.
There were also cost increases during the year ended December 31, 2012 as compared to 2011 related to the infrastructure necessary to support the commercialization of Korlym including (a) $538,000 related to travel,
56
(b) 386,000 related to the expansion of facilities and information technology support, (c) $147,000 related to employee education primarily due to the training of the new medical science liaisons and (d) $95,000 related to donations.
Selling, general and administrative expenses included stock-based compensation expense related to option grants to individuals performing these functions of $4.6 million, $4.8 million and $2.9 million for the years ended December 31, 2013, 2012 and 2011, respectively.
We expect that selling, general and administrative expenses will be higher during 2014 as compared to 2013 in regard to activities directly associated with product commercialization. The level of selling, general and administrative activities and related expenses in 2015 and future years will be largely dependent on our assessment of the staff and other services necessary to support product commercialization and our continued clinical development activities and the availability of additional funds. See also, "Liquidity and Capital Resources."
Interest and other expense – Interest and other expense for the year ended December 31, 2013 was $4.5 million as compared to $1.8 million for the year ended December 31, 2012 and $25,000 for the year ended December 31, 2011. These increases were primarily due to the inclusion in 2013 and 2012 of interest expense related to our Financing Agreement with Biopharma, which was entered into in August 2012. Interest expense for 2014 and future years will decrease from the levels of 2013 due to quarterly payments against the outstanding obligation.
Non-GAAP Financial Measures
Our financial statements and footnotes thereto are prepared in accordance with U.S. Generally Accepted Accounting Principles (GAAP) and are included in Part IV, Item 15 of this Annual Report on Form 10-K. To supplement our financial results presented on a GAAP basis, we use non-GAAP measures of net loss that exclude significant non-cash expenses related to stock-based compensation expense and the accretion of interest expense under our Financing Agreement with Biopharma. We use this non-GAAP measure of net loss to manage our business and believe that it may help investors better evaluate our past financial performance and potential future results. Non-GAAP measures should not be considered in isolation or as a substitute for comparable GAAP accounting and investors should read them in conjunction with our financial statements and notes thereto prepared in accordance with GAAP. The non-GAAP measure of net loss we use may be different from, and not directly comparable to, similarly titled measures used by other companies.
On the following page is a table that reflects the reconciliation of GAAP net loss to non-GAAP net loss for the periods presented.
57
| Year Ended | ||||||||||||
| December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| (in thousands, except per share data) | ||||||||||||
GAAP net loss | $ | 46,011 | $ | 38,048 | $ | 32,354 | ||||||
Significant non-cash expenses: | ||||||||||||
Stock-based compensation | ||||||||||||
Research and development | 618 | 546 | 547 | |||||||||
Selling, general and administrative | 4,578 | 4,764 | 2,888 | |||||||||
|
|
|
|
|
| |||||||
Total stock-based compensation | 5,196 | 5,310 | 3,435 | |||||||||
Accretion of interest expense related to long-term obligation | 4,410 | 1,680 | - | |||||||||
|
|
|
|
|
| |||||||
Non-GAAP net loss, as adjusted for significant non-cash expenses | $ | 36,405 | $ | 31,058 | $ | 28,919 | ||||||
|
|
|
|
|
| |||||||
GAAP basic and diluted net loss per share | $ | (0.46) | $ | (0.41) | $ | (0.39) | ||||||
|
|
|
|
|
| |||||||
Non-GAAP basic and diluted net loss per share, as adjusted for significant non-cash expenses | $ | (0.36) | $ | (0.33) | $ | (0.35) | ||||||
|
|
|
|
|
| |||||||
Shares used in computing basic and diluted net loss per share | 99,819 | 93,015 | 83,309 | |||||||||
|
|
|
|
|
| |||||||
Liquidity and Capital Resources
We have incurred operating losses since inception, and at December 31, 2013, we had an accumulated deficit of $292.6 million. Since our inception, we have relied primarily on the proceeds from public and private sales of our equity securities and our Financing Agreement with Biopharma to fund our operations.
At December 31, 2013, we had cash and cash equivalents of $54.9 million, compared to $93.0 million at December 31, 2012. Net cash used in operating activities for the years ended December 31, 2013, 2012 and 2011 was $37.1 million, $36.0 million and $27.4 million, respectively. We used cash in each period primarily for the commercialization of Korlym and for research and development activities. In addition, we made payments under the Biopharma Financing Agreement of $1.0 million during 2013.
We expect net cash used during 2014 will be lower than in 2013 as we expect that the cash expected to be generated from increasing sales of Korlym will be greater than expenditures related to the continued commercialization of Korlym, the continuation of our Phase 3 clinical trial of mifepristone for the treatment of psychotic depression, the initiation of our Phase 1 trial of mifepristone for triple-negative breast cancer, the continued development of our selective GR-II antagonists and payments under our Biopharma Financing Agreement.
Our funding requirements for operating activities may increase in 2015 and beyond as the cost of continuing our development programs for Cushing's syndrome, psychotic depression, oncology and our selective GR-II antagonists, commercial activities, and selling, general and administrative expenses may be only partially offset by revenues from sales of Korlym.
As discussed below under the caption Contractual Obligations and Commercial Commitments, we are required to make aggregate payments under the Biopharma Financing Agreement of $45.0 million, of which $1.0 million was paid during 2013, with an additional payment of $995,000 in February 2014. Future individual payment amounts will be variable.
58
We may choose to raise additional funds to finance our strategic priorities. We cannot be certain that additional funding will be available on acceptable terms or at all. Further, any additional equity financing may be dilutive to stockholders, and any debt financing, if available, may involve restrictive covenants. If we obtain funds through collaborations with others, these arrangements may be on unfavorable terms or may require us to relinquish certain rights to our technologies or product candidates that we would otherwise seek to develop on our own.
While we monitor the cash balance in our checking account and transfer the funds in only as needed, these cash balances and our money market fund could be influenced if the underlying financial institution were to fail or were subject to other adverse conditions in the financial markets. To date, we have experienced no loss or lack of access to cash in our checking account or money market fund.
Contractual Obligations and Commercial Commitments
The following table presents our estimates of obligations under contractual agreements as of December 31, 2013.
Contractual Obligations | Total | Less than 1 year | 1-3 Years | 3-5 Years | More than 5 Years | |||||||||||||||
| (in thousands) | ||||||||||||||||||||
Long-term obligation (1) | $ | 43,975 | ||||||||||||||||||
|
| |||||||||||||||||||
Other contractual obligations: | ||||||||||||||||||||
Research and development studies (2 to 4) | $ | 5,703 | $ | 4,053 | $ | 1,650 | $ | - | $ | - | ||||||||||
Operating lease (5) | 428 | 428 | - | - | - | |||||||||||||||
Minimum royalty payments (6) | 65 | 130 | 130 | 65 per year | ||||||||||||||||
|
|
|
|
|
|
|
| |||||||||||||
Total other contractual obligations | $ | 4,546 | $ | 1,780 | $ | 130 | $ | 65 per year | ||||||||||||
|
|
|
|
|
|
|
| |||||||||||||
| (1) | As discussed above under the caption "Liquidity and Capital Resources", in August 2012, we entered into a Financing Agreement with Biopharma under which we received $30.0 million from Biopharma. In consideration of the $30.0 million payment, we are obligated to make payments to Biopharma totaling $45.0 million, of which $1.0 million was paid during 2013. The remaining payment obligations will be calculated as follows: |
| • | 20 percent of our net product sales of Covered Products, subject to quarterly payment caps of $3.0 million during 2014 and $3.75 million during 2015. There is no quarterly cap on payments with respect to net product sales in 2016 and later. Payments are due within 30 days of quarter-end for the first, second and third calendar quarters and within 45 days of year-end. |
| • | 20 percent of payments received for upfront, milestone or other contingent fees under co-promotion and out-license agreements for Covered Products (without application of quarterly caps). |
| • | The percentage used to calculate our payments to Biopharma would increase to 50 percent and any applicable payment caps would lapse if we (i) fail to provide Biopharma with certain information regarding our promotion and sales of Covered Products, (ii) do not devote a commercially reasonable amount of resources to the promotion and marketing of the Covered Products or (iii) violate the indebtedness covenant by incurring indebtedness greater than the sum of earnings before interest, taxes, depreciation and amortization, including such items as non-cash stock-based compensation, (EBITDA) for the four calendar quarters preceding such incurrence and, in each case, fail to cure within the applicable cure period. |
| • | Upon the occurrence of a Corcept change of control transaction or the licensing of Korlym to a third-party for promotion and sale in the United States, the entire $45 million, less any amounts already paid by us, would become due. |
Under the terms of the Financing Agreement, our payments are entirely variable, with no fixed minimums. The timing of our payments is determined by future sales and other receipts. If there are no net sales, upfront, milestone or other contingent payments in a period with respect to Covered Products, then no payment will be due for that period. During the year ended December 31, 2013, we made payments to Biopharma of $1.0 million with an additional payment in the amount of $995,000 in February 2014. Biopharma's right to receive payments will expire once it has received cumulative payments of $45 million.
| (2) | Amounts reflected for research and development studies exclude amounts included in accounts payable and accrued clinical costs reflected on the balance sheet as of December 31, 2013. |
59
| (3) | Through 2013, we entered into and amended agreements for services in connection with our ongoing Phase 3 trial to confirm the utility of mifepristone for the treatment of the psychotic features of psychotic depression. The total commitment under these agreements, including amendments through 2013, is now estimated to be $19.9 million over the course of the trial. We expensed $17.1 million of these costs through December 31, 2013, with the remainder to be incurred over the course of the trial. Under the master services agreements with these vendors, the project contracts may be terminated upon thirty to sixty days notice. If terminated early, we would be responsible for the costs incurred by the vendors through the effective date of termination plus cancellation charges as stipulated in the agreements. |
| (4) | In December 2013, we entered into an agreement with Ockham to assist in the management and conduct of a clinical trial evaluating mifepristone for treatment of triple-negative breast cancer. The total commitment under this agreement is $2.9 million, but the actual amount to be paid is dependent on actual services provided under this agreement. Approximately $18,000 of the costs under this agreement were incurred during 2013, with the remainder to be incurred over the course of the trial. |
| (5) | In June 2013, we exercised our option to extend the lease for our office space through December 2014. At December 31, 2013, the remaining minimum rental payments under this operating lease were $428,000. |
| (6) | Under our cancellable license agreements with Stanford University and the University of Chicago, we are obligated to make nonrefundable minimum royalty payments aggregating $65,000 annually for as long as we maintain these licenses; however, a portion of these payments are creditable against future royalties. |
We also have other contractual payment obligations and purchase commitments, the timing of which are contingent on future events. In November 2006, we entered into an agreement with PCAS for the manufacture of mifepristone, the API in Korlym, for our development and commercial needs which has been extended through March 31, 2014. We are currently in discussions for a new contract to continue the relationship thereafter. The current agreement calls for us to purchase from PCAS at least 75 percent of our requirements through the expiration of the agreement. If PCAS is unable to manufacture the product for a consecutive six-month period, we have the right to terminate the agreement without penalty.
Net Operating Loss Carryforwards
At December 31, 2013 we had net operating loss carryforwards available to offset any future taxable income that we may generate for federal income tax purposes of $145.4 million, which expire in the years 2019 through 2033, and California net operating loss carryforwards of $111.7 million, which expire in the years 2014 through 2033. We also had federal and California research and development tax credits of $19.7 million and $2.3 million, respectively. The federal research credits will expire in the years 2019 through 2033 and the California research credits have no expiration date. Our deferred tax assets have been offset by a full valuation allowance as the realization of such assets is uncertain. Utilization of our net operating losses and tax credit carryforwards may be subject to substantial annual limitation due to the ownership change limitations provided by the Internal Revenue Code and similar state provisions. Such limitations could result in the expiration of the net operating losses and tax credit carryforwards before utilization.
Off-Balance Sheet Arrangements
None.
Critical Accounting Policies and Estimates
Our financial statements have been prepared in accordance with U.S. generally accepted accounting principles. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
60
Net Product Sales
From our initial launch in April 2012 through June 30, 2013, we sold Korlym primarily to a specialty pharmacy and a specialty distributor, which subsequently resold Korlym to patients and healthcare providers. Korlym is not available in retail pharmacies. In July 2013, we began using a specialty pharmacy that operates on a consignment basis, without carrying any Korlym inventory, resulting in product sales being made directly to patients. (See discussion in forth in Part IV – Item 15(1) - Financial Statements, Notes to Financial Statements, Note 2, Significant Agreements – Commercial Agreements .)
We recognize product revenues from sales of Korlym upon delivery to our customers as long as (i) there is persuasive evidence that an arrangement exists between ourselves and the customer, (ii) collectability is reasonably assured and (iii) the price is fixed or determinable. Prior authorization or confirmation of coverage level by the patient's private insurance plan or government payor is a prerequisite to the shipment of product to a patient. In order to conclude that the price is fixed or determinable, we must be able to (i) calculate gross product revenues from the sales to our customers and (ii) reasonably estimate net product revenues.
We provide cash donations to a non-profit third party organization that supports patients who meet certain eligibility requirements with financial assistance for the treatment of Cushing's syndrome, which treatment may include Korlym. We do not include as net product revenues sales of Korlym tablets funded through this source.
We calculate gross product revenues based on the price that we charge our customers. We estimate our net product revenues by deducting from our gross product revenues (a) trade allowances, such as discounts for prompt payment and distributor fees, (b) estimated government rebates and chargebacks, (c) reserves for expected product returns and (d) estimated costs of patient assistance programs. We initially record estimates for these deductions at the time we recognize the gross revenue. We update our estimates on a recurring basis as new information becomes available.
Trade Allowances: We offer our specialty distributor customer a discount on Korlym sales for payment within 30 days. We also offer them a small discount for the provision of data services. We expect our customers to earn these discounts and accordingly deduct them in full from gross product revenues and trade receivables at the time we recognize such revenues.
Rebates and Chargebacks: We contract with Medicaid and other government agencies so that Korlym will be eligible for purchase by, or qualify for partial or full reimbursement from, such government programs. We estimate the rebates and chargebacks that we will be obligated to provide to government programs and deduct these estimated amounts from our gross product sales at the time the revenues are recognized. We base our estimates of these rebates and chargebacks upon (i) the discount amounts applicable to government-funded programs and (ii) information obtained from our vendors regarding the percentage of sales by our customers to patients who are covered by entities or programs that are eligible for such rebates and chargebacks.
Allowances for Patient Assistance Program: We provide financial assistance to eligible patients whose insurance policies require them to pay high deductibles and co-pays. We estimate the cost of assistance to be provided under this program by applying our actual experience regarding such assistance to our estimate of the percentage of our sales in the period that will be provided to patients covered by the program.
