UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark one)
| ☑ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2009
or
| o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number 0-16132
CELGENE CORPORATION
(Exact name of registrant as specified in its charter)
| Delaware | 22-2711928 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | |
| 86 Morris Avenue Summit, New Jersey | 07901 | |
| (Address of principal executive offices) | (Zip Code) |
(Registrant's telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Name of each exchange on which registered | |
| Common Stock, par value $.01 per share | NASDAQ Global Select Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes ☑ No o
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
Yes o No ☑
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes ☑ No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Yes ☑ No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ☑ | Accelerated filer o | Non-accelerated filer o | Smaller reporting company o | |||
| (Do not check if a smaller reporting company) |
Indicate by check mark whether the registrant is a shell company (as defined in Rule12b-2 of the Act).
Yes o No ☑
The aggregate market value of voting stock held by non-affiliates of the registrant on June 30, 2009, the last business day of the registrant's most recently completed second quarter was $21,935,672,339 based on the last reported sale price of the registrant's Common Stock on the NASDAQ Global Select Market on that date.
There were 459,730,918 shares of Common Stock outstanding as of February 11, 2010.
Documents Incorporated by Reference
The registrant intends to file a definitive proxy statement pursuant to Regulation 14A within 120 days of the end of the fiscal year ended December 31, 2009. The proxy statement is incorporated herein by reference into the following parts of the Form 10-K:
Part II, Item 5, Equity Compensation Plan Information
Part III, Item 10, Directors, Executive Officers and Corporate Governance;
Part III, Item 11, Executive Compensation;
Part III, Item 12, Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters;
Part III, Item 13, Certain Relationships and Related Transactions, and Director Independence;
Part III, Item 14, Principal Accountant Fees and Services.
CELGENE CORPORATION
ANNUAL REPORT ON FORM 10-K
TABLE OF CONTENTS
| Item No. | Page | |||||||
| ||||||||
Part I | ||||||||
| ||||||||
1. Business | 1 | |||||||
| ||||||||
1A. Risk Factors | 21 | |||||||
| ||||||||
1B. Unresolved Staff Comments | 34 | |||||||
| ||||||||
2. Properties | 34 | |||||||
| ||||||||
3. Legal Proceedings | 35 | |||||||
| ||||||||
4. Submission of Matters to a Vote of Security Holders | 39 | |||||||
| ||||||||
Part II | ||||||||
| ||||||||
5. Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | 40 | |||||||
| ||||||||
6. Selected Financial Data | 42 | |||||||
| ||||||||
7. Management's Discussion and Analysis of Financial Condition and Results of Operations | 43 | |||||||
| ||||||||
7A. Quantitative and Qualitative Disclosures About Market Risk | 65 | |||||||
| ||||||||
8. Financial Statements and Supplementary Data | 68 | |||||||
| ||||||||
9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure | 118 | |||||||
| ||||||||
9A. Controls and Procedures | 118 | |||||||
| ||||||||
9B. Other Information | 121 | |||||||
| ||||||||
Part III | ||||||||
| ||||||||
10. Directors, Executive Officers and Corporate Governance | 121 | |||||||
| ||||||||
11. Executive Compensation | 121 | |||||||
| ||||||||
12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 121 | |||||||
| ||||||||
13. Certain Relationships and Related Transactions, and Director Independence | 121 | |||||||
| ||||||||
14. Principal Accountant Fees and Services | 121 | |||||||
| ||||||||
Part IV | ||||||||
| ||||||||
15. Exhibits, Financial Statement Schedules | 122 | |||||||
| ||||||||
Signatures and Power of Attorney | 128 | |||||||
| ||||||||
| Exhibit 3.2 | ||||||||
| Exhibit 21.1 | ||||||||
| Exhibit 23.1 | ||||||||
| Exhibit 31.1 | ||||||||
| Exhibit 31.2 | ||||||||
| Exhibit 32.1 | ||||||||
| Exhibit 32.2 | ||||||||
| EX-101 INSTANCE DOCUMENT | ||||||||
| EX-101 SCHEMA DOCUMENT | ||||||||
| EX-101 CALCULATION LINKBASE DOCUMENT | ||||||||
| EX-101 LABELS LINKBASE DOCUMENT | ||||||||
| EX-101 PRESENTATION LINKBASE DOCUMENT | ||||||||
| EX-101 DEFINITION LINKBASE DOCUMENT | ||||||||
PART I
ITEM 1. BUSINESS
Celgene Corporation and its subsidiaries (collectively "we" or "our") is a global integrated biopharmaceutical company primarily engaged in the discovery, development and commercialization of innovative therapies designed to treat cancer and immune-inflammatory related diseases. We are dedicated to innovative research and development which is designed to bring new therapies to market and are involved in research in several scientific areas that may deliver proprietary next-generation therapies, targeting areas such as intracellular signaling pathways in cancer and immune cells, immunomodulation in cancer and autoimmunity and placental cell, including stem and progenitor cell, research. The drug and cell therapies we develop are designed to treat life-threatening diseases or chronic debilitating conditions. Building on our growing knowledge of the biology underlying hematological and solid tumor cancers as well as in immune-inflammatory diseases, we are investing in a range of innovative therapeutic programs that are investigating ways to treat and manage chronic diseases by targeting the disease source through multiple mechanisms of action.
Our commercial stage products include REVLIMID ® , THALOMID ® (inclusive of Thalidomide Celgene TM and Thalidomide Pharmion TM , subsequent to the acquisition of Pharmion Corporation, or Pharmion), VIDAZA ® and FOCALIN ® . FOCALIN ® is sold exclusively to Novartis Pharma AG, or Novartis. We also derive revenues from a licensing agreement with Novartis, which entitles us to royalties on FOCALIN XR ® and the entire RITALIN ® family of drugs, and sales of bio-therapeutic products and services through our Cellular Therapeutics subsidiary. ALKERAN ® was licensed from GlaxoSmithKline, or GSK, and sold under our label through March 31, 2009, the conclusion date of the ALKERAN ® license with GSK. For the ensuing two years, we will continue to earn residual payments based upon GSK's ALKERAN ® revenues.
In 1986, we were spun off from Celanese Corporation and, in July 1987, completed an initial public offering. Our initial operations focused on the research and development of chemical and biotreatment processes for the chemical and pharmaceutical industries. We subsequently completed the following strategic acquisitions to strengthen our research and manufacturing capabilities in addition to enhancing our commercialized products:
| • | In August 2000, we acquired Signal Pharmaceuticals, Inc., currently Signal Pharmaceuticals, LLC, a privately held biopharmaceutical company focused on the discovery and development of drugs that regulate genes associated with disease. |
| • | In December 2002, we acquired Anthrogenesis Corp., a privately held New Jersey-based biotherapeutics company and cord blood banking business, developing technologies for the recovery of stem cells from human placental tissues following the completion of full-term, successful pregnancies. Anthrogenesis d/b/a Celgene Cellular Therapeutics, or CCT, now operates as our wholly owned subsidiary engaged in the research, recovery, culture-expansion, preservation, development and distribution of placental cells, including stem and progenitor cells, as therapeutic agents. |
| • | In October 2004, we acquired Penn T Limited, a UK-based global supplier of THALOMID ® . This acquisition expanded our corporate capabilities and enabled us to control manufacturing for THALOMID ® worldwide. |
| • | In December 2006, we acquired an active pharmaceutical ingredient, or API, manufacturing facility from Siegfried Ltd. and Siegfried Dienste AG (together "Siegfried") located in Zofingen, Switzerland. The manufacturing facility has the capability to produce multiple drug substances and is being used to produce REVLIMID ® and THALOMID ® API to supply global markets. The facility may also be used to produce drug substance for our future drugs and drug candidates. |
1
| • | In March 2008, we acquired Pharmion Corporateion, or Pharmion, a global biopharmaceutical company that acquired, developed and commercialized innovative products for the treatment of hematology and oncology patients. Pharmion was acquired to enhance our portfolio of therapies for patients with life-threatening illnesses worldwide with the addition of Pharmion's marketed products, and several products in development for the treatment of hematological and solid tumor cancers. By combining this new product portfolio with our existing operational and financial capabilities, we enlarged our global market share through increased product offerings and expanded clinical, regulatory and commercial capabilities. |
| • | In January 2010, we acquired Gloucester Pharmaceuticals Inc., or Gloucester, a privately held pharmaceutical company, for $340.0 million in cash plus $300.0 million in contingent regulatory milestone payments. The acquisition is expected to advance our leadership position in the development of disease-altering therapies through innovative approaches for patients with rare and debilitating blood cancers. Gloucester developed ISTODAX ® (romidepsin), which was approved in November 2009 by the U.S. Food and Drug Administration, or FDA, for the treatment of cutaneous T-cell lymphoma, or CTCL, in patients who have received at least one prior systemic therapy. Additionally, ISTODAX ® has received both orphan drug designation for the treatment of non-Hodgkin's T-cell lymphomas, which includes CTCL and peripheral T-cell lymphoma, or PTCL, and fast track status in PTCL from the FDA. The European Agency for the Evaluation of Medicinal Products, or EMEA, has granted orphan status designation for ISTODAX ® for the treatment of both CTCL and PTCL. |
For the year ended December 31, 2009, we reported revenue of $2.690 billion, net income of $776.7 million and diluted earnings per share of $1.66. Revenue increased by $435.1 million in 2009 compared to 2008 primarily due to our continuing expansion into international markets and growth of REVLIMID ® and VIDAZA ® , which more than offset decreases in revenues from THALOMID ® and ALKERAN ® . The decrease in THALOMID ® was primarily due to lower unit volumes in the United States resulting from the increased use of REVLIMID ® , while the decrease in ALKERAN ® was due to the March 31, 2009 conclusion of the ALKERAN ® license with GSK. Net income and earnings per share for 2009 reflect the earnings contributions from higher REVLIMID ® and VIDAZA ® revenues, partly offset by increased spending for new product launches, research and development and expansion of our international operations. The year ended December 31, 2008 included a $1.740 billion charge for acquired in-process research and development, or IPR&D, related to the Pharmion acquisition in March 2008.
Our future growth and operating results will depend on the continued acceptance of our currently marketed products, regulatory approvals and successful commercialization of new products and new product indications, depth of our product pipeline, competition with our marketed products and challenges to our intellectual property. See also Forward-Looking Statements and Risk Factors contained in Part I, Item 1A of this Annual Report on Form 10-K.
2
COMMERCIAL STAGE PRODUCTS
REVLIMID ® (lenalidomide) : REVLIMID ® is an oral immunomodulatory drug approved by the FDA and a number of other regulatory agencies in Europe, Latin America, Middle East and Asia/Pacific for treatment in combination with dexamethasone for multiple myeloma patients who have received at least one prior therapy and in Australia and New Zealand in combination with dexamethasone for the treatment of patients whose disease has progressed after one therapy.