Sales Returns: Our specialty distribution customer has the right to return Korlym beginning six months before the labeled expiration date and ending 12 months after the labeled expiration date. This right of return is extended to our specialty distributor channel's hospital customers who generally have the right to return only unopened bottles. The expiration date for our Korlym product sold to date to the specialty distributor will not occur until November 2014. We estimate the amount of Korlym that we believe will be returned and deduct that estimated amount from gross revenue at the time we recognize such revenue. When estimating future returns, we analyze quantitative and qualitative information including, but not limited to, actual return rates, the amount of
61
product in the distribution channel, the expected shelf life of such product, current and projected product demand, the introduction of competing products that may erode demand, and broad economic and industry-wide indicators. If we cannot reasonably estimate product returns with respect to a particular sale, we defer recognition of revenue from that sale until we can make a reasonable estimate.
Inventory and Cost of Sales
We consider regulatory approval of product candidates to be uncertain, and product manufactured prior to regulatory approval may not be sold unless regulatory approval is obtained. We expense manufacturing costs for product candidates incurred prior to regulatory approval as research and development expenses as we incur them. When regulatory approval of a product is obtained, we begin capitalizing manufacturing costs related to the approved product into inventory, provided such product is produced by a facility the FDA has approved to manufacture Korlym for distribution as commercial product.
We value our inventories at the lower of cost or net realizable value. We determine the cost of inventory using the specific identification method, which approximates a first-in, first-out basis. We analyze our inventory levels quarterly and write down inventory that has become obsolete or has a cost basis in excess of its expected net realizable value, as well as any inventory quantities in excess of expected requirements. Any expired inventory is disposed of and the related costs are recognized as cost of sales.
Cost of sales includes the cost of product (the cost to manufacture Korlym, which includes material, third-party manufacturing costs and indirect personnel and other overhead costs) based on units for which revenue is recognized in the current period, as well as costs of stability testing, logistics and distribution of the product. We began capitalizing Korlym production costs as inventory following approval by the FDA in February 2012. Prior to receiving FDA approval for Korlym, we expensed all costs related to the manufacturing of the product (including stability costs and manufacturing overhead) as incurred; we classified these costs as research and development expense. A portion of the product manufactured prior to FDA approval is available for us to use commercially.
Inventory amounts that are not expected to be consumed within twelve months following the balance sheet date are classified as a noncurrent asset.
Accruals of Research and Development Costs
We recorded accruals for estimated costs of research, pre-clinical and clinical studies, and manufacturing development, which activities represent a significant component of our research and development expenses. We make significant judgments and estimates in determining the accrual balance in each reporting period. Accrued clinical trial costs are based on estimates of the work completed under the service agreements, milestones achieved, patient enrollment and past experience with similar contracts and service providers. Our estimate of the work completed, and associated costs to be accrued, includes our assessment of the information received from our third-party contract research organizations and the overall status of our clinical trial activities. In the past, we have not experienced any material deviations between accrued and actual clinical trial expenses. However, actual services performed, number of patients enrolled and the rate of patient enrollment may vary from our estimates, resulting in adjustments to clinical trial expense in future periods.
Stock-based compensation
Stock-based compensation arises from the granting of stock options to employees and directors, as well as to non-employees.
62
Employees and directors
Our accounting practices and the estimates and judgments that are considered in determining fair value in regard to stock option grants to employees and directors are as follows:
| • | We base the expected volatility of our common stock used in determining the fair value-based measurement of option grants to employees, officers and directors on a weighted-average combination of the volatility of our own stock price and that of a group of peer companies for those grants with expected terms longer than the period of time that we have been a public company. For stock options granted to employees with expected terms of less than the period of time that we have been a public company, the volatility is based on historical data of the price for our common stock for periods of time equivalent to the expected term of these grants. |
| • | For service-based awards, we recognize the expense over the requisite service period utilizing the straight-line amortization method. For options with performance-based vesting criteria, we recognize the expense at such time as there is a high degree of probability (i.e., greater than 70%) of achieving the required vesting criteria. |
| • | Because we have a limited base of employees and directors and have experienced minimal turnover, we apply a forfeiture rate of zero. When an employee terminates, we will record a change in accounting estimate that represents the difference between the expense recorded in the financial statements and the expense that would have been recorded based upon the rights to options that vested during the individual's service as an employee. |
As of December 31, 2013, we had $9.0 million of unrecognized compensation expense for employee and director options outstanding as of that date, which had a remaining weighted-average vesting period of 2.7 years.
Non-employees
All stock option grants to consultants vest solely based upon continuing service, with the exception of a performance-based award granted during 2010 for 50,000 shares, and an award granted in December 2012 for 10,000 shares. Stock-based compensation related to service-based option grants to non-employees is charged to expense on a straight line basis over the vesting period of the options, which approximates the period over which the related services are rendered, based on the fair value-based measurement of the options using the Black-Scholes option pricing model. The assumptions used in these calculations are similar to those used for the determination of fair value-based measurements for options granted to employees, with the exception that, for non-employee options, we are required to use the remaining contractual term as the life of the option and the fair value-based measurement related to unvested non-employee options is re-measured quarterly, based on the then current stock price as reflected on the NASDAQ Capital Market. For options with performance-based vesting criteria, we recognize expense based on the minimum number of shares that will vest over time as the criteria are met based on the Black-Scholes valuation of the vested shares.
Long-term obligation
The accounting for the Financing Agreement with Biopharma requires us to make certain estimates and assumptions, including the timing of royalty payments due to Biopharma, the expected rate of return to Biopharma, the split between current and long-term portions of the obligation, and the accretion of related interest expense. Korlym has only been marketed since April 2012 and the magnitude and timing of Korlym revenue is difficult to predict. Therefore, these estimates and assumptions are subject to significant variability and are likely to change as we gain experience marketing Korlym, which will result in changes in our classification of the current and long-term portions of the amounts payable pursuant to this financing agreement, as well as the internal rate of return paid to Biopharma and the accretion of interest expense related to this obligation. Actual payment amounts will be based on Korlym receipts over the term of the Financing Agreement but in no event will the total amount paid to Biopharma exceed $45.0 million.
63
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Quantitative and Qualitative Disclosures About Market Risk
Interest Rate Risk
The primary objective of our investment activities is to preserve principal while at the same time maximizing the income we receive from our investments without significantly increasing risk of loss. As of December 31, 2013, the fair value of our cash and cash equivalents was $54.9 million and consisted primarily of a money market fund maintained at a major U.S. financial institution that invests primarily in short-term U.S. Treasury notes and bills. To minimize our exposure to interest rate risk, we have limited the maturities of our investments to less than two years with an average maturity not to exceed one year. Due to the short-term nature of these instruments, a 10% increase or decrease in market interest rates would not have a material impact on the total value of our portfolio as of December 31, 2013.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The financial statements required by this item are set forth beginning at page F-1 of this report and are incorporated herein by reference.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
(a) Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our periodic and current reports that we file with the SEC is recorded, processed, summarized and reported within the time periods specified in the SEC's rules and forms, and that such information is accumulated and discussed with our management, including our Chief Executive Officer, Chief Financial Officer and Chief Accounting Officer, as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable and not absolute assurance of achieving the desired control objectives. In reaching a reasonable level of assurance, management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures. In addition, the design of any system of controls also is based in part upon certain assumptions about the likelihood of future events, and we cannot assure you that any design will succeed in achieving its stated goals under all potential future conditions; over time, control may become inadequate because of changes in conditions, or the degree of compliance with policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
As of December 31, 2013, our Chief Executive Officer, Chief Financial Officer and Chief Accounting Officer have evaluated our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) of the Exchange Act) which were designed to ensure that the information required to be disclosed by us in this Annual Report on Form 10-K was recorded, processed, summarized and reported within the time periods specified in the SEC's rules and on Form 10-K. Our disclosure controls and procedures are designed to provide reasonable, not absolute, assurance that the objectives of our disclosure control system are met. Based on the evaluation, our Chief Executive Officer, Chief Financial Officer and Chief Accounting Officer have concluded that our disclosure controls and procedures are effective.
64
There were no changes in our internal controls over financial reporting during the quarter ended December 31, 2013 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
(b) Management's Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). Our internal control system is designed to provide reasonable assurance regarding the preparation and fair presentation of financial statements for external purposes in accordance with generally accepted accounting principles. All internal control systems, no matter how well designed, have inherent limitations and can provide only reasonable assurance that the objectives of the internal control system are met.
Our management, including our Chief Executive Officer, Chief Financial Officer and Chief Accounting Officer, conducted an evaluation of the effectiveness of our internal control over financial reporting, based on criteria established in Internal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in 1992. Based on this evaluation, our management concluded that our internal control over financial reporting was effective as of December 31, 2013.
Our independent registered public accounting firm has issued an attestation report on our internal control over financial reporting as included below.
(c) Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of Corcept Therapeutics Incorporated
We have audited Corcept Therapeutics Incorporated's internal control over financial reporting as of December 31, 2013, based on criteria established in Internal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (1992 framework) (the COSO criteria). Corcept Therapeutics Incorporated's management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management's Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company's internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
65
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Corcept Therapeutics Incorporated maintained, in all material respects, effective internal control over financial reporting as of December 31, 2013, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the 2013 financial statements of Corcept Therapeutics Incorporated and our report dated March 14, 2014 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Redwood City, California
March 14, 2014
ITEM 9B. OTHER INFORMATION
None.
66
PART III
Certain information required by Part III is omitted from this Annual Report on Form 10-K because we expect to file with the U.S. Securities and Exchange Commission, not later than 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, a definitive proxy statement (the Proxy Statement), pursuant to Regulation 14A in connection with the solicitation of proxies for our 2014 Annual Meeting of Stockholders, and certain information included therein is incorporated herein by reference.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this Item concerning our executive officers is set forth in Part I of this Annual Report on Form 10-K. The remaining information required by this Item will be included in the Proxy Statement and is incorporated herein by reference.
ITEM 11. EXECUTIVE COMPENSATION
Compensation Discussion and Analysis
The information required by this Item will be included in the Proxy Statement and is incorporated herein by reference.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this Item will be included in the Proxy Statement and is incorporated herein by reference.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this Item will be included in the Proxy Statement and is incorporated herein by reference.
| ITEM 14. | PRINCIPAL ACCOUNTING FEES AND SERVICES |
The information required by this Item will be included in the Proxy Statement and is incorporated herein by reference.
67
PART IV
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
The following documents are filed as part of this Form 10-K
(1) Financial Statements:
| Page | ||||
Report of Independent Registered Public Accounting Firm | F-2 | |||
Audited Financial Statements | ||||
Balance Sheets | F-3 | |||
Statements of Comprehensive Loss | F-4 | |||
Statement of Stockholders' Equity | F-5 | |||
Statements of Cash Flows | F-6 | |||
Notes to Financial Statements | F-7 | |||
(2) Financial Statement Schedules:
All schedules have been omitted because the information required to be set forth therein is not applicable or is shown in the financial statements or notes thereto.
(3) Exhibits:
Item 601 of Regulation S-K requires the exhibits listed below. Each management contract or compensatory plan or arrangement required to be filed as an exhibit to this Form 10-K has been identified.
(A) EXHIBITS
Exhibit | Description of Document | |
| 3.1 | Amended and Restated Certificate of Incorporation, as amended (incorporated by reference to Exhibit 3.1 to the registrant's Quarterly Report on Form 10-Q filed on August 9. 2012). | |
| 3.2 | Amended and Restated Bylaws (incorporated by reference to Exhibit 3.1 to the registrant's Current Report on Form 8-K filed on September 27, 2007). | |
| 4.1 | Specimen Common Stock Certificate (incorporated by reference to Exhibit 4.1 to the registrant's Registration Statement on Form S-1 (Registration No. 333-112676) filed on February 10, 2004). | |
| 4.2 | Registration Rights Agreement by and among Corcept Therapeutics Incorporated and the investors signatory thereto, dated March 14, 2008 (incorporated by reference to Exhibit 10.25 to the registrant's Annual Report on Form 10-K filed on March 31, 2008). | |
| 4.3 | Amendment to Registration Rights Agreement by and among Corcept Therapeutics Incorporated and the investors signatory thereto, dated November 11, 2008 (incorporated by reference to Exhibit 10.30 to the registrant's Annual Report on Form 10-K filed on March 31, 2009). | |
| 4.4 | Registration Rights Agreement dated as of April 21, 2010 by and among Corcept Therapeutics Incorporated and the investors signatory thereto (incorporated by reference to Exhibit 4.2 to the registrant's Current Report on Form 8-K filed on April 23, 2010). | |
| 4.5 | Registration Rights Agreement, dated as of March 29, 2012, by and among Corcept Therapeutics Incorporated and the investors signatory thereto (incorporated by reference to Exhibit 4.2 to the registrant's Current Report on Form 8-K filed on March 29, 2012). | |
68
Exhibit | Description of Document | |
| 4.6 | Form of Warrant issued in connection with the Securities Purchase Agreement by and among Corcept Therapeutics Incorporated and the purchasers named therein, dated March 14, 2008 (incorporated by reference to Exhibit 4.4 to the registrant's Annual Report on Form 10-K filed on March 31, 2008). | |
| 4.7 | Form of Warrant issued in connection with Warrant Purchase Agreement dated as of March 25, 2012 by and among Corcept Therapeutics Incorporated and the purchasers named therein (incorporated by reference to Exhibit 4.2 to the registrant's Current Report on Form 8-K filed on March 29. 2012). | |
| 10.1 † | 2000 Stock Option Plan (incorporated by reference to Exhibit 10.1 to the registrant's Registration Statement on Form S-1 (Registration No. 333-112676) filed on February 10, 2004). | |
| 10.2 | License Agreement by and between The Board of Trustees of the Leland Stanford Junior University and Corcept Therapeutics Incorporated, dated as of July 1, 1999 (incorporated by reference to Exhibit 10.6 to the registrant's Registration Statement on Form S-1 (Registration No. 333-112676) filed on February 10, 2004). | |
| 10.3 | Master Services Agreement by and between Corcept Therapeutics Incorporated and PPD Development, LP, dated as of January 17, 2003 (incorporated by reference to Exhibit 10.12 to the registrant's Registration Statement on Form S-1/A (File No. 333-112676) filed on March 19, 2004). | |
| 10.4# | Manufacturing Agreement with Produits Chimiques Auxiliaires et de Synthese SA, dated November 8, 2006 (incorporated by reference to Exhibit 10.15 to the registrant's Annual Report on Form 10-K filed on April 2, 2007). | |
| 10.5 † | Form of Indemnification Agreement for directors and officers approved by the Board of Directors on September 24, 2007 (incorporated by reference to Exhibit 10.7 to the registrant's Quarterly Report on Form 10-Q filed on November 14, 2007). | |
| 10.6 | Securities Purchase Agreement by and among Corcept Therapeutics Incorporated and the purchasers named therein, dated March 14, 2008 (incorporated by reference to Exhibit 10.24 to the registrant's Annual Report on Form 10-K filed on March 31, 2008). | |
| 10.7# | Master Service Agreement by and among Corcept Therapeutics Incorporated and ICON Clinical Research, L.P., signed on June 4, 2008 (incorporated by reference to Exhibit 10.5 to the registrant's Quarterly Report on Form 10-Q filed on August 14, 2008). | |
| 10.8 † | Amended and Restated Severance and Change in Control Agreement by and between Corcept Therapeutics Incorporated and Joseph K. Belanoff, M. D., dated September 19, 2008 (incorporated by reference to Exhibit 10.25 to the registrant's Annual Report on Form 10-K filed on March 31, 2009). | |
| 10.9 † | Amended and Restated Severance and Change in Control Agreement by and between Corcept Therapeutics Incorporated and Robert L. Roe, M. D., dated September 19, 2008 (incorporated by reference to Exhibit 10.26 to the registrant's Annual Report on Form 10-K filed on March 31, 2009). | |
| 10.10 † | Amended and Restated Severance and Change in Control Agreement by and between Corcept Therapeutics Incorporated and Anne M. LeDoux, dated September 19, 2008 (incorporated by reference to Exhibit 10.27 to the registrant's Annual Report on Form 10-K filed on March 31, 2009). | |
| 10.11 † | Amended and Restated Severance and Change in Control Agreement by and between Corcept Therapeutics Incorporated and James N. Wilson, dated September 19, 2008 (incorporated by reference to Exhibit 10.28 to the registrant's Annual Report on Form 10-K filed on March 31, 2009). | |
| 10.12 | Securities Purchase Agreement by and among Corcept Therapeutics Incorporated and the purchasers named therein, dated October 12, 2009 (incorporated by reference to Exhibit 10.1 to the registrant's Quarterly Report on Form 10-Q filed on November 12, 2009). | |
| 10.13 † | Amended and Restated 2004 Equity Incentive Plan (incorporated by reference to the registrant's Proxy Statement on Schedule 14A filed on May 7, 2009). | |
| 10.14 † | Form of Option Agreement for options granted pursuant to the Amended and Restated 2004 Equity Incentive Plan (incorporated by reference to Exhibit 10.25 to the registrant's Annual Report on Form 10-K filed on March 15, 2011). | |
69
Exhibit | Description of Document | |
| 10.15 # | Development Agreement by and between Corcept Therapeutics Incorporated and Formulation Technologies L.L.C. d/b/a PharmaForm, dated as of December 14, 2006 (incorporated by reference to Exhibit 10.28 to the registrant's Annual Report on Form 10-K filed on March 15, 2011). | |
| 10.16 # | Master Services Agreement by and between Corcept Therapeutics Incorporated and United BioSource Corporation, dated as of June 29, 2010 (incorporated by reference to Exhibit 10.29 to the registrant's Annual Report on Form 10-K filed on March 15, 2011). | |
| 10.17 † | Employment offer letter to Steven Lo, dated August 9, 2010 (incorporated by reference to Exhibit 10.1 to the registrant's Quarterly Report on Form 10-Q filed on November 12, 2010). | |
| 10.18 † | Severance and Change in Control Agreement by and between Corcept Therapeutics Incorporated and Steven Lo, dated September 15, 2010 (incorporated by reference to Exhibit 10.2 to the registrant's Quarterly Report on Form 10-Q filed on November 12, 2010). | |
| 10.19 † | Severance and Change in Control Agreement by and between Corcept Therapeutics Incorporated and G. Charles Robb, dated September 1, 2011 (incorporated by reference to Exhibit 10.2 to the registrant's Quarterly Report on Form 10-Q filed on November 8, 2011). | |
| 10.20 † | Employment offer letter to G. Charles Robb dated August 12, 2011 (incorporated by reference to Exhibit 10.1 to the registrant's Quarterly Report on Form 10-Q filed on November 8, 2011). | |
| 10.21 # | Manufacturing and Supply Agreement with Formulation Technologies, LLC D/B/A PharmaForm, LLC, dated March 21, 2012 (incorporated by reference to Exhibit 10.1 to the registrant's Quarterly Report on Form 10-Q filed on May 10, 2012). | |
| 10.22 | Warrant Purchase Agreement, dated as of March 25, 2012, by and among Corcept Therapeutics Incorporated and the purchasers named therein (incorporated by reference to Exhibit 10.1 to the registrant's Current Report on Form 8-K filed on March 29, 2012). | |
| 10.23 # | Commercial Outsourcing Services Agreement with Integrated Commercialization Solutions, Inc., dated as of April 14, 2011 (incorporated by reference to Exhibit 10.1 to the registrant's Quarterly Report on Form 10-Q filed on August 9, 2012). | |
| 10.24 # | Amended and Restated Exclusive Pharmacy Product Purchase and Services Agreement with CuraScript, Inc., dated August 8, 2012 (incorporated by reference to Exhibit 10.2 to the registrant's Quarterly Report on Form 10-Q filed on August 9, 2012). | |
| 10.25 † | Corcept Therapeutics Incorporated 2012 Incentive Award Plan (incorporated by reference to Appendix A to the registrant's Definitive Proxy Statement on Schedule 14A filed with the SEC on May 21, 2012). | |
| 10.26 † | Form of 2012 Incentive Award Plan Stock Option Grant Notice and Agreement (incorporated by reference to Exhibit 4.5 to the registrant's Registration Statement on Form S-8 filed with the SEC on August 13, 2012). | |
| 10.27 # | Purchase and Sale Agreement with between Corcept Therapeutics Incorporated and Biopharma Secured Debt Fund II Sub, S.à r.l,, dated as of August 2, 2012 (incorporated by reference to Exhibit 10.4 to the registrant's Quarterly Report on Form 10-Q filed on November 8, 2012). | |
| 10.28 | Amendment to Manufacturing Agreement with Produits Chimiques Auxiliaires et de Synthese SA, dated February 21, 2013 (incorporated by reference to Exhibit 10.31 to the registrant's Annual Report on Form 10-K filed on March 15, 2013). | |
| 10.29 # | Pharmaceutical Manufacturer Services Agreement with Centric Health Resources, Inc., dated May 21, 2013 (incorporated by reference to Exhibit 10.1 to the registrant's Quarterly Report on Form 10-Q filed on August 9, 2013). | |
| 10.30 † | Letter agreement with Robert L. Roe, M.D. regarding terms of retirement and consulting arrangement, dated June 21, 2013 (incorporated by reference to Exhibit 10.2 to the registrant's Quarterly Report on Form 10-Q filed on August 9, 2013). | |
70
Exhibit | Description of Document | |
| 10.31 # | Amendment to Pharmaceutical Manufacturer Services Agreement with Centric Health Resources, Inc., dated July 22, 2013 (incorporated by reference to Exhibit 10.3 to the registrant's Quarterly Report on Form 10-Q filed on August 9, 2013). | |
| 10.32 | Amendment to Manufacturing Agreement with Produits Chimiques Auxiliaires et de Synthese SA, dated August 1, 2013 (incorporated by reference to Exhibit 10.4 to the registrant's Quarterly Report on Form 10-Q filed on August 9, 2013). | |
| 10.33 | Amendment to Manufacturing Agreement with Produits Chimiques Auxiliaires et de Synthese SA, dated November 7, 2013 (incorporated by reference to Exhibit 10.1 to the registrant's Quarterly Report on Form 10-Q filed on November 12, 2013). | |
| 10.34 | Amendment to Manufacturing Agreement with Produits Chimiques Auxiliaires et de Synthese SA, dated January 27, 2014. | |
| 23.1 | Consent of Independent Registered Public Accounting Firm | |
| 24.1 | Power of Attorney (See signature page) | |
| 31.1 | Certification pursuant to Rule 13a-14(a) under the Securities Exchange Act of 1934 of Joseph K. Belanoff, M.D. | |
| 31.2 | Certification pursuant to Rule 13a-14(a) under the Securities Exchange Act of 1934 of G. Charles Robb | |
| 32.1 | Certification pursuant to 18 U.S.C. Section 1350 of Joseph K. Belanoff, M.D. | |
| 32.2 | Certification pursuant to 18 U.S.C. Section 1350 of G. Charles Robb | |
| 101 | The following materials from the registrant's Annual Report on Form 10-K for the year ended December 31, 2013, formatted in Extensible Business Reporting Language (XBRL): (i) Balance Sheets at December 31, 2013 and 2012, (ii) Statements of Comprehensive Loss for the Years Ended December 31, 2013, 2012 and 2011, (iii) Statements of Stockholders' Equity for the Years Ended December 31, 2013, 2012 and 2011, (iv) Statements of Cash Flows for the Years Ended December 31, 2013, 2012 and 2011, and (v) Notes to Financial Statements. | |
| # | Confidential treatment granted |
| † | Management contract or compensatory plan or arrangement |
71
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| CORCEPT THERAPEUTICS INCORPORATED | ||
| By: | / S / JOSEPH K. BELANOFF | |
Joseph K. Belanoff, M.D., Chief Executive Officer and President | ||
| Date: | March 14, 2014 | |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below hereby constitutes and appoints Joseph K. Belanoff and G. Charles Robb, and each of them acting individually, as his or her true and lawful attorneys-in-fact and agents, each with full power of substitution, for him or her in any and all capacities, to sign any and all amendments to this report on Form 10-K and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, with full power of each to act alone, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully for all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or his or their substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Exchange Act, this Annual Report on Form 10-K has been signed by the following persons on behalf of the registrant and in the capacities and on the dates indicated:
Signature | Title | Date | ||
/ S / JOSEPH K. BELANOFF Joseph K. Belanoff, M.D. | Chief Executive Officer, President and Director (Principal Executive Officer) | March 14, 2014 | ||
/ S / G. CHARLES ROBB G. Charles Robb | Chief Financial Officer and Secretary (Principal Financial Officer) | March 14, 2014 | ||
/ S / ANNE M. LEDOUX Anne M. LeDoux | Vice President and Controller (Principal Accounting Officer) | March 14, 2014 | ||
/ S / JAMES N. WILSON James N. Wilson | Director and Chairman of the Board of Directors | March 14, 2014 | ||
/ S / G. LEONARD BAKER, JR. G. Leonard Baker, Jr. | Director | March 14, 2014 | ||
/ S / DANIEL M. BRADBURY Daniel M. Bradbury | Director | March 14, 2014 | ||
/ S / JOSEPH C. COOK, JR. Joseph C. Cook, Jr. | Director | March 14, 2014 | ||
/ S / PATRICK G. ENRIGHT Patrick G. Enright | Director | March 14, 2014 | ||
/ S / DAVID L. MAHONEY David L. Mahoney | Director | March 14, 2014 | ||
/ S / JOSEPH L. TURNER Joseph L. Turner | Director | March 14, 2014 | ||
72
CORCEPT THERAPEUTICS INCORPORATED
INDEX TO FINANCIAL STATEMENTS
Page | ||||
Report of Independent Registered Public Accounting Firm | F-2 | |||
Audited Financial Statements | ||||
Balance Sheets | F-3 | |||
Statements of Comprehensive Loss | F-4 | |||
Statement of Stockholders' Equity | F-5 | |||
Statements of Cash Flows | F-6 | |||
Notes to Financial Statements | F-7 | |||
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of Corcept Therapeutics Incorporated
We have audited the accompanying balance sheets of Corcept Therapeutics Incorporated as of December 31, 2013 and 2012 and the related statements of comprehensive loss, stockholders' equity and cash flows for each of the three years in the period ended December 31, 2013. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States.) Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Corcept Therapeutics Incorporated at December 31, 2013 and 2012, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2013, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Corcept Therapeutics Incorporated's internal control over financial reporting as of December 31, 2013, based on criteria established in Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (1992 framework) and our report dated March 14, 2014 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Redwood City, California
March 14, 2014
F-2
CORCEPT THERAPEUTICS INCORPORATED
BALANCE SHEETS
(in thousands, except per share amounts)
| December 31, | ||||||||
| 2013 | 2012 | |||||||
Assets | ||||||||
Current assets: | ||||||||
Cash and cash equivalents | $ | 54,877 | $ | 93,032 | ||||
Trade receivables | 1,428 | 557 | ||||||
Inventory | 1,096 | 853 | ||||||
Prepaid expenses and other current assets | 910 | 620 | ||||||
|
|
|
| |||||
Total current assets | 58,311 | 95,062 | ||||||
Strategic inventory | 4,450 | 3,810 | ||||||
Property and equipment, net of accumulated depreciation | 203 | 150 | ||||||
Other assets | 113 | 144 | ||||||
|
|
|
| |||||
Total assets | $ | 63,077 | $ | 99,166 | ||||
|
|
|
| |||||
Liabilities and stockholders' equity | ||||||||
Current liabilities: | ||||||||
Accounts payable | $ | 2,381 | $ | 3,804 | ||||
Accrued clinical expenses | 3,288 | 843 | ||||||
Other accrued liabilities | 1,301 | 1,046 | ||||||
Long-term obligation - current portion | 5,743 | 2,650 | ||||||
Deferred revenue | 25 | 16 | ||||||
|
|
|
| |||||
Total current liabilities | 12,738 | 8,359 | ||||||
Long-term obligation, net of current portion | 29,322 | 29,030 | ||||||
Commitments | ||||||||
Stockholders' equity: | ||||||||
Preferred stock, $0.001 par value, 10,000 shares authorized and no shares outstanding at December 31, 2013 or 2012 | - | - | ||||||
Common stock, $0.001 par value, 280,000 shares authorized and 99,849 and 99,814 shares issued and outstanding at December 31, 2013 and 2012, respectively | 100 | 100 | ||||||
Additional paid-in capital | 313,534 | 308,283 | ||||||
Accumulated deficit | (292,617) | (246,606) | ||||||
|
|
|
| |||||
Total stockholders' equity | 21,017 | 61,777 | ||||||
|
|
|
| |||||
Total liabilities and stockholders' equity | $ | 63,077 | $ | 99,166 | ||||
|
|
|
| |||||
The accompanying notes are an integral part of these financial statements.
F-3
CORCEPT THERAPEUTICS INCORPORATED
STATEMENTS OF COMPREHENSIVE LOSS
(in thousands, except per share amounts)
| Year ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
Product sales, net | $ | 10,357 | $ | 3,307 | $ | - | ||||||
|
|
|
|
|
| |||||||
Operating expenses: | ||||||||||||
Cost of sales | 143 | 91 | - | |||||||||
Research and development | 20,470 | 14,074 | 21,001 | |||||||||
Selling, general and administrative | 31,240 | 25,414 | 11,331 | |||||||||
|
|
|
|
|
| |||||||
Total operating expenses | 51,853 | 39,579 | 32,332 | |||||||||
|
|
|
|
|
| |||||||
Loss from operations | (41,496) | (36,272) | (32,332) | |||||||||
Interest and other income | - | - | 3 | |||||||||
Interest and other expense | (4,515) | (1,776) | (25) | |||||||||
|
|
|
|
|
| |||||||
Net loss and comprehensive loss | $ | (46,011) | $ | (38,048) | $ | (32,354) | ||||||
|
|
|
|
|
| |||||||
Basic and diluted net loss per share | $ | (0.46) | $ | (0.41) | $ | (0.39) | ||||||
|
|
|
|
|
| |||||||
Shares used in computing basic and diluted net loss per share | 99,819 | 93,015 | 83,309 | |||||||||
|
|
|
|
|
| |||||||
The accompanying notes are an integral part of these financial statements.