In addition, REVLIMID ® was approved by the regulatory agencies in the United States, Canada, and a number of countries in Latin America for treatment of patients with transfusion-dependent anemia due to low- or intermediate-1-risk myelodysplastic syndromes, or MDS, associated with a deletion 5q cytogenetic abnormality with or without additional cytogenetic abnormalities.
We continue to launch REVLIMID ® in European markets and are preparing to launch in Latin America, the Middle East and Asia/Pacific. REVLIMID ® has obtained orphan drug designation for the treatment of MDS in the United States and a number of international markets.
REVLIMID ® is distributed in the United States primarily through contracted pharmacies under the RevAssist ® program, which is a proprietary risk-management distribution program tailored specifically to help ensure the safe and appropriate distribution and use of REVLIMID ® . Internationally, REVLIMID ® is also distributed under mandatory risk-management distribution programs tailored to meet local competent authorities' specifications to help ensure the safe and appropriate distribution and use of REVLIMID ® . These programs may vary by country and, depending upon the country and the design of the risk-management program, the product may be sold through hospitals or retail pharmacies.
REVLIMID ® continues to be evaluated in numerous clinical trials worldwide either alone or in combination with one or more other therapies in the treatment of a broad range of hematological malignancies, including multiple myeloma, MDS, non-Hodgkin's lymphoma, or NHL, chronic lymphocytic leukemia, or CLL, other cancers and other diseases.
THALOMID ® (thalidomide) : THALOMID ® has been approved by the FDA for use in combination with dexamethasone for the treatment of patients with newly diagnosed multiple myeloma and for the acute treatment of the cutaneous manifestations of moderate to severe erythema nodosum leprosum, or ENL, and as maintenance therapy for prevention and suppression of the cutaneous manifestation of ENL recurrence. The Australian Therapeutic Goods Administration, or TGA, approved a supplemental filing granting THALOMID ® marketing approval for use in combination with melphalan and prednisone for patients with untreated multiple myeloma or ineligible for high dose chemotherapy, and also granted THALOMID ® marketing approval in combination with dexamethasone for induction therapy prior to high dose chemotherapy with autologous stem cell rescue, for the treatment of patients with untreated multiple myeloma. In addition, THALOMID ® was granted full marketing authorization by the European Commission, or EC, for use in combination with melphalan and prednisone as a treatment for patients with newly diagnosed multiple myeloma.
THALOMID ® is distributed in the United States under our " System for Thalidomide Education and Prescribing Safety ," or S.T.E.P.S. ® , program which we developed and is a proprietary comprehensive education and risk-management distribution program with the objective of providing for the safe and appropriate distribution and use of THALOMID ® . Internationally, THALOMID ® is also distributed under mandatory risk-management distribution programs tailored to meet local competent authorities' specifications to help ensure the safe and appropriate distribution and use of THALOMID ® . These programs may vary by country and, depending upon the country and the design of the risk-management program, the product may be sold through hospitals or retail pharmacies.
3
VIDAZA ® (azacitidine for injection) : VIDAZA ® is a pyrimidine nucleoside analog that has been shown to reverse the effects of DNA hypermethylation and promote subsequent gene re-expression. VIDAZA ® was licensed from Pharmacia & Upjohn, now part of Pfizer Inc., or Pfizer, and was approved by the FDA for the treatment of all subtypes of MDS. Additionally, VIDAZA ® was granted orphan drug designation by the FDA for the treatment of acute myeloid leukemia, or AML. In December 2008, VIDAZA ® was granted full marketing authorization by the EC for the treatment of adult patients who are not eligible for haematopoietic stem cell transplantation with Intermediate-2 and high-risk MDS according to the International Prognostic Scoring System, or IPSS, or chronic myelomonocytic leukaemia, or CMML, with 10-29 percent marrow blasts without myeloproliferative disorder, or AML with 20-30 percent blasts and multi-lineage dysplasia, according to World Health Organization, or WHO, classification. In November 2009, the TGA also granted VIDAZA ® approval for the same treatment.
VIDAZA ® is distributed through the more traditional pharmaceutical industry supply chain. VIDAZA ® is not subjected to the same risk-management distribution programs as THALOMID ® and REVLIMID ® .
RITALIN ® Family of Drugs : In April 2000, we licensed to Novartis the worldwide rights (excluding Canada) to FOCALIN ® and FOCALIN XR ® , which are approved for the treatment of attention deficit hyperactivity disorder, or ADHD. We retained the rights to these products for the treatment of oncology-related disorders. We sell FOCALIN ® exclusively to Novartis and receive royalties on all of Novartis' sales of FOCALIN XR ® and RITALIN ® family of ADHD-related products. FOCALIN ® is formulated with the active d-isomer of methylphenidate and contains only the more active isomer responsible for the effective management of the symptoms of ADHD.
ALKERAN ® (melphalan) : ALKERAN ® was licensed from GSK and sold under the Celgene label through March 31, 2009, the conclusion date of the ALKERAN ® license with GSK. ALKERAN ® was approved by the FDA for the palliative treatment of multiple myeloma and of carcinoma of the ovary.
PRECLINICAL AND CLINICAL-STAGE PIPELINE
Our preclinical and clinical-stage pipeline of new drug candidates and cell therapies, is highlighted by multiple classes of small molecule, orally administered therapeutic agents designed to selectively regulate disease-associated genes and proteins. The product candidates in our pipeline are at various stages of preclinical and clinical development. Successful results in preclinical or Phase I/II clinical studies may not be an accurate predictor of the ultimate safety or effectiveness of a drug or product candidate.
| • | Phase I Clinical Trials |
Phase I human clinical trials begin when regulatory agencies allow a request to initiate clinical investigations of a new drug or product candidate to become effective and usually involve between 20 to 80 healthy volunteers or patients. The tests study a drug's safety profile, and may include preliminary determination of a drug or product candidate's safe dosage range. The Phase I clinical studies also determine how a drug is absorbed, distributed, metabolized and excreted by the body, and therefore potentially the duration of its action. |
| • | Phase II Clinical Trials |
Phase II clinical trials are conducted on a limited number of patients with the targeted disease. An initial evaluation of the drug's effectiveness on patients is performed and additional information on the drug's safety and dosage range is obtained. |
4
| • | Phase III Clinical Trials |
Phase III clinical trials typically include controlled multi-center trials and involve a larger target patient population to ensure that study results are statistically significant. During Phase III clinical trials, physicians monitor patients to determine efficacy and to gather further information on safety. |
IMiDs ® COMPOUNDS : IMiDs ® compounds are proprietary novel small molecule, orally available compounds that modulate the immune system and other biologically important targets through multiple processes. The IMiDs ® compound CC-4047 (pomalidomide) is being evaluated in Phase I and Phase II clinical trials for various disease indications. CC-4047 is one of the most potent IMiDs ® compounds that we are developing. Our initial Investigational New Drug, or IND, application was to evaluate CC-4047 in a U.S. proof-of-principle study in sickle cell anemia. We are also evaluating CC-4047 for treatment of other diseases including myelofibrosis and multiple myeloma. Additional compounds are in preclinical development. Our IMiDs ® compounds are covered by an extensive and comprehensive intellectual property estate of U.S. and foreign-issued patents and pending patent applications including composition-of-matter, use and other patents and patent applications.
ORAL ANTI-INFLAMMATORY AGENTS: Our oral PDE-4 inhibitor, CC-10004 (apremilast), is a member of a proprietary pipeline of novel small molecules with anti-inflammatory activities that impede the production of multiple proinflammatory mediators by inhibiting PDE-4, also causing reductions in TNF- a as well as interleukin-2 (IL-2), IL-17 and IL-23, interferon-gamma, leukotrienes and nitric oxide synthase. Apremilast is our lead investigational drug in this class of anti-inflammatory compounds and a current Phase II clinical trial for psoriasis and psoriatic arthritis has exhibited encouraging results. We are also exploring the use of CC-10004 in additional rheumatic, dermatologic and inflammatory diseases to determine the potential of apremilast. In addition, we are investigating our next oral PDE-4 inhibitor, CC-11050, which has completed Phase I trials, towards evaluating its safety and efficacy in a number of inflammatory conditions and are moving forward with its development.
KINASE INHIBITORS : We have generated valuable intellectual property in the identification of kinases that regulate pathways critical in inflammation and oncology. Our kinase inhibitor platform includes inhibitors of the c-Jun N-terminal kinase, or JNK, including CC-401, which has successfully completed a Phase I trial in healthy volunteers and in AML patients to determine safety and tolerability. No further studies with CC-401 are planned at this time as we intend to advance our new second generation JNK inhibitors, specifically CC-930, which recently completed a Phase Ib multiple dose study. We are also planning to investigate CC-930 in fibrotic conditions assuming safety and tolerability continue to be acceptable.
SMALL CELL LUNG CANCER : Amrubicin is a third-generation fully synthetic anthracycline molecule with potent topoisomerase II inhibition and is currently being studied as a single agent and in combination with anti-cancer therapies for solid tumors. In 2008, the FDA granted amrubicin orphan drug designation for the treatment of small cell lung cancer and fast track product designation for the treatment of small cell lung cancer after first-line chemotherapy. A drug designated as a fast track product is intended for the treatment of a serious or life-threatening condition and demonstrates the potential to provide a therapy where none exists or provide a therapy which may offer a significant improvement in safety and/or effectiveness over existing therapy.
CELLULAR THERAPIES : At CCT, we are researching stem cells derived from the human placenta as well as from the umbilical cord. CCT is our state-of-the-art research and development division dedicated to fulfilling the promise of cellular technologies by developing cutting-edge products and therapies to significantly benefit patients. Our goal is to develop proprietary cell therapy products for the treatment of unmet medical needs.
5
Stem cell based therapies offer the potential to provide disease-modifying outcomes for serious diseases which lack adequate therapy. We have developed proprietary technology for collecting, processing and storing placental stem cells with potentially broad therapeutic applications in cancer, auto-immune diseases, including Crohn's disease and multiple sclerosis, neurological disorders including stroke and amyotrophic lateral sclerosis, or ALS, graft-versus-host disease, or GVHD, and other immunological / anti-inflammatory, rheumatologic and bone disorders. We have completed enrollment into a Phase I study for our human placenta derived cell product (PDA-001), which is a multi-center clinical trial in patients with moderate-to-severe Crohn's disease refractory to oral corticosteroids and immune suppressants.
We also maintain an IND with the FDA for a trial with human umbilical cord blood in sickle cell anemia and an IND for human placental-derived stem cells, or HPDSC, to support a study to assess the safety of its transplantation with umbilical cord blood obtained from fully or partially matched related donors in subjects with certain malignant hematological diseases and non-malignant disorders. We are continuing additional preclinical and clinical research to define further the potential of placental-derived stem cells and to characterize other placental-derived products.
ACTIVIN INHIBITORS: We have a collaboration with Acceleron Pharma, or Acceleron, and have initiated Phase I and II clinical trials of ACE-011 for treatment of chemotherapy induced anemia in metastatic breast cancer, metastatic bone disease and renal anemia. ACE-011 is an inhibitor of activin, a member of the growth and differentiation factor, or GDF, family of proteins responsible for the growth and repair of a number of systems in the body. ACE-011 acts as a decoy receptor for activin, blocking activin's effects upon growth and repair of various tissues including bone and red blood cells, as well as breast, ovary and other reproductive tissues.
6
CELGENE LEADING PRODUCT CANDIDATES
The development of our leading new drug candidates and their targeted disease indications are outlined in the following table:
| Disease | ||||
| Product | Indication | Status | ||
| ||||
IMiDs ® Compounds: | ||||
CC-4047 (pomalidomide) | Myelofibrosis | Phase II trial ongoing, pivotal trial planned | ||
| Multiple myeloma | Phase II trial ongoing, pivotal trial planned | ||
| ||||
Oral Anti-Inflammatory: | ||||
CC-10004 (apremilast) | Psoriasis | Phase II trial ongoing, phase III trials planned | ||
| Psoriatic arthritis | Phase II trial ongoing, phase III trials planned | ||
| Rheumatoid arthritis | Phase II trial planned | ||
CC-11050 | Cutaneous lupus | Phase II trial planned | ||
| ||||
Kinase Inhibitors: | ||||
JNK CC-930 | Idiopathic pulmonary fibrosis | Phase II trial planned | ||
| ||||
Small Cell Lung Cancer: | ||||
Amrubicin | Small cell lung cancer | Phase III study ongoing | ||
| ||||
Cellular Therapies: | ||||
PDA-001 | Crohn's disease | Phase I study ongoing, phase II trial planned | ||
| Multiple sclerosis | Phase II trial planned | ||
| Ulcerative colitis | Phase II trial planned | ||
| ||||
Activin Biology: | ||||
ACE-011 | Multiple myeloma/bone loss | Phase II ongoing | ||
| Renal anemia | Phase II trial planned | ||
| Chronic kidney disease | Phase II trial planned |
7
PATENTS AND PROPRIETARY TECHNOLOGY
Patents and other proprietary rights are important to our business. It is our policy to seek patent protection for our inventions, and also to rely upon trade secrets, know-how, continuing technological innovations and licensing opportunities to develop and maintain our competitive position.
We own or have exclusively licensed over 175 issued U.S. patents. In addition, approximately 210 additional pending patent applications are owned by or exclusively licensed to us. We have a policy to seek worldwide patent protection for our inventions and have foreign patent rights corresponding to most of our U.S. patents.
In August 2001, we entered into an agreement, termed the "New Thalidomide Agreement," with EntreMed, Inc., or EntreMed, Children's Medical Center Corporation, or CMCC, and Bioventure Investments kft relating to patents and patent applications owned by CMCC, which agreement superceded several agreements already in place between CMCC, EntreMed and us. Pursuant to the New Thalidomide Agreement, CMCC directly granted to us an exclusive worldwide license under the relevant patents and patent applications relating to thalidomide. Several U.S. and European patents have been issued to CMCC in this patent family and certain of these patents expire in 2013 and 2014. We have applied for and received Supplementary Protection Certificates, or SPCs, in Europe relative to certain of these issued CMCC thalidomide patents. These SPCs extend the terms of these patents relative to uses of thalidomide to 2019. Corresponding foreign patent applications and additional U.S. patent applications are still pending.
In addition to the New Thalidomide Agreement, we entered into an agreement, entitled the "New Analog Agreement," with CMCC and EntreMed in December 2002, pursuant to which we have been granted an exclusive worldwide license to certain CMCC patents and patent applications relating to thalidomide analogs. Under the New Analog Agreement, CMCC exclusively licensed to us these patents and patent applications, which relate to analogs, metabolites, precursors and hydrolysis products of thalidomide, and stereoisomers thereof. Under the New Analog Agreement, we are obligated to comply with certain milestones and other obligations, including those relating to REVLIMID ® brand drug sales. The New Analog Agreement grants us control over the prosecution and maintenance of the licensed thalidomide analog patent rights.
Our research leads us to seek patent protection for molecular targets and drug discovery technologies, as well as therapeutic and diagnostic products and processes. More specifically, proprietary technology has been developed for use in molecular target discovery, the identification of regulatory pathways in cells, assay design and the discovery and development of pharmaceutical product candidates. An increasing percentage of our recent patent applications have been related to potential product candidates or compounds. As of December 2009, included in those inventions described above, we owned, in whole or in part, over 70 issued U.S. patents and have filed over 90 U.S. pending patent applications, including pending provisional applications, some of which are related to sponsored or collaborative research relationships.
In addition, we pursue, where appropriate, patent term extension and patent term adjustment strategies. For example, we have applied for and received SPCs in Europe relative to certain in-licensed CMCC thalidomide patents. These SPCs extend the terms of these patents relative to certain uses of thalidomide to 2019. In addition, we have applied for and received SPCs to 2022 in Europe, and patent term extensions in Australia, relative to certain of our patents claiming lenalidomide. In the United States, we have been granted a patent term extension of our REVLIMID ® compositions of matter patent to 2019. In the United States, we have been granted patent term adjustment with respect to a REVLIMID ® polymorph patent; this patent is presently scheduled to expire in 2026.
Our patents are regularly subject to challenge by generic drug companies and manufacturers. See Part I, Item 3, "Legal Proceedings." We rely on several different types of patents to protect our products, including, without limitation, compound, polymorph, formulation and method of use patents. We do not know whether any of these patents will be circumvented, invalidated or found unenforceable as a result of challenge by generic companies or manufacturers.
8
CCT, our cellular therapeutics subsidiary, seeks patent protection for the collection, processing, composition, formulation and uses of mammalian placental and umbilical cord tissue and placental and umbilical cord stem cells, as well as cells and biomaterials derived from the placenta. As of December 2009, CCT owned, in whole or in part, eight U.S. patents, including claims to novel cells and cellular compositions. In addition, CCT has approximately 50 U.S. patent applications, including pending provisional applications.
Our success will depend, in part, on our ability to obtain and enforce patents, protect trade secrets and obtain licenses to technology owned by third parties where it is necessary to conduct our business without infringing upon the proprietary rights of others. The patent positions of pharmaceutical and biotechnology firms, including ours, can be uncertain and involve complex legal and factual questions. In addition, the coverage sought in a patent application can be significantly reduced before the patent is issued. Further, our competitors, including generic drug companies regularly attack or design around patents, particularly use and formulation patents.
Consequently, we do not know whether any of our owned or licensed pending patent applications, which have not already been allowed, will result in the issuance of patents or, if any patents are issued, whether they will be dominated by third-party patent rights, whether they will provide significant proprietary protection or commercial advantage or whether they will be circumvented, opposed, invalidated, rendered unenforceable or infringed by others. Further, we are aware of third-party U.S. patents that relate to the use of certain stem cell technologies and cannot be assured as to any impact to our potential products, or guarantee that our patents or pending applications will not be involved in, or be defeated as a result of, opposition proceedings before a foreign patent office or any interference proceedings before the United States Patent & Trademark Office, or PTO.
With respect to patents and patent applications we have licensed-in, there can be no assurance that additional patents will be issued to any of the third parties from whom we have licensed patent rights, or that, if any new patents are issued, such patents will not be opposed, challenged, invalidated, infringed or dominated or provide us with significant proprietary protection or commercial advantage. Moreover, there can be no assurance that any of the existing licensed patents will provide us with proprietary protection or commercial advantage. Nor can we guarantee that these licensed patents will not be either infringed, invalidated or circumvented by others, or that the relevant agreements will not be terminated. Any termination of the licenses granted to us by CMCC could have a material adverse effect on our business, financial condition and results of operations.
Because 1) patent applications filed in the United States on or before November 28, 2000 are maintained in secrecy until patents issue, 2) patent applications filed in the United States on or after November 29, 2000 are not published until approximately 18 months after their earliest claimed priority date, 3) United States patent applications that are not filed outside the United States may not publish at all until issued, and 4) publication of discoveries in the scientific or patent literature often lag behind actual discoveries, we cannot be certain that we, or our licensors, were the first to make the inventions covered by each of the issued patents or pending patent applications or that we, or our licensors, were the first to file patent applications for such inventions. In the event a third party has also filed a patent for any of our inventions, we, or our licensors, may have to participate in interference proceedings before the PTO to determine priority of invention, which could result in the loss of a U.S. patent or loss of any opportunity to secure U.S. patent protection for the invention. Even if the eventual outcome is favorable to us, such interference proceedings could result in substantial cost to us.
We are aware of U.S. patents that have been issued to third parties claiming subject matter relating to the NFκB pathway, including U.S. patents which could overlap with technology claimed in some of our owned or licensed patents or patent applications, and a U.S. patent that has been asserted against certain pharmaceutical companies. We are also aware of third-party U.S. patents that relate to the use of certain TNF- a inhibitors to treat inflammation or conditions such as asthma. Further, third parties may, from time to time, assert patents claimed to relate to commercially relevant uses of our products and should such a claim be asserted, we will vigorously and appropriately defend against such action.
9
We may in the future have to prove that we are not infringing patents or we may be required to obtain licenses to such patents. However, we do not know whether such licenses will be available on commercially reasonable terms, or at all. Prosecution of patent applications and litigation to establish the validity and scope of patents, to assert patent infringement claims against others and to defend against patent infringement claims by others can be expensive and time-consuming. There can be no assurance that, in the event that claims of any of our owned or licensed patents are challenged by one or more third parties, any court or patent authority ruling on such challenge will determine that such patent claims are valid and enforceable. An adverse outcome in such litigation could cause us to lose exclusivity relating to the subject matter delineated by such patent claims and may have a material adverse effect on our business. If a third party is found to have rights covering products or processes used by us, we could be forced to cease using the products or processes covered by the disputed rights, be subject to significant liabilities to such third party and/or be required to license technologies from such third party. Also, different countries have different procedures for obtaining patents, and patents issued by different countries provide different degrees of protection against the use of a patented invention by others. There can be no assurance, therefore, that the issuance to us in one country of a patent covering an invention will be followed by the issuance in other countries of patents covering the same invention or that any judicial interpretation of the validity, enforceability or scope of the claims in a patent issued in one country will be similar to the judicial interpretation given to a corresponding patent issued in another country. Competitors have chosen and in the future may choose to file oppositions to patent applications, which have been deemed allowable by foreign patent examiners. Furthermore, even if our owned or licensed patents are determined to be valid and enforceable, there can be no assurance that competitors will not be able to design around such patents and compete with us using the resulting alternative technology. Additionally, for these same reasons, we cannot be sure that patents of a broader scope than ours may be issued and thereby create freedom to operate issues. If this occurs we may need to reevaluate pursuing such technology, which is dominated by others' patent rights, or alternatively, seek a license to practice our own invention, whether or not patented.