F-4
CORCEPT THERAPEUTICS INCORPORATED
STATEMENT OF STOCKHOLDERS' EQUITY
(in thousands)
| Common Stock | Additional Paid-in Capital | Notes Receivable from Stockholders | Accumulated Deficit | Total Stockholders' Equity | ||||||||||||||||||||
Shares | Amount | |||||||||||||||||||||||
Balance at December 31, 2010 | 72,404 | $ | 72 | $ | 197,473 | $ | (97) | $ | (176,204) | $ | 21,244 | |||||||||||||
Sale of common stock in public financing transaction | 11,500 | 12 | 41,771 | - | - | 41,783 | ||||||||||||||||||
Issuance of common stock upon exercise of options | 246 | - | 371 | - | - | 371 | ||||||||||||||||||
Issuance of common stock upon exercise of warrants | 81 | - | 231 | - | - | 231 | ||||||||||||||||||
Stock-based compensation related to employee and director options | - | - | 3,016 | - | - | 3,016 | ||||||||||||||||||
Stock-based compensation related to an option to a consultant | - | - | 419 | - | - | 419 | ||||||||||||||||||
Repayment of notes receivable from stockholders | - | - | - | 97 | - | 97 | ||||||||||||||||||
Net loss and comprehensive loss | (32,354) | (32,354) | ||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||
Balance at December 31, 2011 | 84,231 | 84 | 243,281 | - | (208,558) | 34,807 | ||||||||||||||||||
Sale of common stock in public financing transaction | 11,000 | 11 | 46,119 | - | - | 46,130 | ||||||||||||||||||
Issuance of common stock upon exercise of warrants and issuance of new warrants in private equity transaction | 4,202 | 4 | 12,815 | - | - | 12,819 | ||||||||||||||||||
Issuance of common stock upon exercise of warrants | 216 | - | 470 | - | - | 470 | ||||||||||||||||||
Issuance of common stock upon exercise of options | 165 | 1 | 288 | - | - | 289 | ||||||||||||||||||
Stock-based compensation related to employee and director options | - | - | 5,102 | - | - | 5,102 | ||||||||||||||||||
Stock-based compensation related to consultant options | - | - | 208 | - | - | 208 | ||||||||||||||||||
Net loss and comprehensive loss | (38,048) | (38,048) | ||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||
Balance at December 31, 2012 | 99,814 | 100 | 308,283 | - | (246,606) | 61,777 | ||||||||||||||||||
Issuance of common stock upon exercise of an option | 35 | - | 55 | - | - | 55 | ||||||||||||||||||
Stock-based compensation related to employee and director options | - | - | 5,069 | - | - | 5,069 | ||||||||||||||||||
Stock-based compensation related to consultant options | - | - | 127 | - | - | 127 | ||||||||||||||||||
Net loss and comprehensive loss | (46,011 | ) | (46,011 | ) | ||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||
Balance at December 31, 2013 | 99,849 | $ | 100 | $ | 313,534 | $ | - | $ | (292,617) | $ | 21,017 | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||
The accompanying notes are an integral part of these financial statements
F-5
CORCEPT THERAPEUTICS INCORPORATED
STATEMENTS OF CASH FLOWS
(in thousands)
| Year ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
Operating activities | ||||||||||||
Net loss | $ | (46,011) | $ | (38,048) | $ | (32,354) | ||||||
Adjustments to reconcile net loss to net cash used in operations: | ||||||||||||
Stock-based compensation | 5,196 | 5,310 | 3,435 | |||||||||
Accretion of interest expense | 4,410 | 1,680 | - | |||||||||
Amortization of debt financing costs | 35 | 17 | - | |||||||||
Depreciation and amortization of property and equipment | 74 | 27 | 3 | |||||||||
Changes in operating assets and liabilities: | ||||||||||||
Trade receivables | (871) | (557) | - | |||||||||
Inventory | (883) | (4,663) | - | |||||||||
Prepaid expenses and other current assets | (290) | (480) | 278 | |||||||||
Other assets | (4) | 11 | 72 | |||||||||
Accounts payable | (1,423) | 193 | 2,794 | |||||||||
Accrued clinical expenses | 2,445 | 199 | (171) | |||||||||
Accrued compensation and other liabilities | 255 | 275 | (1,457) | |||||||||
Deferred revenue | 9 | 16 | - | |||||||||
|
|
|
|
|
| |||||||
Net cash used in operating activities | (37,058) | (36,020) | (27,400) | |||||||||
|
|
|
|
|
| |||||||
Investing activities | ||||||||||||
Purchases of property and equipment | (127) | (151) | (25) | |||||||||
|
|
|
|
|
| |||||||
Cash used in investing activities | (127) | (151) | (25) | |||||||||
|
|
|
|
|
| |||||||
Financing activities | ||||||||||||
Proceeds from issuance of common stock and warrants, including collection of stockholder notes receivable, net of cash paid for issuance costs | 55 | 59,708 | 42,482 | |||||||||
Proceeds from issuance of long-term obligation, net of cash paid for issuance costs | - | 29,860 | - | |||||||||
Payments related to long-term obligation | (1,025) | - | - | |||||||||
|
|
|
|
|
| |||||||
Net cash (used in) provided by financing activities | (970) | 89,568 | 42,482 | |||||||||
|
|
|
|
|
| |||||||
Net (decrease) increase in cash and cash equivalents | (38,155) | 53,397 | 15,057 | |||||||||
Cash and cash equivalents at beginning of period | 93,032 | 39,635 | 24,578 | |||||||||
|
|
|
|
|
| |||||||
Cash and cash equivalents at end of period | $ | 54,877 | $ | 93,032 | $ | 39,635 | ||||||
|
|
|
|
|
| |||||||
The accompanying notes are an integral part of these financial statements
F-6
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO FINANCIAL STATEMENTS
1. Basis of Presentation and Summary of Significant Accounting Policies
Description of Business
Corcept Therapeutics Incorporated was incorporated in the state of Delaware in May 1998, and our facilities are located in Menlo Park, California. Corcept is a pharmaceutical company engaged in the discovery, development and commercialization of drugs for the treatment of severe metabolic, psychiatric and oncologic disorders. Since our inception, we have been developing our lead product, Korlym ® . Mifepristone, the active ingredient in Korlym, is a potent competitive antagonist of the glucocorticoid receptor II (GR-II), which means that it competitively blocks the effects of cortisol throughout the body at one of its two receptors. In February 2012, the United States Food and Drug Administration (FDA) approved Korlym (mifepristone) 300 mg Tablets in the United States as a once-daily oral medication for treatment of hyperglycemia secondary to hypercortisolism in adult patients with endogenous Cushing's syndrome who have type 2 diabetes mellitus or glucose intolerance and have failed surgery or are not candidates for surgery. We released Korlym for sale in the United States in April 2012. We also have a clinical program for the use of mifepristone for the treatment of the psychotic features of psychotic depression and are currently conducting a phase 3 study for this indication. In December 2013, we initiated a study of mifepristone for the treatment of triple-negative breast cancer. In addition, we have discovered and patented three series of novel selective GR-II antagonists. Unless otherwise stated, all references in these financial statements to "we," "us," "our," "Corcept," the "Company," "our company" and similar designations refer to Corcept Therapeutics Incorporated.
Use of Estimates
The preparation of financial statements in conformity with U.S. generally accepted accounting principles requires management to use assumptions and make estimates to form judgments that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ materially from those estimates.
We evaluate our estimates and assumptions on an ongoing basis, including those related to our discounts for prompt payment of sales invoices, chargebacks and rebates, patient assistance, potential product returns, excess/obsolete inventories, allowances for doubtful accounts, accruals of clinical and preclinical expenses, contingent liabilities, and the timing of payments with respect to our long-term financing agreement, which determine its effective interest rate. We base our estimates on relevant experience and on other specific assumptions that we believe are reasonable.
We update our assumptions and estimates on a recurring basis as new information becomes available. Any changes in estimates are recorded in the period of the change.
Cash and Cash Equivalents
We invest our excess cash in bank deposits, money market accounts, corporate debt securities, and/or obligations of the U.S. government and U.S. government sponsored entities. We consider all highly liquid investments purchased with maturities of three months or less from the date of purchase to be cash equivalents. Cash equivalents are carried at fair value, which approximates cost and, as of December 31, 2013 and 2012, all of our funds were invested in cash and cash equivalents that consist of a money market fund maintained at a major U.S. financial institution.
Credit Risks and Concentrations
We have a concentration of credit risk related to our cash and cash equivalents. We are exposed to credit risk in the event of default by the financial institution holding these funds or by the entity or entities that issued
F-7
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO FINANCIAL STATEMENTS, Continued
the securities held by the fund to the extent of the amount recorded on our balance sheet. We mitigate this risk by investing in a money market fund that invests primarily in short-term U.S. Treasury notes and bills. We experienced no loss or lack of access to cash and cash equivalents in our operating or investment accounts during the years ended December 31, 2013, 2012 and 2011.
Since the commercialization of Korlym in April 2012, we have been exposed to credit risk in regard to our trade receivables. From the launch of Korlym through June 30, 2013, 97% of our sales were to one specialty pharmacy customer, from whom we have fully collected all receivables. As discussed in Note 2, Significant Agreements – Commercial Agreements , in mid-2013 we transitioned all of our specialty pharmacy business to a new provider, Centric Health Resources, Inc. (Centric). Among other services, Centric dispenses Korlym to patients for us, with title to the medicine passing from us to the patient upon the patient's receipt of the drug. Accordingly, our receivables risk is spread among various third-party payors – pharmacy benefit managers, insurance companies, private charities, government programs – and individual patients. We extend credit to third-party payors based on their creditworthiness. We monitor our exposure and will record a reserve against uncollectible trade receivables as necessary.
We have a concentration of risk in regard to the manufacture of our product. As of December 31, 2013, we had one tablet manufacturer for Korlym with an operational facility – AAI Pharma Services Corp. (AAI), which was approved by the FDA in November 2012 for the manufacture of our commercial tablets, subject to the successful manufacture of validation batches. We plan to undertake this validation process at AAI early in 2014 and are currently in negotiations for a long-term commercial manufacturing agreement with AAI. If AAI is unable to prepare Korlym tablets in the quantities and time frame required, we may not be able to manufacture our product in a timely manner. In addition, we have a single-source manufacturer of mifepristone, the active pharmaceutical ingredient (API), in Korlym. In order to mitigate these risks related to the manufacture of our product, we placed orders for additional quantities of mifepristone API and Korlym tablets, which are now in inventory.
Fair Value Measurements
We categorize financial instruments in a fair value hierarchy that prioritizes the information used to develop assumptions for measuring fair value. The fair value hierarchy gives the highest priority to quoted prices in active markets for identical assets or liabilities (Level 1 input), then to quoted prices in non-active markets or in active markets for similar assets or liabilities, inputs other than quoted prices that are observable for the asset or liability, and inputs that are not directly observable, but that are corroborated by observable market data for the asset or liability (Level 2 input), then the lowest priority to unobservable inputs, for example, our own data about the assumptions that market participants would use in pricing an asset or liability (Level 3 input). Fair value is a market-based measurement, not an entity-specific measurement, and a fair value measurement should therefore be based on the assumptions that market participants would use in pricing the asset or liability.
No assets or liabilities in our financial statements are required to be reported at fair value other than our cash equivalents and the obligation under our Financing Agreement with Biopharma Secured Debt Fund II Sub, S.àr.l (Biopharma).
Trade Receivables
Trade receivables are recorded net of customer allowances for prompt payment and data services, doubtful accounts and sales returns. See the discussion below under "Net Product Sales" regarding the methods for estimation of these allowances and sales returns. We determine our allowance for doubtful accounts based on existing contractual payment terms, actual payment patterns of our customers and individual customer circumstances. To date, we have determined that an allowance for uncollectible trade receivables is not required.
F-8
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO FINANCIAL STATEMENTS, Continued
Inventory
We consider regulatory approval of product candidates to be uncertain, and product manufactured prior to regulatory approval may not be sold unless regulatory approval is obtained. We expense the manufacturing costs for product candidates incurred prior to regulatory approval as research and development expense as we incur them. When regulatory approval of a product is obtained, we begin capitalizing manufacturing costs related to the approved product into inventory, provided such product is produced by a facility the FDA has approved to manufacture Korlym for distribution as commercial product.
We value our inventories at the lower of cost or net realizable value. We determine the cost of inventory using the specific identification method, which approximates a first-in, first-out basis. We analyze our inventory levels quarterly and write down inventory that has become obsolete or has a cost basis in excess of its expected net realizable value, as well as any inventory quantities in excess of expected requirements. Any expired inventory is disposed of and the related costs are recognized as cost of sales in the statement of comprehensive loss.
Inventory amounts that are not expected to be consumed within 12 months following the balance sheet date are classified as strategic inventory, a noncurrent asset.
Property and Equipment
We state property and equipment at cost less accumulated depreciation. Property and equipment are depreciated using the straight-line method over the estimated useful lives of the assets, ranging from three to five years.
Long-term Obligation
In August 2012, we entered into a Purchase and Sale Agreement (Financing Agreement) with Biopharma Secured Debt Fund II Sub, S.à r.l. (Biopharma), a private limited liability company organized under the laws of Luxembourg. Under the terms of the Financing Agreement, we received $30.0 million from Biopharma and are obligated to make payments calculated as a percentage of (i) any licensing or other contingent payments arising from Korlym and any other products containing mifepristone or any of our proprietary selective GR-II antagonists (Covered Products) and (ii) net Covered Product revenues earned in the calendar quarter ended June 30, 2013 and thereafter (together, Korlym Receipts), until such time as we have paid Biopharma a total of $45.0 million.
The accounting for the Financing Agreement requires us to make certain estimates and assumptions, including the timing of royalty payments due to Biopharma, the expected rate of return to Biopharma, the split between current and long-term portions of the obligation and the accretion of related interest expense. Korlym has only been marketed since April 2012 and the magnitude and timing of Korlym revenue is difficult to predict. Therefore, these estimates and assumptions are subject to significant variability and are likely to change as we gain experience marketing Korlym, which will result in changes in our classification of the current and long-term portions of the amounts payable pursuant to the Financing Agreement, as well as the internal rate of return paid to Biopharma and the accretion of interest expense related to this obligation. Actual payment amounts will be based on Korlym Receipts over the term of the Financing Agreement but in no event will the total amount paid to Biopharma exceed $45.0 million.
The amount shown as the current portion of the obligation is an estimate of the total amount under the Financing Agreement that would be paid to Biopharma within 12 months following December 31, 2013. Under the Financing Agreement, our first payment to Biopharma was made in July 2013.
F-9
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO FINANCIAL STATEMENTS, Continued
See Note 6, Long-term Obligation , for additional information regarding this agreement.