We also rely upon unpatented, proprietary and trade secret technology that we seek to protect, in part, by confidentiality agreements with our collaborative partners, employees, consultants, outside scientific collaborators, sponsored researchers and other advisors. There can be no assurance that these agreements provide meaningful protection or that they will not be breached, that we would have adequate remedies for any such breach or that our trade secrets, proprietary know-how and technological advances will not otherwise become known to others. In addition, there can be no assurance that, despite precautions taken by us, others have not and will not obtain access to our proprietary technology or that such technology will not be found to be non-proprietary or not a trade secret.
GOVERNMENTAL REGULATION/EXCLUSIVITIES AFFORED BY REGULATORY AUTHORITIES
Regulation by governmental authorities in the United States and other countries is a significant factor in the manufacture and marketing of pharmaceuticals and in our ongoing research and development activities. Most, if not all, of our therapeutic products require regulatory approval by governmental agencies prior to commercialization. In particular, human therapeutic products are subject to rigorous preclinical testing and clinical trials and other pre-marketing approval requirements by the FDA and regulatory authorities in other countries. In the United States, various federal and, in some cases, state statutes and regulations also govern or impact upon the manufacturing, testing for safety and effectiveness, labeling, storage, record-keeping and marketing of such products. The lengthy process of seeking required approvals, and the continuing need for compliance with applicable statutes and regulations, requires the expenditure of substantial resources. Regulatory approval, if and when obtained, may be limited in scope which may significantly limit the indicated uses for which a product may be marketed. Further, approved drugs, as well as their manufacturers, are subject to ongoing review and discovery of previously unknown problems with such products or the manufacturing or quality control procedures used in their production may result in restrictions on their manufacture, sale or use or in their withdrawal from the market. Any failure by us, our suppliers of manufactured drug product, collaborators or licensees to obtain or maintain, or any delay in obtaining, regulatory approvals could adversely affect the marketing of our products and our ability to receive product revenue, license revenue or profit sharing payments.
10
The activities required before a product may be marketed in the United States begin with preclinical testing not involving human subjects. Preclinical tests include laboratory evaluation of a product candidate's chemistry and its biological activities and the conduct of animal studies to assess the potential safety and efficacy of a product candidate and its formulations. The results of these studies must be submitted to the FDA as part of an IND which must be reviewed by the FDA primarily for safety considerations before proposed clinical trials in humans can begin.
Typically, clinical trials involve a three-phase process as previously described. In some cases, further studies (Phase IV) are required as a condition for a new drug application, or NDA, or biologics license application, or BLA, approval, to provide additional information concerning the drug or product. The FDA requires monitoring of all aspects of clinical trials, and reports of all adverse events must be made to the agency before drug approval. After approval, we have ongoing reporting obligations concerning adverse reactions associated with the drug, including expedited reports for serious and unexpected adverse events. Additionally, we may have limited control over studies conducted with our proprietary compounds or biologics if such studies are performed by others (e.g., cooperative groups).
The results of the preclinical testing and clinical trials are submitted to the FDA as part of an NDA or BLA for evaluation to determine if the product is sufficiently safe and effective for approval to commence commercial sales. In responding to an NDA or BLA, the FDA may grant marketing approval, request additional information or deny the application if it determines that the application does not satisfy its regulatory approval criteria. When an NDA or BLA is approved, the NDA or BLA holder must a) employ a system for obtaining reports of experience and side effects associated with the drug and make appropriate submissions to the FDA and b) timely advise the FDA if any marketed product fails to adhere to specifications established by the NDA or BLA internal manufacturing procedures.
Pursuant to the Orphan Drug Act, a sponsor may request that the FDA designate a drug intended to treat a "rare disease or condition" as an "orphan drug." The term "orphan drug" can refer to either a drug or biologic. A rare disease or condition is defined as one which affects less than 200,000 people in the United States, or which affects more than 200,000 people, but for which the cost of developing and making available the product is not expected to be recovered from sales of the product in the United States. Upon the approval of the first NDA or BLA for a drug designated as an orphan drug for a specified indication, the sponsor of that NDA or BLA is entitled to seven years of exclusive marketing rights in the United States for such drug or product containing the active ingredient for the same indication unless the sponsor cannot assure the availability of sufficient quantities of the drug to meet the needs of persons with the disease. However, orphan drug status is particular to the approved indication and does not prevent another company from seeking approval of other labeled indications. The period of orphan exclusivity is concurrent with any patent exclusivity that relates to the drug or biologic. Orphan drugs may also be eligible for federal income tax credits for costs associated with the drugs' development. Possible amendment of the Orphan Drug Act by the U.S. Congress and possible reinterpretation by the FDA has been discussed by regulators and legislators. FDA regulations reflecting certain definitions, limitations and procedures for orphan drugs initially went into effect in January 1993 and were amended in certain respects in 1998. Therefore, there is no assurance as to the precise scope of protection that may be afforded by orphan drug status in the future or that the current level of exclusivity and tax credits will remain in effect. Moreover, even if we have an orphan drug designation for a particular use of a drug, there can be no assurance that another company also holding orphan drug designation will not receive approval prior to us for the same indication. If that were to happen, our applications for that indication could not be approved until the competing company's seven-year period of exclusivity expired. Even if we are the first to obtain approval for the orphan drug indication, there are certain circumstances under which a competing product may be approved for the same indication during our seven-year period of exclusivity. Further, particularly in the case of large molecule drugs or biologics, a question can be raised whether the competing product is really the "same drug" as that which was approved. In addition, even in cases in which two products appear to be the same drug, the agency may approve the second product based on a showing of clinical superiority compared to the first product. In order to increase the development and marketing of drugs for rare disorders, regulatory bodies outside the United States have enacted regulations similar to the Orphan Drug Act. REVLIMID ® brand drug has been granted orphan medicinal product designation by the EC for treatment of CLL following the favorable opinion of the European Medicines Agency's, or EMA, Committee for Orphan Medicinal Products.
11
Among the conditions for NDA or BLA approval is the requirement that the prospective manufacturer's quality control and manufacturing procedures continually conform with the FDA's current Good Manufacturing Practice, or cGMP, regulations (which are regulations established by the FDA governing the manufacture, processing, packing, storage and testing of drugs and biologics intended for human use). In complying with cGMP, manufacturers must devote extensive time, money and effort in the area of production and quality control and quality assurance to maintain full technical compliance. Manufacturing facilities and company records are subject to periodic inspections by the FDA to ensure compliance. If a manufacturing facility is not in substantial compliance with these requirements, regulatory enforcement action may be taken by the FDA, which may include seeking an injunction against shipment of products from the facility and recall of products previously shipped from the facility.
Under the Hatch-Waxman Amendments to the Federal Food, Drug, and Cosmetic Act, products covered by approved NDAs or supplemental NDAs may be protected by periods of patent and/or non-patent exclusivity. During the exclusivity periods, the FDA is generally prevented from granting effective approval of an abbreviated NDA, or ANDA. Further, NDAs submitted under 505(b)(2) of the Food, Drug and Cosmetic Act may not reference data contained in the NDA for a product protected by an effective and unexpired exclusivity. ANDAs and 505(b)(2) applications are generally less burdensome than full NDAs in that, in lieu of new clinical data, the applications rely in whole, or in part, upon the safety and efficacy findings of the referenced approved drug in conjunction with bridging data, typically bioequivalence data. Upon the expiration of the applicable exclusivities, through passage of time or successful legal challenge, the FDA may grant effective approval of an ANDA for a generic drug, or may accept reference to a previously protected NDA in a 505(b)(2) application. Depending upon the scope of the applicable exclusivities, any such approval could be limited to certain formulations and/or indications/claims, i.e., those not covered by any outstanding exclusivities. While the Food, Drug and Cosmetic Act, or the Act, provides for ANDA and 505(b)(2) abbreviated approval pathways for drugs earlier submitted as NDAs and approved under section 505 of the Act, there are presently no similar provisions for biologics submitted as BLAs and approved under the Public Health Service, or PHS, Act. That is, there is currently no abbreviated application that would permit approval of a generic or "follow-on" biologic based on the FDA's earlier approval of another manufacturer's application under section 351 of the PHS Act.
Failure to comply with applicable FDA regulatory requirements can result in enforcement actions such as warning letters, recalls or adverse publicity issued by the FDA or in legal actions such as seizures, injunctions, fines based on the equitable remedy of disgorgement, restitution and criminal prosecution.
Approval procedures similar to those in the United States must be undertaken in virtually every other country comprising the market for our products before any such product can be commercialized in those countries. The approval procedure and the time required for approval vary from country to country and may involve additional testing. There can be no assurance that approvals will be granted on a timely basis or at all. In addition, regulatory approval of drug and biologics pricing is required in most countries other than the United States. There can be no assurance that the resulting pricing of our products would be sufficient to generate an acceptable return to us.
12
KEY PRODUCTS: TABLE OF EXCLUSIVITIES
The following table shows the estimated expiration dates in the United States and in Europe of the last-to-expire period of exclusivity (regulatory or patent) related to the following approved drugs marketed or soon-to-be-marketed by us:
| U.S. | Europe | |||
| ||||
REVLIMID ® brand drug | 2026 | 2022 | ||
(drug substance patents) | ||||
| ||||
THALOMID ® brand drug | 2023 | 2019 | ||
(use and/or drug product patents) | ||||
| ||||
VIDAZA ® brand drug | 2011 | 2018 | ||
(regulatory exclusivity only) | ||||
| ||||
ISTODAX ® brand drug | 2021 | (10 years regulatory exclusivity upon approval) | ||
(U.S. drug substance patents) | ||||
(EMA regulatory exclusivity upon approval) |
13
COMPETITION
The pharmaceutical and biotechnology industries in which we compete are each highly competitive and some of the major companies within these industries have considerably greater financial, scientific, technical and marketing resources than we do. We also compete with universities and research institutions in the development of products and processes and in the acquisition of technology from outside sources.
Competition in the pharmaceutical industry, and specifically in the oncology and immune-inflammatory areas is particularly intense. Numerous pharmaceutical, biotechnology and generic drug companies have extensive anti-cancer and anti-inflammatory drug discovery, development and commercial resources. Amgen Inc., AstraZeneca PLC., Biogen Idec Inc., Bristol-Myers Squibb Co., Eisai Co., Ltd., F.Hoffmann-LaRoche Ltd, Johnson and Johnson, Merck and Co., Inc., Novartis AG, Pfizer and Takeda Pharmaceutical Co. Ltd. are among some of the companies researching and developing new compounds in the oncology, inflammation and immunology fields. We, along with other pharmaceutical brand-name makers, face the challenges brought on by generic drug manufacturers in their pursuit of obtaining bulk quantities of certain drugs in order for them to be able to develop similar versions of these products and be ready to market as soon as permitted.