Net Product Sales
From our initial launch in April 2012 through June 30, 2013, we sold Korlym primarily to a specialty pharmacy and a specialty distributor, which subsequently resold Korlym to patients and healthcare providers. Korlym is not available in retail pharmacies. As discussed in Note 5, Significant Agreements – Commercial Agreements , Centric, the specialty pharmacy we began using July 1, 2013, operates on a consignment basis, without carrying any Korlym inventory. Accordingly, all of our sales through Centric are made directly to patients.
We recognize product revenues from sales of Korlym upon delivery to patients as long as (i) there is persuasive evidence that an arrangement exists between ourselves and the customer, (ii) collectability is reasonably assured and (iii) the price is fixed or determinable. Prior authorization or confirmation of coverage level by the patient's private insurance plan or government payor is a prerequisite to the shipment of product to a patient. In order to conclude that the price is fixed or determinable, we must be able to (i) calculate gross product revenues from the sales to our customers and (ii) reasonably estimate net product revenues.
We provide cash donations to a non-profit third party organization that supports patients who meet certain eligibility requirements with financial assistance for the treatment of Cushing's syndrome, which treatment may include Korlym. We do not include as net product revenues sales of Korlym tablets funded through this source.
We calculate gross product revenues based on the price that we charge our customers. We estimate our net product revenues by deducting from our gross product revenues (a) trade allowances, such as discounts for prompt payment and distributor fees, (b) estimated government rebates and chargebacks, (c) reserves for expected product returns and (d) estimated costs of our patient co-pay assistance program. We initially record estimates for these deductions at the time we recognize the gross revenue. We update our estimates on a recurring basis as new information becomes available.
Trade Allowances: Through June 30, 2013, we offered our specialty pharmacy and specialty distributor customers a discount on Korlym sales for payment within 30 days. We also offered them a small discount for providing data services. We expected these customers to earn these discounts and, accordingly, deducted them in full from gross product revenues and trade receivables at the time we recognized such revenues. Beginning in the third quarter of 2013, we ceased incurring a prompt-payment discount to our specialty pharmacy.
Rebates and Chargebacks: We contract with Medicaid and other government programs so that Korlym will be eligible for purchase by, or qualify for partial or full reimbursement from, such government programs. We estimate the rebates and chargebacks that we are obligated to provide to government programs and deduct these estimated amounts from our gross product sales at the time the revenues are recognized. We base our estimates of these rebates and chargebacks upon (i) the discount amounts applicable to government-funded programs and (ii) information obtained from our vendors regarding the percentage of sales by our customers to patients who are covered by entities or programs that are eligible for such rebates and chargebacks.
Allowances for Patient Assistance Program: We provide financial assistance to eligible patients whose insurance policies require them to pay high deductibles and co-pays. We estimate the cost of assistance to be provided under this program by applying our actual experience regarding such assistance to our estimate of the percentage of our sales in the period that will be provided to patients covered by the program.
Sales Returns: Our specialty distribution customer has the right to return Korlym beginning six months before the labeled expiration date and ending 12 months after the labeled expiration date. This right of return is
F-10
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO FINANCIAL STATEMENTS, Continued
extended to hospital customers of our specialty distribution customer who, generally, have the right to return only unopened bottles. Individual patients do not have the right to return product. We have the right to resell returned product, provided the bottles have not been opened or damaged and the product has not expired. The expiration date for the Korlym product sold to date to the specialty distributor will not occur until November 2014. We estimate the amount of Korlym that we believe will be returned and deduct that estimated amount from gross revenue at the time we recognize such revenue. When estimating future returns, we analyze quantitative and qualitative information including, but not limited to, actual return rates, the amount of product in the distribution channel, the expected shelf life of such product, current and projected product demand, the introduction of competing products that may erode demand, and broad economic and industry-wide indicators. If we cannot reasonably estimate product returns with respect to a particular sale, we defer recognition of revenue from that sale until we can make a reasonable estimate.
See Note 2, Significant Agreements – Commercial Agreements , for additional information regarding our specialty pharmacy agreement signed in May 2013 with Centric, termination of our previous agreement with CuraScript Specialty Pharmacy (CuraScript) and the product return reserve recorded as of June 30, 2013 as a result of termination of our CuraScript agreement. Because Centric operates on a consignment model and does not carry inventory, our exposure to product returns is now limited to the specialty distributor channel and is not expected to be material.
Cost of Sales
Cost of sales includes the cost of product (the cost to manufacture Korlym, which includes material, third-party manufacturing costs and indirect personnel and other overhead costs) based on units for which revenue is recognized in the current period, as well as costs of stability testing, logistics and distribution of the product. We began capitalizing Korlym production costs as inventory following approval by the FDA in February 2012. Prior to receiving FDA approval for Korlym, we expensed all costs related to the manufacturing of the product as incurred; we classified these costs as research and development expense. A portion of the product manufactured prior to FDA approval is available for us to use commercially.
Research and Development
Research and development expenses consist of costs incurred for research and development activities that we sponsor. These costs include direct expenses, such as the cost of clinical trials, pre-clinical studies, manufacturing development, preparations for submissions to the FDA and other regulatory bodies and efforts to prosecute and defend those submissions and the development of second-generation compounds, as well as research and development-related overhead expenses. We also expense as incurred nonrefundable payments to third parties and our cost of acquiring technologies and materials used in research and development that have no alternative future use.
We base our cost accruals for clinical trials, research and preclinical activities on estimates of work completed under service agreements, milestones achieved, patient enrollment and past experience with similar contracts. Our estimates of work completed and associated cost accruals include our assessments of information from third-party contract research organizations and the overall status of clinical trial and other development and administrative activities.
Segment Reporting
We determine our operating segments based on the way we organize our business to make operating decisions and assess performance. We have only one operating segment, which concerns the discovery, development and commercialization of pharmaceutical products.
F-11
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO FINANCIAL STATEMENTS, Continued
Stock-Based Compensation
Stock-based compensation for employee and director options
We account for stock-based compensation related to option grants to employees and directors under the fair value method, based on the fair value-based measurement of the award at the grant date as determined utilizing the Black-Scholes option valuation model. For service-based awards, we recognize expense over the requisite service period. For options with performance-based vesting criteria, we begin to recognize expense when we believe there is a high degree of probability (i.e., greater than 70%) of achieving the vesting criteria.
Stock-based compensation expense related to non-employees
We recognize the expense of options granted to non-employees based on the fair-value based measurement of the option grants at the time of vesting. For service-based awards, we recognize expense over the requisite service period. For options with performance-based vesting criteria, we recognize expense based on the minimum number of shares that will vest over time as the criteria are met based on the Black-Scholes valuation of the vested shares.
See Note 9 for a detailed discussion of stock-based compensation expense.
Income Taxes
We determine deferred tax assets and liabilities based on the differences between the financial reporting and tax bases of assets and liabilities, measured using the enacted tax rates that will be in effect when the differences are expected to reverse. A valuation allowance is recorded when it is more likely than not that the deferred tax asset will not be realized.
No amounts have been recognized as interest or penalties on income tax related matters. The determination of an accounting policy as to the classification of such costs has been deferred until such time as any such costs are incurred.
2. Significant Agreements
Commercial Agreements
On May 21, 2013, we entered into a services agreement with Centric Health Resources, Inc. (Centric) to provide exclusive specialty pharmacy and patient services programs for Korlym beginning July 1, 2013. Under the terms of this agreement, Centric acts as the exclusive specialty pharmacy distributor of Korlym in the United States, subject to certain exceptions. Among other services, Centric provides services related to pharmacy operations; patient intake, access and reimbursement; patient support; claims management and accounts receivable; and data and reporting. We provide Korlym to Centric, which it dispenses to patients. Centric does not take title to the product, which passes directly from us to the patient at the time the patient receives the medicine.
The initial term of the agreement is a period of three years, with successive automatic renewal terms of three years unless either party gives at least 180 days' prior notice of non-renewal. The agreement contains customary termination provisions, representations, warranties and covenants. Subject to certain limitations, we have agreed to indemnify Centric for certain third party claims related to the product, and we have each agreed to indemnify the other for certain breaches of representations, warranties, covenants and other specified matters.
F-12
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO FINANCIAL STATEMENTS, Continued
As of May 20, 2013, we gave notice to CuraScript, our previous specialty pharmacy provider, of our intent to terminate our agreement with them effective July 20, 2013. As of June 30, 2013, we recorded a return reserve estimate of $300,000 for inventory that CuraScript had purchased from us but had the right to return as a result of this termination. This amount was reflected as an adjustment to net revenue in our Statement of Comprehensive Loss for the year ended December 31, 2013. Our exposure to product returns is now limited to the specialty distributor channel and is not expected to be material.
In April 2011, we signed an agreement with Integrated Commercialization Solution for the provision of warehousing and distribution of Korlym for an initial term of three years that may be extended by mutual agreement. The majority of the costs under this agreement are variable and dependent on the volume of material handled and transactions processed. Either party may terminate this agreement for non-performance upon 30 days written notice.
Manufacturing Agreements Related to Korlym
Active Pharmaceutical Ingredient
We have an agreement with Produits Chimiques Auxiliaires et de Synthese SA (PCAS) for the manufacture of the API in Korlym, for our development and commercial needs that expires on March 31, 2014. We intend to continue the relationship and are in the process of negotiations for a new agreement. The current agreement calls for us to purchase from PCAS at least 75 percent of our requirements until the termination of the agreement. If PCAS is unable to manufacture the product for a consecutive six-month period, we have the right to terminate the agreement without penalty.
Tablet Manufacture
We have one tablet manufacturer for Korlym with an operational facility – AAI Pharma, which was approved by the FDA in November 2012 for the manufacture of our commercial tablets, subject to the successful manufacture of validation batches. We plan to undertake this validation process at AAI early in 2014 and are currently in negotiations for a long-term commercial manufacturing agreement with AAI.
Our original tablet manufacturer, Formex LLC (formerly PharmaForm, L.L.C. and referred to herein as PharmaForm), has temporarily suspended manufacturing operations for relocation to a new facility. We have not taken steps to qualify PharmaForm's new facility for the manufacture of Korlym. Our agreement with PharmaForm was executed in March 2012 for an initial period of two years. The agreement will be automatically extended for additional one year periods unless one Party gives six months prior written notice that it does not want such an extension. The agreement with PharmaForm may be terminated by either party upon 180 days written notice; we may terminate projects initiated under this agreement with 30 days written notice. There are no minimum purchase amounts under this agreement.
See the discussion above in Note 1, Basis of Presentation and Summary of Significant Accounting Policies - Credit Risks and Concentrations, for a further discussion of the business risks and mitigation measures taken in regard to tablet manufacture.
Research and Development Agreements
In 1998, we entered into an agreement with The Board of Trustees of Leland Stanford Junior University (Stanford) in which Stanford granted us an exclusive option to acquire an exclusive license for inventions and patents related to "Mifepristone for Psychotic Major Depression" and "Mifepristone and Alzheimer's Disease"
F-13
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO FINANCIAL STATEMENTS, Continued
owned by Stanford. ("Psychotic major depression" is referred to in this document as "psychotic depression.") In 1999, we exercised our option to acquire an exclusive license to patents covering the use of glucocorticoid receptor antagonists for the treatment of psychotic depression, early dementia, and cocaine-induced psychosis, as specified in the license agreement. This license agreement expires upon the expiration of the related patents or upon notification by us to Stanford. In exchange for the license, we paid Stanford an initial non-refundable fee, immediately issued 30,000 shares of our common stock to Stanford and are obligated to pay Stanford $50,000 per year as a nonrefundable royalty payment. In addition, we are obligated to pay additional milestone payments in the future, which are not material and which are creditable against future royalties and will pay a royalty based on net revenue generated by any product arising from the patent until its expiration.
In 2003, we entered into a contract research agreement with Argenta Discovery Limited (Argenta) in which Argenta agreed to conduct research toward identifying a novel small molecule glucocorticoid receptor antagonist for the treatment of psychotic depression, Alzheimer's disease, and other metabolic and psychiatric disorders. We continued our relationship with Argenta through the end of 2011, requesting them to conduct research projects on a regular basis. Under the agreements with Argenta, we may be obligated to make milestone payments upon the occurrence of certain events, the amounts of which are not material. These obligations remain in force after the conclusion of work under the agreement. In January 2012, we entered into a Master Services Agreement with Sygnature Discovery Limited, a contract research company located in the United Kingdom, which does not obligate us to any milestone payments.
Through 2013, we entered into agreements for services in connection with our ongoing Phase 3 trial of psychotic depression with ICON Clinical Research, L.P. (ICON) and MedAvante, Inc. (MedAvante) to manage the trial and conduct patient screening and evaluation services, which have been amended from time to time. The total commitment under these agreements, including amendments through December 31, 2013, is estimated to be $19.9 million over the course of the trial. We expensed $17.1 million of these costs through December 31, 2013, with the remainder to be incurred over the course of the trial. Under the master services agreements with these vendors, the project contracts may be terminated upon thirty to sixty days notice. If terminated early, we would be responsible for the costs incurred by the vendors through the effective date of termination plus cancellation charges as stipulated in the agreements.
In November 2013, we licensed from the University of Chicago exclusive rights to certain claims contained in the University's U.S. Patent Application No. 13/071,363 "Methods and Compositions Related to Glucocorticoid Receptor Antagonists and Breast Cancer". In exchange for the license, we paid an initial non-refundable fee to the University of Chicago and are committed to additional annual and milestone payments in the future, which are not material and which are creditable against future royalties and will pay a royalty based on net revenue generated by any product arising from the patent until its expiration.
In December 2013, we entered into an agreement with Ockham Development Group Inc. to assist in the management and conduct of a clinical trial evaluating mifepristone for treatment of triple-negative breast cancer. The total commitment under this agreement is $2.9 million, but the actual amount to be paid is dependent on actual services provided under this agreement. Approximately $18,000 of the costs under this agreement were incurred during 2013, with the remainder to be incurred over the course of the trial.
3. Fair Value of Financial Instruments
As of December 31, 2013 and 2012, we had invested our financial assets in a money market fund that can be converted to cash at par on demand. We measured these funds, which totaled $52.9 million and $92.5 million as of December 31, 2013 and 2012, respectively, at fair value, which approximates cost, as of the respective dates and classified them as Level 1 assets in the fair value hierarchy for financial assets.
F-14
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO FINANCIAL STATEMENTS, Continued
All cash equivalents and short-term investments held as of December 31, 2013 and 2012 were in active markets and valued based upon their quoted prices.
4. Financial Instruments
The following tables present a summary of cash and cash equivalents.
| December 31, 2013 | Cost | Unrealized Gain | Unrealized Loss | Fair Value | ||||||||||||
| (in thousands) | ||||||||||||||||
Cash | $ | 2,023 | $ | - | $ | - | $ | 2,023 | ||||||||
Money market fund | 52,854 | - | - | 52,854 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
| $ | 54,877 | $ | - | $ | - | $ | 54,877 | |||||||||
|
|
|
|
|
|
|
| |||||||||
Reported as: | ||||||||||||||||
Cash and cash equivalents | $ | 54,877 | $ | - | $ | - | $ | 54,877 | ||||||||
|
|
|
|
|
|
|
| |||||||||
| December 31, 2012 | Cost | Unrealized Gain | Unrealized Loss | Fair Value | ||||||||||||
| (in thousands) | ||||||||||||||||
Cash | $ | 579 | $ | - | $ | - | $ | 579 | ||||||||
Money market fund | 92,453 | - | - | 92,453 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
| $ | 93,032 | $ | - | $ | - | $ | 93,032 | |||||||||
|
|
|
|
|
|
|
| |||||||||
Reported as: | ||||||||||||||||
Cash and cash equivalents | $ | 93,032 | $ | - | $ | - | $ | 93,032 | ||||||||
|
|
|
|
|
|
|
| |||||||||
As of December 31, 2013 and 2012, all cash and cash equivalents were classified as available-for-sale securities. We did not invest in mortgage-backed securities or auction rate securities at any time during 2013 or 2012. We did not recognize any realized gains or losses on sales of available-for-sale investments for any period presented.
5. Composition of Certain Balance Sheet Items
Inventory
The composition of inventory was as follows:
| December 31, 2013 | December 31, 2012 | |||||||
| (in thousands) | ||||||||
Raw materials | $ | 4,318 | $ | 3,478 | ||||
Work in progress | 2 | 1,165 | ||||||
Finished goods | 1,226 | 20 | ||||||
|
|
|
| |||||
Total inventory | 5,546 | 4,663 | ||||||
Less strategic inventory classified as non-current | (4,450) | (3,810) | ||||||
|
|
|
| |||||
Total inventory classified as current | $ | 1,096 | $ | 853 | ||||
|
|
|
| |||||
The finished goods inventory as of December 31, 2013 includes all costs of manufacture and packaging with the exception of the cost of raw materials that were expensed prior to FDA approval. The finished goods
F-15
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO FINANCIAL STATEMENTS, Continued
inventory as of December 31, 2012 consists of tablets that were manufactured prior to FDA approval. The inventory value for this material includes only the costs of bottling, packaging and labeling as the costs of raw materials and tablet manufacture were expensed prior to approval.
In order to be prepared for potential demand for Korlym and because we have single-source manufacturers of both the API for Korlym and Korlym tablets, we have invested in inventory of both of these materials. Inventory amounts that are not expected to be consumed within twelve months following the balance sheet date are referred to as "Strategic Inventory" and classified as a noncurrent asset.
Property and Equipment
Property and equipment consisted of the following:
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| (in thousands) | ||||||||
Furniture and equipment | $ | 183 | $ | 157 | ||||
Vehicles | 38 | 41 | ||||||
Software | 116 | 15 | ||||||
Leasehold improvements | 14 | 14 | ||||||
|
|
|
| |||||
| 351 | 227 | |||||||
Less: accumulated depreciation | (148 | ) | (77 | ) | ||||
|
|
|
| |||||
| $ | 203 | $ | 150 | |||||
|
|
|
| |||||
Other Accrued Liabilities
Other accrued liabilities consisted of the following:
| December 31, | ||||||||
2013 | 2012 | |||||||
| (in thousands) | ||||||||
Accrued vacation | $ | 466 | $ | 351 | ||||
Professional fees | 369 | 311 | ||||||
Commercialization costs | 288 | 159 | ||||||
Government rebates | 40 | 78 | ||||||
Legal fees | 110 | 31 | ||||||
Other | 28 | 116 | ||||||
|
|
|
| |||||
| $ | 1,301 | $ | 1,046 | |||||
|
|
|
| |||||
6. Long-Term Obligation
As discussed in Note 1, Basis of Presentation and Summary of Significant Accounting Policies - Long-term Obligation , in August 2012, we entered into a Financing Agreement with Biopharma under which we received $30.0 million from Biopharma. In return, we are obligated to make payments, calculated as a percentage of our net sales of Korlym, any future mifepristone-based products, our selective GR-II antagonists (together referred to as Covered Products) and any upfront, milestone or other contingent payments with respect to
F-16
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO FINANCIAL STATEMENTS, Continued
Covered Products. Biopharma's right to receive payments will expire once it has received cumulative payments of $45.0 million. During the year ended December 31, 2013, we made aggregate payments to Biopharma in the amount of $1.0 million, with an additional payment in the amount of $995,000 made in February 2014.
Under the terms of the Financing Agreement, our payments are entirely variable, with no fixed minimums. If there are no net sales, upfront, milestone or other contingent payments in a period with respect to Covered Products, then no payment will be due for that period.
We are obligated to make payments as follows:
| • | 20 percent of our net product sales of Covered Products, beginning with the calendar quarter ended June 30, 2013, subject to quarterly payment caps of $3.0 million during 2014, and $3.75 million during 2015. There is no quarterly cap on payments with respect to net product sales in 2016 and later. |
| • | 20 percent of payments received for upfront, milestone or other contingent fees under co-promotion and out-license agreements for Covered Products (without application of quarterly caps). |
| • | The percentage used to calculate our payments to Biopharma would increase to 50 percent and any applicable payment caps would lapse if we (i) fail to provide Biopharma with certain information regarding our promotion and sales of Covered Products, (ii) do not devote a commercially reasonable amount of resources to the promotion and marketing of the Covered Products or (iii) violate the indebtedness covenant by incurring indebtedness greater than the sum of earnings before interest, taxes, depreciation and amortization, including such items as non-cash stock-based compensation, (EBITDA) for the four calendar quarters preceding such incurrence and, in each case, fail to cure within the applicable cure period. |
| • | Upon the occurrence of a Corcept change of control transaction or the licensing of Korlym to a third-party for promotion and sale in the United States, the entire $45.0 million, less any amounts already paid by us, would become due. |
To secure our obligations in connection with this Financing Agreement, we granted Biopharma a security interest in our rights in patents, trademarks, trade names, domain names, copyrights, know-how and regulatory approvals related to the Covered Products, all books and records relating to the foregoing and all proceeds of the foregoing (together, the Collateral). If we (i) fail to deliver a royalty payment when due and do not remedy that failure within 30 days, (ii) fail to maintain a first-priority perfected security interest in the Collateral in the United States and do not remedy that failure within five business days of receiving notice of such failure or (iii) become subject to an event of bankruptcy, then Biopharma may attempt to recover up to $45.0 million (after deducting any payments we have already made). In addition, pursuant to this agreement, we are not allowed to pay a dividend or other cash distribution, unless we will have cash and cash equivalents in excess of $50.0 million after such payment.
The cash payment of $30.0 million received from Biopharma was recorded as a long-term obligation at issuance in August 2012. As discussed in Note 1, Basis of Presentation and Summary of Significant Accounting Policies, Long-term Obligation , we make estimates of the timing of payments during the term of this agreement for purposes of calculating the expected rate of return to Biopharma, the accretion of related interest expense and the current portion of our obligation. Interest expense of $4.4 million for the year ended December 31, 2013 and total accreted interest of $6.1 million for the period from August 16, 2012, the date of funding of the Financing Agreement, through December 31, 2013, was calculated based on the internal interest rate to Biopharma that would result from these assumed payment streams. Korlym has only been marketed since April 2012 and the magnitude and timing of Korlym revenue is difficult to predict. Therefore, these estimates and assumptions are subject to significant variability and are likely to change as we gain experience marketing
F-17
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO FINANCIAL STATEMENTS, Continued
Korlym. The timing of payment amounts will be based on actual Korlym Receipts recorded in the financial statements over the term of this agreement and may differ from these estimates. While changes in the timing of Korlym revenue may affect the timing of recognition of interest expense and the split between the current and long-term portions of the obligation at any balance sheet date, the aggregate amount to be repaid to Biopharma is fixed.
The carrying value of the long-term obligation was $35.1 million and $31.7 million as of December 31, 2013 and December 31, 2012, respectively. The long-term obligation, including accreted interest, is presented on the balance sheet in two components; the Long-term obligation - current portion, which equates to the estimated amount due under the agreement to be paid within twelve months following the balance sheet date, and the remaining amount, which is included in Long-term obligation, net of current portion.
The following table provides a summary of the payment obligations under the Financing Agreement as of December 31, 2013 and 2012, utilizing the payment assumptions discussed above.
| December 31, 2013 | December 31, 2012 | |||||||
| (in thousands) | ||||||||
Total repayment obligation | $ | 45,000 | $ | 45,000 | ||||
Less interest to be accreted in future periods | (8,910) | (13,320) | ||||||
Less payments made | (1,025) | - | ||||||
Less current portion | (5,743) | (2,650) | ||||||
|
|
|
| |||||
Long-term obligation, net of current portion | $ | 29,322 | $ | 29,030 | ||||
|
|
|
| |||||
The estimated fair value of the long-term obligation, as measured using Level 3 inputs, approximates the carrying amounts as presented on the balance sheet as of December 31, 2013 and 2012. The estimated fair value was calculated using the income method of valuation. The key assumptions required for the calculation were an estimate of the amount and timing of future product revenues and an estimated cost of capital. Management's estimate of the future product revenues is subject to significant uncertainty due to the fact that Korlym has been available for less than two years and the extended time period associated with the Financing Agreement.
We capitalized $140,000 of issuance costs related to the Financing Agreement, which are being amortized over the estimated term of the obligation, based on the assumptions discussed above. At December 31, 2013, the unamortized issuance costs were $87,000, and are included in other assets on our balance sheet.
7. Lease Obligations
As of June 28, 2013, we exercised our option to extend the lease for our office space through December 2014. At December 31, 2013, the remaining minimum rental payments under this operating lease were $428,000.
Rent expense amounted to $428,000, $360,000 and $285,000, for the years ended December 31, 2013, 2012 and 2011, respectively.
8. Related Party Transactions
See discussion below in Note 9, Preferred Stock and Stockholders' Equity , under the captions Stockholder Notes Receivable and Common Stock, regarding the sale of securities in March 2012 to various investors, including members of our Board of Directors and related entities, and a Note Receivable from one of our officers.
F-18
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO FINANCIAL STATEMENTS, Continued
9. Preferred Stock and Stockholders' Equity
Preferred Stock
The board of directors is authorized, subject to any limitations prescribed by law, without stockholder approval, to issue up to an aggregate of 10,000,000 shares of preferred stock at $0.001 par value in one or more series and to fix the rights, preferences, privileges and restrictions granted to or imposed upon the preferred stock, including voting rights, dividend rights, conversion rights, redemption privileges and liquidation preferences. The rights of the holders of common stock will be subject to the rights of holders of any preferred stock that may be issued in the future.
As of December 31, 2013 and 2012, we had no outstanding shares of preferred stock.
Common Stock
Significant stock transactions
There were no significant transactions relating to the sale and issuance of common stock or the exercise and issuance of warrants during the year ended December 31, 2013. The following paragraphs describe significant transactions relating to the sale and issuance of common stock and the exercise and issuance of warrants during the years ended December 31, 2012 and 2011. Information regarding the issuance of common stock upon the exercise of stock options is discussed below under the caption, Stock Option Plans.
Transactions during 2012
On March 29, 2012, we issued 4.2 million shares of our common stock upon the exercise of warrants that we had issued in a private placement transaction in April 2010 at an exercise price of $2.96 per share and sold new warrants to the same investors to purchase 4.2 million shares of common stock at an exercise price of $4.05 per share. The new warrants are exercisable through March 29, 2015. We generated net proceeds in these transactions of $12.8 million, after the deduction of issuance costs. Venture capital funds, trusts and other entities affiliated with members of our Board of Directors purchased 40 percent of the securities sold in this transaction, with the remainder being purchased by other qualified investors.
On July 6, 2012, we sold 11.0 million shares of our common stock in an underwritten public offering at a price to the public of $4.49 per share, generating net proceeds of $46.1 million after deducting expenses of the offering.
During the year ended December 31, 2012, investors exercised additional warrants for the purchase of our common stock with exercise prices ranging from $1.66 to $2.96 per share. As a result, we issued an aggregate of 216,000 shares of common stock and generated aggregate proceeds of $470,000.
Transactions during 2011
On January 26, 2011, we sold 11.5 million shares of our common stock in an underwritten public offering at a price to the public of $3.90 per share for aggregate net proceeds of $41.8 million after deducting the underwriter's discount and commissions and other expenses of the offering. Longitude Venture Partners, L.P. purchased 750,000 (6.5%) of the shares sold in this transaction. Patrick Enright, who is a member of our board of directors, is a managing member of Longitude Capital Partners, LLC, the general partner of Longitude Venture Partners, L.P.
F-19
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO FINANCIAL STATEMENTS, Continued
On July 13, 2011, we issued 80,991 shares of common stock to an investor upon the exercise of warrants that had been issued in our April 2010 warrant transaction and our March 2008 financing, for an average exercise price of $2.85 per share, receiving aggregate proceeds of $231,000.
Registration Rights related to March 2008 Financing
In March 2008, we sold 8.9 million shares of our common stock and warrants to purchase 4.5 million shares of our common stock in a private placement (the March 2008 Financing). The registration rights agreement covering securities issued in the March 2008 Financing provides that if we do not fulfill certain of our obligations under the registration rights agreement, we will be required to pay liquidated damages to the holders of the shares and warrants. We filed the registration statement covering the resale of the shares sold and shares underlying the warrants sold in this transaction with the Securities and Exchange Commission (SEC) on April 11, 2008, and it was declared effective by the SEC on November 10, 2008. During 2008, we recorded $1.3 million in liquidated damages to other non-operating expense because of the delay in the effectiveness of the registration statement, which represented 5% of the purchase price. No separate contingent obligation has been recorded since that time as no additional liquidated damages have become probable of payment.
No dividends have been declared or paid by us.
Shares of common stock reserved for future issuance as of December 31, 2013 are as follows:
Common stock: | (in thousands) | |||
Exercise of outstanding options | 14,712 | |||
Exercise of warrants | 8,574 | |||
Shares available for grant under stock option plans | 4,926 | |||
|
| |||
| 28,212 | ||||
|
|
On February 6, 2014, our Board of Directors authorized an increase of 4.0 million shares in the number of shares available under the 2012 Incentive Award Plan (the 2012 Plan), which was equivalent to 4% of the shares of our common stock outstanding as of December 31, 2013, pursuant to the terms of the 2012 Plan.
Stock Option Plans
We have three stock option plans – the 2000 Stock Option Plan (the 2000 Plan), the 2004 Equity Incentive Plan (the 2004 Plan) and the 2012 Plan. As of December 31, 2013, all option grants under the 2000 Plan were fully vested and grants covering 40,000 shares remained outstanding with contractual lives expiring in March 2014.
In 2004, our board of directors and stockholders approved the 2004 Plan, which became effective upon the completion of our Initial Public Offering (IPO), after which time, no additional options have been or will be issued under the 2000 Plan. Under the 2004 Plan, options, stock purchase and stock appreciation rights and restricted stock awards can be issued to our employees, officers, directors and consultants. The 2004 Plan provided that the exercise price for incentive stock options will be no less than 100% of the fair value of the Company's common stock, as of the date of grant. Options granted under the 2004 Plan vest over periods ranging from one to five years. The vesting period of the options is generally equivalent to the requisite service period.