The pharmaceutical and biotechnology industries have undergone, and are expected to continue to undergo, rapid and significant technological change. Consolidation and competition are expected to intensify as technical advances in each field are achieved and become more widely known. In order to compete effectively, we will be required to continually upgrade and expand our scientific expertise and technology, identify and retain capable personnel and pursue scientifically feasible and commercially viable opportunities.
Our competition will be determined in part by the indications and geographic markets for which our products are developed and ultimately approved by regulatory authorities. The relative speed with which we develop new products, complete clinical trials, obtain regulatory approvals, receive pricing and reimbursement approvals, finalize agreements with outside contract manufacturers when needed and market our products are critical factors in gaining a competitive advantage. Competition among products approved for sale will include product efficacy, safety, convenience, reliability, availability, price, third-party reimbursement and patent and non-patent exclusivity.
SIGNIFICANT ALLIANCES
From time to time we enter into strategic alliances with third parties whereby we either grant rights to certain of our compounds in exchange for rights to receive payments, or acquire rights to compounds owned by other pharmaceutical or biotechnology companies in exchange for obligations to make payments to the partnering companies. Payments either to or from third parties may be in the form of cash, upfront payments, milestone payments contingent upon the achievement of pre-determined criteria, research and development funding, product supply contracts and royalty payments based on net product sales.
Novartis Pharma AG: We entered into an agreement with Novartis in which we granted to Novartis an exclusive worldwide license (excluding Canada) to develop and market FOCALIN ® (d-methylphenidate, or d - MPH) and FOCALIN XR ® , the long-acting drug formulation. We have retained the exclusive commercial rights to FOCALIN ® and FOCALIN XR ® for oncology-related disorders, such as chronic fatigue associated with chemotherapy. We also granted Novartis rights to all of its related intellectual property and patents, including formulations of the currently marketed RITALIN LA ® . We also sell FOCALIN ® to Novartis and receive royalties on sales of all of Novartis' FOCALIN XR ® and RITALIN ® family of ADHD-related products.
14
Array BioPharma Inc.: We have a research collaboration agreement with Array BioPharma Inc., or Array, focused on the discovery, development and commercialization of novel therapeutics in cancer and inflammation. As part of this agreement, we made an upfront payment in September 2007 to Array of $40.0 million, which was recorded as research and development expense, in return for an option to receive exclusive worldwide rights for compounds developed against two of the four research targets defined in the agreement, except for Array's limited U.S. co-promotional rights. In June 2009, we made an additional payment of $4.5 million to expand the research targets defined in the agreement, which was also recorded as research and development expense. Array will be responsible for all discovery and clinical development through Phase I or Phase IIa and be entitled to receive, for each compound, potential milestone payments of approximately $200.0 million, if certain discovery, development and regulatory milestones are achieved and $300.0 million if certain commercial milestones are achieved, as well as royalties on net sales.
Our option will terminate upon the earlier of either a termination of the agreement, the date we have exercised our options for compounds developed against two of the four research targets defined in the agreement, or September 21, 2012, unless the term is extended. We may unilaterally extend the option term for two additional one-year terms until September 21, 2014 and the parties may mutually extend the term for two additional one-year terms until September 21, 2016. Upon exercise of an option, the agreement will continue until we have satisfied all royalty payment obligations to Array. Upon the expiration of the agreement, Array will grant us a fully paid-up, royalty-free license to use certain intellectual properties of Array to market and sell the compounds and products developed under the agreement. The agreement may expire on a product-by-product and country-by-country basis as we satisfy our royalty payment obligation with respect to each product in each country.
Prior to its expiration as described above, the agreement may be terminated by:
| (i) | us at our sole discretion, or |
| (ii) | either party if the other party: |
| (x) | materially breaches any of its material obligations under the agreement, or |
| (y) | files for bankruptcy. |
If the agreement is terminated by us at our sole discretion or by Array for a material breach by us, then our rights to the compounds and products developed under the agreement will revert to Array. If the agreement is terminated by Array for a material breach by us, then we will also grant to Array a non-exclusive, royalty-free license to certain intellectual property controlled by us necessary to continue the development of such compounds and products. If the agreement is terminated by us for a material breach by Array, then, among other things, our payment obligations under the agreement could be either reduced by 50% or terminated entirely.
PTC Therapeutics, Inc.: In September 2007, we invested $20.0 million, of which $1.1 million represented research and development expense, in Series F-2 Convertible Preferred Stock of PTC Therapeutics, Inc., or PTC, and in December 2009, we invested an additional $1.5 million in Series G Convertible Preferred Stock of PTC. In September 2007, we also entered into a separate research and option agreement whereby PTC would perform discovery research activities. Under the agreement, both parties could subsequently agree to advance research on certain discovery targets and enter into a separate pre-negotiated collaboration and license agreement which would replace the original research and option agreement.
On July 16, 2009, we and PTC agreed to advance research on one discovery target and entered into a pre-negotiated collaboration and license agreement under which PTC is eligible to receive quarterly research fees, as defined in the agreement, and is entitled to receive potential milestone payments of approximately $129.0 million if certain development, regulatory and sales-based milestones are achieved. PTC will also receive tiered royalties on worldwide net sales. Under the agreement, we may transfer certain research and development activities from PTC to us and upon such transfer we will no longer fund such quarterly research fees to PTC.
15
The agreement will continue until we have satisfied all royalty payment obligations to PTC. Upon our full satisfaction of our royalty payment obligations to PTC under the agreement, the license granted to us by PTC under the agreement will become a non-exclusive, fully paid-up, sub-licensable, royalty-free license to use certain intellectual property of PTC to market and sell the products developed under the agreement. The agreement may expire on a product-by-product and country-by-country basis as we satisfy our royalty payment obligation with respect to each product in each country.
Prior to its expiration as described above, the agreement may be terminated by:
| (i) | us at our sole discretion following the first anniversary of the agreement, or |
| (ii) | either party if the other party: |
| (x) | materially breaches any of its material obligations under the agreement, or |
| (y) | files for bankruptcy. |
If the agreement is terminated by us at our sole discretion or by PTC for a material breach by us, then all licenses granted to us under the agreement will terminate. If PTC materially breaches any of its obligations under the agreement, we can either terminate the agreement, in which case all licenses and rights granted under the agreement are terminated, or elect to continue the agreement, in which case all milestone obligations cease and future royalties payable by us under the agreement will be reduced by between 50% and 70%.
Acceleron Pharma: We have a worldwide strategic collaboration with Acceleron Pharma, or Acceleron, for the joint development and commercialization of ACE-011, currently being studied for treatment of chemotherapy-induced anemia in metastatic breast cancer, metastatic bone disease and renal anemia. The collaboration combines both companies' resources and commitment to developing products for the treatment of cancer and cancer-related bone loss. The agreement also includes an option for certain discovery stage programs. Under the terms of the agreement, we and Acceleron will jointly develop, manufacture and commercialize Acceleron's products for bone loss. We made an upfront payment to Acceleron in February 2008 of $50.0 million, which included a $5.0 million equity investment in Acceleron, with the remainder recorded as research and development expense. In addition, in the event of an initial public offering of Acceleron, we will purchase a minimum of $7.0 million of Acceleron common stock.
Acceleron will retain responsibility for initial activities, including research and development, through the end of Phase IIa clinical trials, as well as manufacturing the clinical supplies for these studies. In turn, we will conduct the Phase IIb and Phase III clinical studies and will oversee the manufacture of Phase III and commercial supplies. Acceleron will pay a share of the development expenses and is eligible to receive development, regulatory approval and sales-based milestones of up to $510.0 million for the ACE-011 program and up to an additional $437.0 million for each of the three discovery stage programs. The companies will co-promote the products in North America. Acceleron will receive tiered royalties on worldwide net sales.
The agreement will continue until we have satisfied all royalty payment obligations to Acceleron and we have either exercised or forfeited all of our options under the agreement. Upon our full satisfaction of our royalty payment obligations to Acceleron under the agreement, all licenses granted to us by Acceleron under the agreement will become fully paid-up, perpetual, non-exclusive, irrevocable and royalty-free licenses. The agreement may expire on a product-by-product and country-by-country basis as we satisfy our royalty payment obligation with respect to each product in each country.
Prior to its expiration as described above, the agreement may be terminated by:
| (i) | us at our sole discretion, or |
| (ii) | either party if the other party: |
| (x) | materially breaches any of its material obligations under the agreement, or |
| (y) | files for bankruptcy. |
If the agreement is terminated by us at our sole discretion or by Acceleron for a material breach by us, then all licenses granted to us under the agreement will terminate and we will also grant to Acceleron a non-exclusive license to certain of our intellectual property related to the compounds and products. If the agreement is terminated by us for a material breach by Acceleron, then, among other things, (A) the licenses granted to Acceleron under the agreement will terminate, (B) the licenses granted to us will continue in perpetuity, (C) all future royalties payable by us under the agreement will be reduced by 50% and (D) our obligation to make any future milestone payments will terminate.
16
Cabrellis Pharmaceuticals Corp.: As a result of our acquisition of Pharmion, we obtained an exclusive license to develop and commercialize amrubicin in North America and Europe pursuant to a license agreement with Dainippon Sumitomo Pharma Co. Ltd, or DSP. Pursuant to Pharmion's acquisition of Cabrellis Pharmaceutics Corp., or Cabrellis, prior to our acquisition of Pharmion, we will pay $12.5 million for each approval of amrubicin in an initial indication by regulatory authorities in the United States and the European Union, or E.U., to the former shareholders of Cabrellis. Upon approval of amrubicin for a second indication in the United States or E.U., we will pay an additional $10.0 million for each market to the former shareholders of Cabrellis. Under the terms of the license agreement for amrubicin, we are required to make milestone payments of $7.0 million and $1.0 million to DSP upon regulatory approval of amrubicin in the United States and upon receipt of the first approval in the E.U., respectively, and up to $17.5 million upon achieving certain annual sales levels in the United States. Pursuant to the supply agreement for amrubicin, we are to pay DSP a semiannual supply price calculated as a percentage of net sales for a period of ten years. In September 2008, amrubicin was granted fast track product designation by the FDA for the treatment of small cell lung cancer after first-line chemotherapy.
The amrubicin license expires on a country-by-country basis and on a product-by-product basis upon the later of (i) the tenth anniversary of the first commercial sale of the applicable product in a given country after the issuance of marketing authorization in such country and (ii) the first day of the first quarter for which the total number of generic product units sold in a given country exceeds 20% of the total number of generic product units sold plus licensed product units sold in the relevant country during the same calendar quarter.
Prior to its expiration as described above, the amrubicin license may be terminated by:
| (i) | us at our sole discretion, |
| (ii) | either party if the other party: |
| (x) | materially breaches any of its material obligations under the agreement, or |
| (y) | files for bankruptcy, |
| (iii) | DSP if we take any action to challenge the title or validity of the patents owned by DSP, or |
| (iv) | DSP in the event of our change in control. |
If the agreement is terminated by us at our sole discretion or by DSP under circumstances described in clauses (ii)(x) and (iii) above, then we will transfer our rights to the compounds and products developed under the agreement to DSP and will also grant to DSP a non-exclusive, perpetual, royalty-free license to certain intellectual property controlled by us necessary to continue the development of such compounds and products. If the agreement is terminated by us for a material breach by DSP, then, among other things, DSP will grant to us an exclusive, perpetual, paid-up license to all of the intellectual property of DSP necessary to continue the development, marketing and selling of the compounds and products subject to the agreement.
GlobeImmune, Inc.: In September 2007, we made a $3.0 million equity investment in GlobeImmune, Inc., or GlobeImmune. In April 2009 and May 2009, we made additional $0.1 million and $10.0 million equity investments, respectively, in GlobeImmune. In addition, we have a collaboration and option agreement with GlobeImmune focused on the discovery, development and commercialization of novel therapeutics in cancer. As part of this agreement, we made an upfront payment in May 2009 of $30.0 million, which was recorded as research and development expense, to GlobeImmune in return for the option to license compounds and products based on the GI-4000, GI-6200, GI-3000 and GI-10000 oncology drug candidate programs as well as oncology compounds and products resulting from future programs controlled by GlobeImmune. GlobeImmune will be responsible for all discovery and clinical development until we exercise our option with respect to a drug candidate program and GlobeImmune will be entitled to receive potential milestone payments of approximately $230.0 million for the GI-4000 program, $145.0 million for each of the GI-6200, GI-3000 and GI-10000 programs as well as $161.0 million for each additional future program if certain development, regulatory and sales-based milestones are achieved. GlobeImmune will also receive tiered royalties on worldwide net sales.
17
Our options with respect to the GI-4000, GI-6200, GI-3000 and GI-10000 oncology drug candidate programs will terminate if we do not exercise our respective options after delivery of certain reports from GlobeImmune on the completed clinical trials with respect to each drug candidate program, as set forth in the initial development plan specified in the agreement. If we do not exercise our options with respect to any drug candidate program or future program, our option with respect to the oncology products resulting from future programs controlled by GlobeImmune will terminate three years after the last of the options with respect to the GI-4000, GI-6200, GI-3000 and GI-10000 oncology drug candidate programs terminates. Upon exercise of an option, the agreement will continue until we have satisfied all royalty payment obligations to GlobeImmune. Upon the expiration of the agreement, on a product by product, country by country basis, GlobeImmune will grant us an exclusive, fully paid-up, royalty-free perpetual, license to use certain intellectual properties of GlobeImmune to market and sell the compounds and products developed under the agreement. The agreement may expire on a product-by-product and country-by-country basis as we satisfy our royalty payment obligation with respect to each product in each country.
Prior to its expiration as described above, the agreement may be terminated by:
| (i) | us at our sole discretion, or |
| (ii) | either party if the other party: |
| (x) | materially breaches any of its material obligations under the agreement, or |
| (y) | files for bankruptcy. |
If the agreement is terminated by us at our sole discretion or by GlobeImmune for a material breach by us, then our rights to the compounds and products developed under the agreement will revert to GlobeImmune. If the agreement is terminated by us for a material breach by GlobeImmune, then, among other things, our royalty payment obligations under the agreement will be reduced by 50%, our development milestone payment obligations under the agreement will be reduced by 50% or terminated entirely and our sales milestone payment obligations under the agreement will be terminated entirely.
MANUFACTURING
We own and operate an FDA approved API manufacturing facility in Zofingen, Switzerland. The API facility is used to produce REVLIMID ® and THALOMID ® API. We have also contracted with third-party manufacturing service providers in order to maintain backup manufacturing capabilities. These manufacturing service providers manufacture API in accordance with our specifications and are required to meet the FDA's and foreign regulatory authorities' cGMP regulations and guidelines. Our backup API manufacturing service provider is Aptuit Inc. with respect to REVLIMID ® and THALOMID ® .
We own and operate an FDA approved drug product manufacturing facility in Boudry (near Neuchatel), Switzerland to perform formulation, encapsulation, packaging, warehousing and distribution. We maintain backup FDA approved drug product manufacturing service providers for the manufacture of REVLIMID ® and THALOMID ® . These drug product manufacturing service providers include Penn Pharmaceutical Ltd. and Institute of Drug Technology Australia Ltd. Our packaging service providers include Sharp Corporation for worldwide packaging and Acino Holding Ltd. for non-U.S. packaging.
The API for VIDAZA ® is supplied by Ash Stevens, Inc. We also have contract manufacturing agreements with Baxter GmBH and Ben Venue Laboratories, Inc. for VIDAZA ® product formulation, filling vials and packaging. Our packaging service provider for non-U.S. packaging is Catalent Pharma Solutions.
18
The API for FOCALIN ® and FOCALIN XR ® is currently obtained from two suppliers, Johnson Matthey Inc. and Siegfried USA Inc., and we rely on a single manufacturer, Mikart, Inc., for the tableting and packaging of FOCALIN ® finished product.
CCT currently operates an FDA registered facility for the recovery and storage of cord blood and placental stem cells for LifeBankUSA ® . In addition, in our Warren, New Jersey facility we are producing PDA-001, a culture-expanded placenta-derived stem cell under cGMP to supply clinical studies. This is a multi-purpose facility capable of supporting other products.
INTERNATIONAL OPERATIONS
Our international headquarters and a drug product manufacturing facility which performs formulation, encapsulation, packaging, warehousing and distribution are located in Boudry, Switzerland. Our API manufacturing facility located in Zofingen, Switzerland has the capability to produce multiple drug substances and expands our global commercial manufacturing capabilities. We continue to expand our international regulatory, clinical and commercial infrastructure and currently conduct our international operations in over 65 countries and regions including Europe, Latin America, Middle East, Asia/Pacific and Canada. Aside from our international headquarters, the office facilities we maintain in these markets are on a leased basis with terms expiring at various dates between 2010 and 2018.
SALES AND COMMERCIALIZATION
We have a highly trained global pharmaceutical commercial organization with considerable experience in the pharmaceutical industry, specializing products in the areas of oncology and immunology. Our intention is to expand and develop our sales and commercialization capabilities internally as needed in order to support our existing products. We are also positioned to expand our sales and marketing resources as appropriate to take advantage of product acquisition and in-licensing opportunities, and may also consider partnering with other pharmaceutical companies on future products with indications involving large patient populations.
EMPLOYEES
As of December 31, 2009, we had 2,813 full-time company-wide employees, 1,574 engaged primarily in research and development activities, 767 engaged in sales and commercialization activities and the remainder were engaged in executive and general and administrative activities. The number of full-time employees in our international operations has grown from 789 at the end of 2008 to 1,051 at the end of 2009. We also employ a number of part-time employees and maintain consulting arrangements with a number of researchers at various universities and other research institutions around the world.
19
FORWARD-LOOKING STATEMENTS
Certain statements contained or incorporated by reference in this Annual Report are forward-looking statements concerning our business, results of operations, economic performance and financial condition based on our current expectations. Forward-looking statements within the meaning of Section 27A of the Securities Act of 1933 as amended and within the meaning of Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act, are included, for example, in the discussions about:
| • | strategy; |
| • | new product discovery and development; |
| • | current or pending clinical trials; |
| • | our products' ability to demonstrate efficacy or an acceptable safety profile; |
| • | actions by the FDA; |
| • | product manufacturing, including our arrangements with third party suppliers; |
| • | product introduction and sales; |
| • | royalties and contract revenues; |
| • | expenses and net income; |
| • | credit and foreign exchange risk management; |
| • | liquidity; |
| • | asset and liability risk management; and |
| • | operational and legal risks. |
From time to time, we also provide forward-looking statements in other materials we release to the public, as well as oral forward-looking statements. All our forward-looking statements give our then current expectations or forecasts of future events. None of our forward-looking statements are guarantees of future performance, although we believe we have been prudent in our plans and assumptions. Each forward-looking statement involves risks, uncertainties and potentially inaccurate assumptions that could cause actual results to differ materially from those implied by our forward-looking statement. Should known or unknown risks or uncertainties materialize, or should underlying assumptions prove inaccurate, actual results could differ materially from past results and those anticipated, estimated or projected. You should bear this in mind as you consider our forward-looking statements. Given these risks, uncertainties and assumptions, you are cautioned not to place undue reliance on any forward-looking statements.
We have tried, wherever possible, to identify these forward-looking statements by using words such as "forecast," "project," "anticipate," "plan," "strategy," "intend," "potential," "outlook," "target," "seek," "continue," "believe," "could," "estimate," "expect," "may," "probable," "should," "will" or other words of similar meaning in conjunction with, among other things, discussions of our future operations, business plans and prospects, prospective products or product approvals, our strategies for growth, product development and regulatory approval, our expenses, the impact of foreign exchange rates, the outcome of contingencies, such as legal proceedings, and our financial performance and results generally. You also can identify our forward-looking statements by the fact that they do not relate strictly to historical or current facts.
20
We provide in this report a cautionary discussion of risks and uncertainties relevant to our business under the headings "Item 1A. Risk Factors" and "Item 7. Management's Discussion and Analysis of Financial Condition and Results of Operations." We note these factors as permitted by the Private Securities Litigation Reform Act of 1995. These are factors that, individually or in the aggregate, we think could cause our actual results to differ materially from expected and historical results. You should understand, however, that it is not possible to predict or identify all such factors. Consequently, you should not consider the factors that are noted to be a complete discussion of all potential risks or uncertainties.
Except as required under the federal securities laws and the rules and regulations of the Securities and Exchange Commission, or SEC, we disclaim and do not undertake any obligations to update or revise publicly any of our forward-looking statements, including forward-looking statements in this report, whether as a result of new information, future events, changes in assumptions, or otherwise. You are advised, however, to consult any further disclosure we make on related subjects in our Quarterly Reports on Form 10-Q and Current Reports on Form 8-K filed with or furnished to the SEC.
ITEM 1A. RISK FACTORS
The statements in this section describe the major risks to our business and should be considered carefully. Any of the factors described below could significantly and negatively affect our business, prospects, financial condition, operating results or credit ratings, which could cause the trading price of our common stock to decline. The risks described below are not the only risks we may face. Additional risks and uncertainties not presently known to us, or risks that we currently consider immaterial, could also negatively affect our business, our results and operations.
We may experience significant fluctuations in our quarterly operating results which could cause our financial results to be below expectations and cause our stock price to be volatile.