In February 2012, our Board of Directors and stockholders approved the 2012 Plan, which became effective upon its approval at our Annual Meeting of Stockholders on June 13, 2012. As of the effective date of the 2012
F-20
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO FINANCIAL STATEMENTS, Continued
Plan, 5.3 million shares that remained available for issuance of new grants under the 2004 Plan were transferred to the 2012 Plan. After that date, no additional options were or will be issued under the 2004 Plan. Vested options under the 2000 Plan and the 2004 Plan that are not exercised within the remaining contractual life and any options under the 2004 Plan that do not vest because of terminations after the effective date of the 2012 Plan will be added to the pool of shares available for future grants under the 2012 Plan.
Under the 2012 Plan, we can issue options, stock purchase and stock appreciation rights and restricted stock awards to our employees, officers, directors and consultants. The 2012 Plan provides that the exercise price for incentive stock options will be no less than 100 percent of the fair value of our common stock, as of the date of grant. Options granted under the 2012 Plan are expected to vest over periods ranging from one to four years. We expect the vesting period of the options that we grant under the 2012 Plan to be generally equivalent to the requisite service period.
Upon exercise of options, new shares are issued.
On January 30, 2013, our Board of Directors authorized an increase of 4.0 million shares in the number of shares available under the 2012 Plan, which was equivalent to 4% of the shares of our common stock outstanding as of December 31, 2012, pursuant to the terms of the 2012 Plan. As of December 31, 2013, 4.9 million shares remained available for future grants under the 2012 Plan. See the discussion above under Common Stock regarding an additional increase to the shares available for grant under the 2012 Plan that was authorized by the Board of Directors in February 2014.
Option activity during 2011, 2012 and 2013
The following table summarizes all stock plan activity:
| Outstanding Options | ||||||||||||||||||||
| Shares Available For Future Grant | Shares Subject to Options Outstanding | Weighted- Average Exercise Price | Weighted Average Remaining Contractual Life | Aggregate Intrinsic Value | ||||||||||||||||
| (in thousands) | (in thousands) | (in years) | (in thousands) | |||||||||||||||||
Balance at December 31, 2010 | 1,949 | 7,961 | $ | 2.40 | ||||||||||||||||
Increase in shares authorized for grant | 2,896 | - | - | |||||||||||||||||
Shares granted | (2,825) | 2,825 | $ | 3.97 | ||||||||||||||||
Shares exercised | - | (246) | $ | 1.51 | ||||||||||||||||
Shares cancelled and forfeited under 2000 Plan | - | (1) | $ | 0.10 | ||||||||||||||||
Shares cancelled and forfeited under 2004 Plan | 231 | (231) | $ | 1.72 | ||||||||||||||||
|
|
|
| |||||||||||||||||
Balance at December 31, 2011 | 2,251 | 10,308 | $ | 2.86 | ||||||||||||||||
Increase in shares authorized for grant | 3,369 | - | - | |||||||||||||||||
Shares granted | (1,695) | 1,695 | $ | 3.26 | ||||||||||||||||
Shares exercised | - | (165) | $ | 1.91 | ||||||||||||||||
Shares expired under 2000 Plan | 11 | (93) | $ | 7.00 | ||||||||||||||||
Shares cancelled and forfeited under 2004 and 2012 Plans | 119 | (119) | $ | 3.26 | ||||||||||||||||
|
|
|
| |||||||||||||||||
Balance at December 31, 2012 | 4,055 | 11,626 | $ | 2.90 | ||||||||||||||||
Increase in shares authorized for grant | 3,992 | - | - | |||||||||||||||||
Shares granted | (3,565) | 3,565 | $ | 1.98 | ||||||||||||||||
Shares exercised | - | (35) | $ | 1.58 | ||||||||||||||||
Shares cancelled and forfeited | 444 | (444) | $ | 4.58 | ||||||||||||||||
|
|
|
| |||||||||||||||||
Balance at December 31, 2013 | 4,926 | 14,712 | $ | 2.63 | 6.5 | $ | 14,257 | |||||||||||||
|
|
|
| |||||||||||||||||
Options exercisable at December 31, 2013 | 10,049 | $ | 2.65 | 5.4 | $ | 10,249 | ||||||||||||||
|
| |||||||||||||||||||
Options fully vested and expected to vest at December 31, 2013 | 14,712 | $ | 2.63 | 6.5 | $ | 14,257 | ||||||||||||||
|
|
|
| |||||||||||||||||
F-21
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO FINANCIAL STATEMENTS, Continued
The total intrinsic value of options exercised during the years ended December 31, 2013, 2012 and 2011 was $55,000, $303,000 and $702,000, respectively, based on the difference between the closing price of our common stock on the date of exercise of the options and the exercise price.
The total grant date fair value of options to employees and directors that vested during the years ended December 31, 2013, 2012 and 2011 was $5.0 million, $5.0 million and $2.8 million, respectively.
The following is a summary of options outstanding and options exercisable at December 31, 2013.
| Options Outstanding | Options Exercisable | |||||||||||||||||||||||||||
Exercise Prices Of Options | Number of Shares | Weighted Average Remaining Contractual Life | Weighted Average Exercise Price | Aggregate Intrinsic Value | Number of Shares | Weighted Average Exercise Price | Aggregate Intrinsic Value | |||||||||||||||||||||
| (in thousands) | (in years) | (in thousands) | (in thousands) | (in thousands) | ||||||||||||||||||||||||
$ 0.96 - $ 1.49 | 2,521 | 5.4 | $ | 1.15 | $ | 5,191 | 2,365 | $ | 1.13 | $ | 4,922 | |||||||||||||||||
$ 1.50 - $ 3.50 | 7,945 | 7.0 | $ | 2.08 | 9,066 | 4,443 | $ | 2.02 | 5,327 | |||||||||||||||||||
$ 3.51 - $ 5.00 | 3,986 | 6.5 | $ | 4.24 | - | 2,981 | $ | 4.24 | - | |||||||||||||||||||
$ 5.01 - $ 14.50 | 260 | 0.7 | $ | 9.15 | - | 260 | $ | 9.15 | - | |||||||||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||||||
| 14,712 | 6.5 | $ | 2.63 | $ | 14,257 | 10,049 | $ | 2.65 | $ | 10,249 | ||||||||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||||||
The aggregate intrinsic value in the table above represents the total pre-tax intrinsic value that option holders would have received had all option holders exercised their options on December 31, 2013. The aggregate intrinsic value is the difference between our closing stock price on December 31, 2013 and the exercise price, multiplied by the number of in-the-money options.
Stock-Based Compensation related to Employee and Director Options
Assumptions used in determining fair value-based measurements for options to employees and directors
The following table summarizes the weighted-average assumptions and resultant fair value-based measurements for options granted to employees and directors.
| Year Ended December 31, | ||||||
| 2013 | 2012 | 2011 | ||||
Weighted-average assumptions for stock options granted: | ||||||
Risk-free interest rate | 1.76% | 1.06% | 2.65% | |||
Expected term | 8.3 years | 6.7 years | 8.9 years | |||
Expected volatility of stock price | 83.9% | 86.6% | 90.0% | |||
Dividend rate | 0% | 0% | 0% | |||
Weighted average grant date fair value-based measurement | $1.54 | $2.41 | $3.29 | |||
The expected term of options reflected in the table above has been based on a formula that considers the expected service period and expected post-vesting termination behavior differentiated by whether the grantee is an employee, an officer or a director.
The expected volatility of our stock used in determining the fair value-based measurement of option grants to employees, officers and directors is based on a weighted-average combination of the volatility of our own stock price and that of a group of peer companies for those grants with expected terms longer than the period of
F-22
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO FINANCIAL STATEMENTS, Continued
time that we have been a public company. For stock options granted to employees with expected terms of less than the period of time that we have been a public company, the volatility is based on historical data of the price for our common stock for periods of time equivalent to the expected term of these grants.
We apply a forfeiture rate of zero in our stock option expense calculations as we have a limited employee base and have experienced minimal turnover. When an employee terminates, we will record a change in accounting estimate that represents the difference between the expense recorded in the financial statements and the expense that would have been recorded based upon the rights to options that vested during the individual's service as an employee.
Summary of compensation expense related to options to employees and directors
We recognized compensation expense of $5.1 million, $5.1 million and $3.0 million, related to options to employees and directors during the years ended December 31, 2013, 2012 and 2011, respectively. The data for the year ended December 31, 2012 include $1.3 million of expense related to performance-based option awards to officers that vested upon the FDA approval of Korlym in February 2012, which is classified as selling, general and administrative expense.
As of December 31, 2013, we had $9.0 million of unrecognized compensation expense for employee and director options outstanding as of that date, which had a remaining weighted-average vesting period of 2.7 years.
Stock Options to Consultants
We expense stock-based compensation related to service-based option grants to non-employees on a straight line basis over the vesting period of the options, which approximates the period over which the related services are rendered, based on the fair value-based measurement of the options using the Black-Scholes option pricing model. The assumptions used in these calculations are similar to those used for the determination of fair value-based measurement for options granted to employees and directors, with the exception that, for non-employee options, the remaining contractual term is utilized as the expected term of the option and the fair value-based measurement related to unvested non-employee options is re-measured quarterly, based on the then current stock price as reflected on the NASDAQ Capital Market. For options with performance-based vesting criteria, we recognize expense based on the minimum number of shares that will vest over time as the criteria are met based on the Black-Scholes valuation of the vested shares.
We recorded charges to expense for stock options granted to consultants of $127,000, $208,000 and $419,000 for the years ended December 31, 2013, 2012 and 2011, respectively.
As of December 31, 2013, all options that had been granted to consultants were fully vested, with the exception of one award with 42,000 shares unvested as of that date.
Summary of Stock-based Compensation Expense
The following table presents a summary of non-cash stock-based compensation by financial statement classification.
| Year ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| (in thousands) | ||||||||||||
Research and development expense | $ | 618 | $ | 546 | $ | 547 | ||||||
Selling, general and administrative expense | 4,578 | 4,764 | 2,888 | |||||||||
|
|
|
|
|
| |||||||
Total | $ | 5,196 | $ | 5,310 | $ | 3,435 | ||||||
|
|
|
|
|
| |||||||
F-23
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO FINANCIAL STATEMENTS, Continued
Stockholder Notes Receivable
In 2001, we recorded notes receivable from stockholders in the aggregate amount of $438,165 in connection with the exercise of options issued under the 2000 Plan to purchase 585,000 shares of common stock. The notes were secured by the related shares of common stock and were full recourse notes, with interest compounded annually at the rate of 6.5% per year. As of December 31, 2011, all amounts of principal and interest related to these notes had been paid.
Warrants
Outstanding warrants at December 31, 2013 were as follows:
Number of shares | Exercise Price | Expiration Date | ||||||||
| (in thousands) | ||||||||||
March 2008 Financing | 4,372 | $ | 2.77 | 3/25/15 | ||||||
March 2012 Warrant Exchange | 4,202 | $ | 4.05 | 3/29/15 | ||||||
|
| |||||||||
Total warrants outstanding | 8,574 | |||||||||
|
| |||||||||
10. Net Loss Per Share
Basic and diluted net loss per share is computed by dividing the net loss by the weighted-average number of common shares outstanding during the period. The computation of net loss per share for each period, including the number of weighted-average shares outstanding, is shown on the face of the statements of comprehensive loss.
We have excluded the impact of common stock equivalents relating to shares underlying outstanding stock option grants and warrants from the calculation of diluted net loss per common share because all such securities are antidilutive for all periods presented.
The following table presents information on securities outstanding as of the end of each period that could potentially dilute the per share data in the future.
December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| (in thousands) | ||||||||||||
Stock options outstanding | 14,712 | 11,626 | 10,308 | |||||||||
Warrants outstanding | 8,574 | 8,904 | 9,119 | |||||||||
|
|
|
|
|
| |||||||
Total | 23,286 | 20,530 | 19,427 | |||||||||
|
|
|
|
|
| |||||||
F-24
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO FINANCIAL STATEMENTS, Continued
11. Income Taxes
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of our deferred tax assets are as follows:
| December 31, | ||||||||
| 2013 | 2012 | |||||||
Deferred tax assets: | (in thousands) | |||||||
Federal and state net operating losses | $ | 55,955 | $ | 45,954 | ||||
Capitalized research and patent costs | 23,395 | 22,442 | ||||||
Research credits | 21,252 | 19,236 | ||||||
Biopharma Financing Agreement | 13,021 | 12,108 | ||||||
Stock-based compensation costs | 5,398 | 4,320 | ||||||
Other | 1,148 | 189 | ||||||
|
|
|
| |||||
Total deferred tax assets | 120,169 | 104,249 | ||||||
Valuation allowance | (120,169 | ) | (104,249 | ) | ||||
|
|
|
| |||||
Net deferred tax assets | $ | - | $ | - | ||||
|
|
|
| |||||
Realization of deferred tax assets is dependent upon future earnings, if any, the timing and amount of which are uncertain. Accordingly, the net deferred tax assets have been fully offset by a valuation allowance. The valuation allowance increased by $15.9 million, $14.0 million and $16.0 million, respectively, for the years ended December 31, 2013, 2012 and 2011, respectively.
At December 31, 2013 we had net operating loss carryforwards available to offset any future taxable income that we may generate for federal income tax purposes of $145.4 million, which expire in the years 2019 through 2033, and California net operating loss carryforwards of $111.7 million, which expire in the years 2014 through 2033. We also had federal and California research and development tax credits of $19.7 million and $2.3 million, respectively. The federal research credits will expire in the years 2019 through 2033 and the California research credits have no expiration date. Our deferred tax assets have been offset by a full valuation allowance as the realization of such assets is uncertain. Utilization of our net operating losses and tax credit carryforwards may be subject to substantial annual limitation due to the ownership change limitations provided by the Internal Revenue Code and similar state provisions. Such limitations could result in the expiration of the net operating losses and tax credit carryforwards before utilization.
All tax years from inception remain open to examination by the Internal Revenue Service and the California Franchise Tax Board until such time as the net operating losses and research credits are either fully utilized or expire.
The following table presents a reconciliation from the statutory federal income tax rate to the effective rate.
| Year ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| (in thousands) | ||||||||||||
U.S. federal taxes (benefit) at statutory rate | $ | (15,644 | ) | $ | (12,936 | ) | $ | (11,000 | ) | |||
Unutilized, net operating loss | 14,666 | 11,663 | 8,588 | |||||||||
Non-deductible offset of Orphan Drug Credit | 383 | 827 | 2,119 | |||||||||
Non-deductible stock based compensation | 567 | 404 | 280 | |||||||||
Other | 28 | 42 | 13 | |||||||||
|
|
|
|
|
| |||||||
Total | $ | - | $ | - | $ | - | ||||||
|
|
|
|
|
| |||||||
F-25
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO FINANCIAL STATEMENTS, Continued
12. Commitments
We have entered into a number of agreements to conduct clinical trials and pre-clinical studies for further development of our lead product, Korlym, and our proprietary, selective GR-II antagonists. See the discussion in Note 2, Significant Agreements , for further discussion regarding the commitments under these agreements.
In the ordinary course of our business, we make certain indemnities, commitments and guarantees under which we may be required to make payments in relation to certain transactions. These include indemnities of clinical investigators and contract research organizations involved in the development of our clinical stage product candidates, indemnities of contract manufacturers and indemnities to our directors and officers to the maximum extent permitted under the laws of the State of Delaware. The duration of these indemnities, commitments and guarantees varies, and in certain cases, is indefinite. The majority of these indemnities, commitments and guarantees do not provide for any limitation of the maximum potential future payments that we could be obligated to make. We have not recorded any liability for these indemnities, commitments and guarantees in the accompanying balance sheets. However, we would accrue for losses for any known contingent liability, including those that may arise from indemnification provisions, when future payment is probable . No such losses have been recorded to date.
13. Subsequent Events
On February 6, 2014, our Board of Directors approved discretionary, one-time cash bonus payments to our officers and employees in a aggregate amount of $3.2 million, plus related payroll tax expense. The expense related to these bonuses will be recorded in the quarter ending March 31, 2014 in the same financial statement classifications as other compensation expense for these individuals.
14. Quarterly Financial Data (Unaudited)
The following table is in thousands, except per share amounts:
| Quarter Ended | March 31 | June 30 | September 30 | December 31 | ||||||||||||
2013 | ||||||||||||||||
Product sales, net | $ | 1,717 | $ | 1,891 | $ | 2,634 | $ | 4,115 | ||||||||
Gross profit on product sales | 1,697 | 1,868 | 2,594 | 4,055 | ||||||||||||
Net loss | (12,084 | ) | (11,897 | ) | (10,906 | ) | (11,124 | ) | ||||||||
Basic and diluted net loss per share | (0.12 | ) | (0.12 | ) | (0.11 | ) | (0.11 | ) | ||||||||
2012 | ||||||||||||||||
Product sales, net | $ | - | $ | 875 | $ | 1,055 | $ | 1,377 | ||||||||
Gross profit on product sales | - | 827 | 1,031 | 1,358 | ||||||||||||
Net loss | (11,034 | ) | (7,597 | ) | (8,293 | ) | (11,124 | ) | ||||||||
Basic and diluted net loss per share | (0.13 | ) | (0.09 | ) | (0.08 | ) | (0.11 | ) | ||||||||
The table above does not reflect data for product sales and gross profit for the quarter ended March 31, 2012 as we began commercial sales of our first product in April 2012.
F-26
Exhibit Index
Exhibit Number | Description of Document | |
| 3.1 | Amended and Restated Certificate of Incorporation, as amended (incorporated by reference to Exhibit 3.1 to the registrant's Quarterly Report on Form 10-Q filed on August 9. 2012). | |
| 3.2 | Amended and Restated Bylaws (incorporated by reference to Exhibit 3.1 to the registrant's Current Report on Form 8-K filed on September 27, 2007). | |
| 4.1 | Specimen Common Stock Certificate (incorporated by reference to Exhibit 4.1 to the registrant's Registration Statement on Form S-1 (Registration No. 333-112676) filed on February 10, 2004). | |
| 4.2 | Registration Rights Agreement by and among Corcept Therapeutics Incorporated and the investors signatory thereto, dated March 14, 2008 (incorporated by reference to Exhibit 10.25 to the registrant's Annual Report on Form 10-K filed on March 31, 2008). | |
| 4.3 | Amendment to Registration Rights Agreement by and among Corcept Therapeutics Incorporated and the investors signatory thereto, dated November 11, 2008 (incorporated by reference to Exhibit 10.30 to the registrant's Annual Report on Form 10-K filed on March 31, 2009). | |
| 4.4 | Registration Rights Agreement dated as of April 21, 2010 by and among Corcept Therapeutics Incorporated and the investors signatory thereto (incorporated by reference to Exhibit 4.2 to the registrant's Current Report on Form 8-K filed on April 23, 2010). | |
| 4.5 | Registration Rights Agreement, dated as of March 29, 2012, by and among Corcept Therapeutics Incorporated and the investors signatory thereto (incorporated by reference to Exhibit 4.2 to the registrant's Current Report on Form 8-K filed on March 29, 2012). | |
| 4.6 | Form of Warrant issued in connection with the Securities Purchase Agreement by and among Corcept Therapeutics Incorporated and the purchasers named therein, dated March 14, 2008 (incorporated by reference to Exhibit 4.4 to the registrant's Annual Report on Form 10-K filed on March 31, 2008). | |
| 4.7 | Form of Warrant issued in connection with Warrant Purchase Agreement dated as of March 25, 2012 by and among Corcept Therapeutics Incorporated and the purchasers named therein (incorporated by reference to Exhibit 4.2 to the registrant's Current Report on Form 8-K filed on March 29. 2012). | |
| 10.1 † | 2000 Stock Option Plan (incorporated by reference to Exhibit 10.1 to the registrant's Registration Statement on Form S-1 (Registration No. 333-112676) filed on February 10, 2004). | |
| 10.2 | License Agreement by and between The Board of Trustees of the Leland Stanford Junior University and Corcept Therapeutics Incorporated, dated as of July 1, 1999 (incorporated by reference to Exhibit 10.6 to the registrant's Registration Statement on Form S-1 (Registration No. 333-112676) filed on February 10, 2004). | |
| 10.3 | Master Services Agreement by and between Corcept Therapeutics Incorporated and PPD Development, LP, dated as of January 17, 2003 (incorporated by reference to Exhibit 10.12 to the registrant's Registration Statement on Form S-1/A (File No. 333-112676) filed on March 19, 2004). | |
| 10.4# | Manufacturing Agreement with Produits Chimiques Auxiliaires et de Synthese SA, dated November 8, 2006 (incorporated by reference to Exhibit 10.15 to the registrant's Annual Report on Form 10-K filed on April 2, 2007). | |
| 10.5 † | Form of Indemnification Agreement for directors and officers approved by the Board of Directors on September 24, 2007 (incorporated by reference to Exhibit 10.7 to the registrant's Quarterly Report on Form 10-Q filed on November 14, 2007). | |
| 10.6 | Securities Purchase Agreement by and among Corcept Therapeutics Incorporated and the purchasers named therein, dated March 14, 2008 (incorporated by reference to Exhibit 10.24 to the registrant's Annual Report on Form 10-K filed on March 31, 2008). | |
| 10.7# | Master Service Agreement by and among Corcept Therapeutics Incorporated and ICON Clinical Research, L.P., signed on June 4, 2008 (incorporated by reference to Exhibit 10.5 to the registrant's Quarterly Report on Form 10-Q filed on August 14, 2008). | |
| 10.8 † | Amended and Restated Severance and Change in Control Agreement by and between Corcept Therapeutics Incorporated and Joseph K. Belanoff, M. D., dated September 19, 2008 (incorporated by reference to Exhibit 10.25 to the registrant's Annual Report on Form 10-K filed on March 31, 2009). | |
Exhibit Number | Description of Document | |
| 10.9 † | Amended and Restated Severance and Change in Control Agreement by and between Corcept Therapeutics Incorporated and Robert L. Roe, M. D., dated September 19, 2008 (incorporated by reference to Exhibit 10.26 to the registrant's Annual Report on Form 10-K filed on March 31, 2009). | |
| 10.10 † | Amended and Restated Severance and Change in Control Agreement by and between Corcept Therapeutics Incorporated and Anne M. LeDoux, dated September 19, 2008 (incorporated by reference to Exhibit 10.27 to the registrant's Annual Report on Form 10-K filed on March 31, 2009). | |
| 10.11 † | Amended and Restated Severance and Change in Control Agreement by and between Corcept Therapeutics Incorporated and James N. Wilson, dated September 19, 2008 (incorporated by reference to Exhibit 10.28 to the registrant's Annual Report on Form 10-K filed on March 31, 2009). | |
| 10.12 | Securities Purchase Agreement by and among Corcept Therapeutics Incorporated and the purchasers named therein, dated October 12, 2009 (incorporated by reference to Exhibit 10.1 to the registrant's Quarterly Report on Form 10-Q filed on November 12, 2009). | |
| 10.13 † | Amended and Restated 2004 Equity Incentive Plan (incorporated by reference to the registrant's Proxy Statement on Schedule 14A filed on May 7, 2009). | |
| 10.14 † | Form of Option Agreement for options granted pursuant to the Amended and Restated 2004 Equity Incentive Plan (incorporated by reference to Exhibit 10.25 to the registrant's Annual Report on Form 10-K filed on March 15, 2011). | |
| 10.15 # | Development Agreement by and between Corcept Therapeutics Incorporated and Formulation Technologies L.L.C. d/b/a PharmaForm, dated as of December 14, 2006 (incorporated by reference to Exhibit 10.28 to the registrant's Annual Report on Form 10-K filed on March 15, 2011). | |
| 10.16 # | Master Services Agreement by and between Corcept Therapeutics Incorporated and United BioSource Corporation, dated as of June 29, 2010 (incorporated by reference to Exhibit 10.29 to the registrant's Annual Report on Form 10-K filed on March 15, 2011). | |
| 10.17 † | Employment offer letter to Steven Lo, dated August 9, 2010 (incorporated by reference to Exhibit 10.1 to the registrant's Quarterly Report on Form 10-Q filed on November 12, 2010). | |
| 10.18 † | Severance and Change in Control Agreement by and between Corcept Therapeutics Incorporated and Steven Lo, dated September 15, 2010 (incorporated by reference to Exhibit 10.2 to the registrant's Quarterly Report on Form 10-Q filed on November 12, 2010). | |
| 10.19 † | Severance and Change in Control Agreement by and between Corcept Therapeutics Incorporated and G. Charles Robb, dated September 1, 2011 (incorporated by reference to Exhibit 10.2 to the registrant's Quarterly Report on Form 10-Q filed on November 8, 2011). | |
| 10.20 † | Employment offer letter to G. Charles Robb dated August 12, 2011 (incorporated by reference to Exhibit 10.1 to the registrant's Quarterly Report on Form 10-Q filed on November 8, 2011). | |
| 10.21 # | Manufacturing and Supply Agreement with Formulation Technologies, LLC D/B/A PharmaForm, LLC, dated March 21, 2012 (incorporated by reference to Exhibit 10.1 to the registrant's Quarterly Report on Form 10-Q filed on May 10, 2012). | |
| 10.22 | Warrant Purchase Agreement, dated as of March 25, 2012, by and among Corcept Therapeutics Incorporated and the purchasers named therein (incorporated by reference to Exhibit 10.1 to the registrant's Current Report on Form 8-K filed on March 29, 2012). | |
| 10.23 # | Commercial Outsourcing Services Agreement with Integrated Commercialization Solutions, Inc., dated as of April 14, 2011 (incorporated by reference to Exhibit 10.1 to the registrant's Quarterly Report on Form 10-Q filed on August 9, 2012). | |
| 10.24 # | Amended and Restated Exclusive Pharmacy Product Purchase and Services Agreement with CuraScript, Inc., dated August 8, 2012 (incorporated by reference to Exhibit 10.2 to the registrant's Quarterly Report on Form 10-Q filed on August 9, 2012). | |
| 10.25 † | Corcept Therapeutics Incorporated 2012 Incentive Award Plan (incorporated by reference to Appendix A to the registrant's Definitive Proxy Statement on Schedule 14A filed with the SEC on May 21, 2012). | |
| 10.26 † | Form of 2012 Incentive Award Plan Stock Option Grant Notice and Agreement (incorporated by reference to Exhibit 4.5 to the registrant's Registration Statement on Form S-8 filed with the SEC on August 13, 2012). | |
Exhibit Number | Description of Document | |
| 10.27 # | Purchase and Sale Agreement with between Corcept Therapeutics Incorporated and Biopharma Secured Debt Fund II Sub, S.à r.l,, dated as of August 2, 2012 (incorporated by reference to Exhibit 10.4 to the registrant's Quarterly Report on Form 10-Q filed on November 8, 2012). | |
| 10.28 | Amendment to Manufacturing Agreement with Produits Chimiques Auxiliaires et de Synthese SA, dated February 21, 2013 (incorporated by reference to Exhibit 10.31 to the registrant's Annual Report on Form 10-K filed on March 15, 2013). | |
| 10.29 # | Pharmaceutical Manufacturer Services Agreement with Centric Health Resources, Inc., dated May 21, 2013 (incorporated by reference to Exhibit 10.1 to the registrant's Quarterly Report on Form 10-Q filed on August 9, 2013). | |
| 10.30 † | Letter agreement with Robert L. Roe, M.D. regarding terms of retirement and consulting arrangement, dated June 21, 2013 (incorporated by reference to Exhibit 10.2 to the registrant's Quarterly Report on Form 10-Q filed on August 9, 2013). | |
| 10.31 # | Amendment to Pharmaceutical Manufacturer Services Agreement with Centric Health Resources, Inc., dated July 22, 2013 (incorporated by reference to Exhibit 10.3 to the registrant's Quarterly Report on Form 10-Q filed on August 9, 2013). | |
| 10.32 | Amendment to Manufacturing Agreement with Produits Chimiques Auxiliaires et de Synthese SA, dated August 1, 2013 (incorporated by reference to Exhibit 10.4 to the registrant's Quarterly Report on Form 10-Q filed on August 9, 2013). | |
| 10.33 | Amendment to Manufacturing Agreement with Produits Chimiques Auxiliaires et de Synthese SA, dated November 7, 2013 (incorporated by reference to Exhibit 10.1 to the registrant's Quarterly Report on Form 10-Q filed on November 12, 2013). | |
| 10.34 | Amendment to Manufacturing Agreement with Produits Chimiques Auxiliaires et de Synthese SA, dated January 27, 2014. | |
| 23.1 | Consent of Independent Registered Public Accounting Firm | |
| 24.1 | Power of Attorney (See signature page) | |
| 31.1 | Certification pursuant to Rule 13a-14(a) under the Securities Exchange Act of 1934 of Joseph K. Belanoff, M.D. | |
| 31.2 | Certification pursuant to Rule 13a-14(a) under the Securities Exchange Act of 1934 of G. Charles Robb | |
| 32.1 | Certification pursuant to 18 U.S.C. Section 1350 of Joseph K. Belanoff, M.D. | |
| 32.2 | Certification pursuant to 18 U.S.C. Section 1350 of G. Charles Robb | |
| 101 | The following materials from the registrant's Annual Report on Form 10-K for the year ended December 31, 2013, formatted in Extensible Business Reporting Language (XBRL): (i) Balance Sheets at December 31, 2013 and 2012, (ii) Statements of Comprehensive Loss for the Years Ended December 31, 2013, 2012 and 2011, (iii) Statements of Stockholders' Equity for the Years Ended December 31, 2013, 2012 and 2011, (iv) Statements of Cash Flows for the Years Ended December 31, 2013, 2012 and 2011, and (v) Notes to Financial Statements. | |
| # | Confidential treatment granted |
| † | Management contract or compensatory plan or arrangement |