We have historically experienced, and may continue to experience, significant fluctuations in our quarterly operating results. These fluctuations are due to a number of factors, many of which are outside our control, and may result in volatility of our stock price. Future operating results will depend on many factors, including:
| • | demand or lack of demand for our products, including demand that adversely affects our ability to optimize the use of our manufacturing facilities; |
| • | the introduction and pricing of products competitive with ours, including generic competition; |
| • | developments regarding the safety or efficacy of our products; |
| • | regulatory approvals for our products and pricing determinations with respect to our products; |
| • | regulatory approvals for our and our competitor's manufacturing facilities; |
| • | timing and levels of spending for research and development, sales and marketing; |
| • | timing and levels of reimbursement from third-party payors for our products; |
| • | development or expansion of business infrastructure in new clinical and geographic markets; |
| • | the acquisition of new products and companies; |
| • | tax rates in the jurisdictions in which we operate; | ||
| • | timing and recognition of certain research and development milestones and license fees; |
| • | ability to control our costs; |
| • | fluctuations in foreign currency exchange rates; and |
| • | economic and market instability. |
21
We remain dependent on the continued commercial success of our primary products REVLIMID ® , THALOMID ® and VIDAZA ® and a significant decline in demand for or use of these products or our other commercially available products could materially and adversely affect our operating results.
During the next several years, the growth of our business will be largely dependent on the commercial success of REVLIMID ® , THALOMID ® , and VIDAZA ® . We cannot predict whether these or our other existing or new products will be accepted by regulators, physicians, patients and other key opinion leaders as effective drugs with certain advantages over existing or future therapies. We are continuing to introduce our products in additional international markets and to obtain approvals for additional indications both in the United States and internationally. A delay in gaining the requisite regulatory approvals for these markets or indications could negatively impact our growth plans and the value of our stock.
Further, if unexpected adverse experiences are reported in connection with the use of our products, physician and patient comfort with the product could be undermined, the commercial success of such products could be adversely affected and the acceptance of our other products could be negatively impacted. We are subject to adverse event reporting regulations that require us to report to the FDA or similar bodies in other countries if our products are associated with a death or serious injury. These adverse events, among others, could result in additional regulatory controls, such as the performance of costly post-approval clinical studies or revisions to our approved labeling, which could limit the indications or patient population for our products or could even lead to the withdrawal of a product from the market. Similarly, the occurrence of serious adverse events known or suspected to be related to the products could negatively impact product sales. For example, THALOMID ® is known to be toxic to the human fetus and exposure to the drug during pregnancy could result in significant deformities in the baby. REVLIMID ® is also considered fetal toxic and there are warnings against use of VIDAZA ® in pregnant women as well. While we have restricted distribution systems for both THALOMID ® and REVLIMID ® and we endeavor to educate patients regarding the potential known adverse events including pregnancy risks, we can not ensure that all such warnings and recommendations will be complied with or that adverse events resulting from non-compliance will not have a material adverse effect on our business.
It is necessary that our primary products achieve and maintain market acceptance as well as our other products including ISTODAX ® , FOCALIN XR ® and the RITALIN ® family of drugs. A number of factors may adversely impact the degree of market acceptance of our products, including the products' efficacy, safety and advantages, if any, over competing products, as well as the reimbursement policies of third-party payors, such as government and private insurance plans, patent disputes and claims about adverse side effects.
Sales of our products will be significantly reduced if access to and reimbursement for our products by governmental and other third party payors is reduced or terminated.
Sales of our products will depend, in part, on the extent to which the costs of our products will be paid by health maintenance, managed care, pharmacy benefit and similar health care management organizations, or reimbursed by government health administration authorities, private health coverage insurers and other third-party payors. Generally, in Europe and other countries outside the United States, the government-sponsored healthcare system is the primary payor of healthcare costs of patients. These health care management organizations and third-party payors are increasingly challenging the prices charged for medical products and services. Additionally, the containment of health care costs has become a priority of federal and state governments, and the prices of drugs have been a focus in this effort. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. The establishment of limitations on patient access to our drugs, adoption of price controls, and cost-containment measures in new jurisdictions or programs, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could adversely impact our business and our future results. If these organizations and third-party payors do not consider our products to be cost-effective compared to other available therapies, they may not reimburse providers or consumers of our products or, if they do, the level of reimbursement may not be sufficient to allow us to sell our products on a profitable basis.
22
Our ability to sell our products to hospitals in the United States depends in part on our relationships with group purchasing organizations, or GPOs. Many existing and potential customers for our products become members of GPOs. GPOs negotiate pricing arrangements and contracts, sometimes on an exclusive basis, with medical supply manufacturers and distributors, and these negotiated prices are made available to a GPO's affiliated hospitals and other members. If we are not one of the providers selected by a GPO, affiliated hospitals and other members may be less likely to purchase our products, and if the GPO has negotiated a strict sole source, market share compliance or bundling contract for another manufacturer's products, we may be precluded from making sales to members of the GPO for the duration of the contractual arrangement. Our failure to renew contracts with GPOs may cause us to lose market share and could have a material adverse effect on our sales, financial condition and results of operations. We cannot assure you that we will be able to renew these contracts at the current or substantially similar terms. If we are unable to keep our relationships and develop new relationships with GPOs, our competitive position may suffer.
We encounter similar regulatory and legislative issues in most countries outside the United States. International operations are generally subject to extensive governmental price controls and other market regulations, and we believe the increasing emphasis on cost-containment initiatives in Europe and other countries has and will continue to put pressure on the price and usage of our pharmaceutical and medical device products. Although we cannot predict the extent to which our business may be affected by future cost-containment measures or other potential legislative or regulatory developments, additional foreign price controls or other changes in pricing regulation could restrict the amount that we are able to charge for our current and future products, which could adversely affect our revenue and results of operations.
If we do not gain or maintain regulatory approval of our products we will be unable to sell our current products and products in development.
Changes in law, government regulations or policies can have a significant impact on our results of operations. The discovery, preclinical development, clinical trials, manufacturing, risk evaluation and mitigation strategies (such as our S.T.E.P.S. ® and RevAssist ® programs), marketing and labeling of pharmaceuticals and biologics are all subject to extensive laws and regulations, including, without limitation, the U.S. Federal Food, Drug, and Cosmetic Act, the U.S. Public Health Service Act, Medicare Modernization Act, Food and Drug Administration Amendments Act, the U.S. Foreign Corrupt Practices Act, the Sherman Antitrust Act, patent laws, environmental laws, privacy laws and other federal and state statutes, including anti-kickback, antitrust and false claims laws, as well as similar laws in foreign jurisdictions. Enforcement of and changes in laws, government regulations or policies can have a significant adverse impact on our ability to continue to commercialize our products or introduce new products to the market, which would adversely affect our results of operations.
23
If we or our agents, contractors or collaborators are delayed in receiving, or are unable to obtain all, necessary governmental approvals, we will be unable to effectively market our products.
The testing, marketing and manufacturing of our products requires regulatory approval, including approval from the FDA and, in some cases, from the Environmental Protection Agency, or EPA, or governmental authorities outside of the United States that perform roles similar to those of the FDA and EPA, including the EMA, EC, the Swissmedic, the TGA and Health Canada. Certain of our pharmaceutical products, such as FOCALIN ® , fall under the Controlled Substances Act of 1970 that requires authorization by the U.S. Drug Enforcement Agency, or DEA, of the U.S. Department of Justice in order to handle and distribute these products.
The regulatory approval process presents a number of risks to us, principally:
| • | In general, preclinical tests and clinical trials can take many years, and require the expenditure of substantial resources, and the data obtained from these tests and trials can be susceptible to varying interpretation that could delay, limit or prevent regulatory approval; |
| • | Delays or rejections may be encountered during any stage of the regulatory process based upon the failure of the clinical or other data to demonstrate compliance with, or upon the failure of the product to meet, a regulatory agency's requirements for safety, efficacy and quality or, in the case of a product seeking an orphan drug indication, because another designee received approval first or receives approval of other labeled indications; |
| • | Requirements for approval may become more stringent due to changes in regulatory agency policy, or the adoption of new regulations or legislation; |
| • | The scope of any regulatory approval, when obtained, may significantly limit the indicated uses for which a product may be marketed and reimbursed and may impose significant limitations in the nature of warnings, precautions and contra-indications that could materially affect the sales and profitability of the drug; |
| • | Approved products, as well as their manufacturers, are subject to continuing and ongoing review, and discovery of previously unknown problems with these products or the failure to adhere to manufacturing or quality control requirements may result in restrictions on their manufacture, sale or use or in their withdrawal from the market; |
| • | Regulatory authorities and agencies of the United States or foreign governments may promulgate additional regulations restricting the sale of our existing and proposed products, including specifically tailored risk evaluation and mitigation strategies; |
| • | Guidelines and recommendations published by various governmental and non-governmental organizations can reduce the use of our products; |
| • | Once a product receives marketing approval, we may not market that product for broader or different applications, and the FDA may not grant us approval with respect to separate product applications that represent extensions of our basic technology. In addition, the FDA may withdraw or modify existing approvals in a significant manner or promulgate additional regulations restricting the sale of our present or proposed products. The FDA may also request that we perform additional clinical trials or change the labeling of our existing or proposed products if we or others identify side effects after our products are on the market; |
| • | Products, such as REVLIMID ® , that are subject to accelerated approval can be subject to an expedited withdrawal if the post-marketing study commitments are not completed with due diligence, the post-marketing restrictions are not adhered to or are shown to be inadequate to assure the safe use of the drug, or evidence demonstrates that the drug is not shown to be safe and effective under its conditions of use. Additionally, promotional materials for such products are subject to enhanced surveillance, including pre-approval review of all promotional materials used within 120 days following marketing approval and a requirement for the submissions 30 days prior to initial dissemination of all promotional materials disseminated after 120 days following marketing approval; and |
24
| • | Our risk evaluation and mitigation strategies, labeling and promotional activities relating to our products as well as our post-marketing activities are regulated by the FDA, the Federal Trade Commission, The United States Department of Justice, the DEA, state regulatory agencies and foreign regulatory agencies and are subject to associated risks. In addition, individual states, acting through their attorneys general, have become active as well, seeking to regulate the marketing of prescription drugs under state consumer protection and false advertising laws. If we fail to comply with regulations regarding the promotion and sale of our products, appropriate distribution of our products under our restricted distribution systems, prohibition on off-label promotion and the promotion of unapproved products, such agencies may bring enforcement actions against us that could inhibit our commercial capabilities as well as result in significant penalties. |
Other matters that may be the subject of governmental or regulatory action which could adversely affect our business include:
| • | changes in laws and regulations, including without limitation, patent, environmental, privacy, health care and competition laws; |
| • | importation of prescription drugs from outside the U.S. at prices that are regulated by the governments of various foreign countries; |
| • | additional restrictions on interactions with healthcare professionals; and |
| • | privacy restrictions that may limit our ability to share data from foreign jurisdictions. |
We collect placentas and umbilical cord blood for our unrelated allogeneic and private stem cell banking businesses. The FDA's Center for Biologics Evaluation and Research currently regulates human tissue or cells intended for transplantation, implantation, infusion or transfer to a human recipient under 21 CFR Parts 1270 and 1271. Part 1271 requires cell and tissue establishments to screen and test donors, to prepare and follow written procedures for the prevention of the spread of communicable disease and to register the establishment with FDA. This part also provides for inspection by the FDA of cell and tissue establishments. The FDA recently announced that as of October 21, 2011, a BLA will be required to distribute cord blood for unrelated allogeneic use. Currently, we are required to be, and are, licensed to operate in New York, New Jersey, Maryland and California. If other states adopt similar licensing requirements, we would need to obtain such licenses to continue operating our stem cell banking businesses. If we are delayed in receiving, or are unable to obtain at all, necessary licenses, we will be unable to provide services in those states and this could impact negatively on our revenues.
Our products may face competition from lower cost generic or follow-on products and providers of these products may be able to sell them at a substantially lower cost than us.
Generic drug manufactures are seeking to compete with our drugs and will become an important challenge to us. Our success depends, in part, on our ability to obtain and enforce patents, protect trade secrets, obtain licenses to technology owned by third parties and to conduct our business without infringing upon the proprietary rights of others. The patent positions of pharmaceutical and biopharmaceutical companies, including ours, can be uncertain and involve complex legal and factual questions including those related to our risk evaluation and mitigation strategies (such as our S.T.E.P.S. ® and RevAssist ® programs).
25
Furthermore, even if our patent applications, or those we have licensed-in, are issued, our competitors may challenge the scope, validity or enforceability of such patents in court, requiring us to engage in complex, lengthy and costly litigation. Alternatively, our competitors may be able to design around our owned or licensed patents and compete with us using the resulting alternative technology. If any of our issued or licensed patents are infringed or challenged, we may not be successful in enforcing or defending our or our licensor's intellectual property rights and subsequently may not be able to develop or market the applicable product exclusively.
Upon the expiration or loss of patent protection for one of our products, or upon the "at-risk" launch (despite pending patent infringement litigation against the generic product) by a generic manufacturer of a generic version of one of our products, we can quickly lose a significant portion of our sales of that product, which can adversely affect our business. In addition, if generic versions of our competitors' branded products lose their market exclusivity, our patented products may face increased competition which can adversely affect our business.
The FDA approval process allows for the approval of an ANDA or 505(b)(2) application for a generic version of our approved products upon the expiration, through passage of time or successful legal challenge, of relevant patent or non-patent exclusivity protection. Generic manufacturers pursuing ANDA approvals are not required to conduct costly and time-consuming clinical trials to establish the safety and efficacy of their products; rather, they are permitted to rely on the innovator's data regarding safety and efficacy. Thus, generic manufacturers can sell their products at prices much lower than those charged by the innovative pharmaceutical or biotechnology companies who have incurred substantial expenses associated with the research and development of the drug product. Accordingly, while our products currently may retain certain regulatory and or patent exclusivity; our products are or will be subject to ANDA applications to the FDA in light of the Hatch-Waxman Amendments to the Federal Food, Drug, and Cosmetic Act. The ANDA procedure includes provisions allowing generic manufacturers to challenge the effectiveness of the innovator's patent protection prior to the generic manufacturer actually commercializing their products-the so-called "Paragraph IV" certification procedure. In recent years, generic manufacturers have used Paragraph IV certifications extensively to challenge the applicability of Orange Book-listed patents on a wide array of innovative pharmaceuticals, and we expect this trend to continue and to implicate drug products with even relatively modest revenues. During the exclusivity periods, the FDA is generally prevented from granting effective approval of an ANDA. Upon the expiration of the applicable exclusivities, through passage of time or successful legal challenge, the FDA may grant effective approval of an ANDA for a generic drug, or may accept reference to a previously protected NDA in a 505(b)(2) application. Further, upon such expiration event, the FDA may require a generic competitor to participate in some form of risk management system which could include our participation as well. Depending upon the scope of the applicable exclusivities, any such approval could be limited to certain formulations and/or indications/claims, i.e., those not covered by any outstanding exclusivities.
If an ANDA filer or a generic manufacturer were to receive approval to sell a generic or follow-on version of one of our products, that product would become subject to increased competition and our revenues for that product would be adversely affected.
26
If we are not able to effectively compete our business will be adversely affected.
The pharmaceutical and biotech industry in which we operate is highly competitive and subject to rapid and significant technological change. Our present and potential competitors include major pharmaceutical and biotechnology companies, as well as specialty pharmaceutical firms, including, but not limited to:
| • | Takeda and Johnson & Johnson, compete with REVLIMID ® and THALOMID ® in the treatment of multiple myeloma and in clinical trials with our compounds; |
| • | Eisai Co., Ltd., SuperGen, Inc. and Johnson & Johnson compete or may potentially compete with VIDAZA ® ; |
| • | Amgen, which potentially competes with our TNF- a and kinase inhibitors; |
| • | AstraZeneca plc, which potentially competes in clinical trials with our compounds and TNF- a inhibitors; |
| • | Biogen Idec Inc. and Genzyme Corporation, both of which are generally developing drugs that address the oncology and immunology markets; |
| • | Bristol Myers Squibb Co., which potentially competes in clinical trials with our compounds and TNF- a inhibitors; |
| • | F. Hoffman-La Roche Ltd., which potentially competes in clinical trials with our IMiDs ® compounds and TNF- a inhibitors; |
| • | Johnson & Johnson, which potentially competes with certain of our proprietary programs, including our oral anti-inflammatory programs; |
| • | Novartis, which potentially competes with our compounds and kinase programs; and |
| • | Pfizer, which potentially competes in clinical trials with our kinase inhibitors. |
Many of these companies have considerably greater financial, technical and marketing resources than we do. This enables them, among other things, to make greater research and development investments and spread their research and development costs, as well as their marketing and promotion costs, over a broader revenue base. Our competitors may also have more experience and expertise in obtaining marketing approvals from the FDA, and other regulatory authorities. We also experience competition from universities and other research institutions, and in some instances, we compete with others in acquiring technology from these sources. The pharmaceutical industry has undergone, and is expected to continue to undergo, rapid and significant technological change, and we expect competition to intensify as technical advances in the field are made and become more widely known. The development of products, including generics, or processes by our competitors with significant advantages over those that we are seeking to develop could cause the marketability of our products to stagnate or decline.
We may be required to modify our business practices, pay fines and significant expenses or experience losses due to governmental investigations or other litigation.
From time to time, we may be subject to governmental investigation or litigation on a variety of matters, including, without limitation, regulatory, intellectual property, product liability, antitrust, consumer, commercial, securities and employment litigation and claims and other legal proceedings that may arise from the conduct of our business as currently conducted or as conducted in the future.
In particular, we are subject to significant product liability risks as a result of the testing of our products in human clinical trials and for products that we sell after regulatory approval.
Pharmaceutical companies involved in Hatch-Waxman litigation are often subject to follow-on lawsuits and governmental investigations, which may be costly and could result in lower-priced generic products that are competitive with our products being introduced to the market.
27
In the fourth quarter of 2009, we received a civil inquiry and demand from the Federal Trade Commission (FTC). The FTC requested documents and other information relating to requests by generic companies to purchase our patented THALOMID ® and REVLIMID ® brand drugs in order to evaluate whether there is reason to believe that we have engaged in unfair methods of competition. We continue to cooperate with the FTC's request for information.
Litigation and governmental investigations are inherently unpredictable and may:
| • | result in rulings that are materially unfavorable to us, including a requirement that we pay significant damages, fines or penalties or prevent us from operating our business in a certain manner; |
| • | cause us to change our business operations to avoid perceived risks associated with such litigation or investigations; |
| • | have an adverse affect on our reputation and the demand for our products; and |
| • | require the expenditure of significant time and resources, which may divert the attention of our management and interfere with the pursuit of our strategic objectives. |
While we maintain insurance for certain risks, the amount of our insurance coverage may not be adequate to cover the total amount of all insured claims and liabilities. It also is not possible to obtain insurance to protect against all potential risks and liabilities. If any litigation or governmental investigation were to have a material adverse result, there could be a material impact on our results of operations, cash flows, or financial position. See also Legal Proceedings contained in Part I, Item 3 of this Annual Report on Form 10-K.
The development of new biopharmaceutical products involves a lengthy and complex process, and we may be unable to commercialize any of the products we are currently developing.
Many of our drug candidates are in the early or mid-stages of research and development and will require the commitment of substantial financial resources, extensive research, development, preclinical testing, clinical trials, manufacturing scale-up and regulatory approval prior to being ready for sale. This process involves a high degree of risk and takes many years. Our product development efforts with respect to a product candidate may fail for many reasons, including the failure of the product candidate in preclinical studies; adverse patient reactions to the product candidate or indications or other safety concerns; insufficient clinical trial data to support the effectiveness or superiority of the product candidate; our inability to manufacture sufficient quantities of the product candidate for development or commercialization activities in a timely and cost-efficient manner; our failure to obtain, or delays in obtaining, the required regulatory approvals for the product candidate, the facilities or the process used to manufacture the product candidate; or changes in the regulatory environment, including pricing and reimbursement, that make development of a new product or of an existing product for a new indication no longer desirable. Moreover, our commercially available products may require additional studies with respect to approved indications as well as new indications pending approval.
The stem cell products that we are developing through our CCT subsidiary may represent substantial departures from established treatment methods and will compete with a number of traditional products and therapies which are now, or may be in the future, manufactured and marketed by major pharmaceutical and biopharmaceutical companies. Furthermore, public attitudes may be influenced by claims that stem cell therapy is unsafe, and stem cell therapy may not gain the acceptance of the public or the medical community.
28
Due to the inherent uncertainty involved in conducting clinical studies, we can give no assurances that our studies will have a positive result or that we will receive regulatory approvals for our new products or new indications.
Manufacturing and distribution risks including a disruption at certain of our manufacturing sites would significantly interrupt our production capabilities, which could result in significant product delays and adversely affect our results.
We have our own manufacturing facilities for many of our products and we have contracted with third party manufacturers and distributors to provide API, encapsulation, finishing services packaging and distribution services to meet our needs. These risks include the possibility that our or our suppliers' manufacturing processes could be partially or completely disrupted by a fire, natural disaster, terrorist attack, governmental action or military action. In the case of a disruption, we may need to establish alternative manufacturing sources for these products. This would likely lead to substantial production delays as we build or locate replacement facilities and seek and obtain the necessary regulatory approvals. If this occurs, and our finished goods inventories are insufficient to meet demand, we may be unable to satisfy customer orders on a timely basis, if at all. Further, our business interruption insurance may not adequately compensate us for any losses that may occur and we would have to bear the additional cost of any disruption. For these reasons, a significant disruptive event at certain of our manufacturing facilities or sites could materially and adversely affect our business and results of operations. In addition, if we fail to predict market demand for our products, we may be unable to sufficiently increase production capacity to satisfy demand or may incur costs associated with excess inventory that we manufacture.
In all the countries where we sell our products, governmental regulations exist to define standards for manufacturing, packaging, labeling, distribution and storing. All of our suppliers of raw materials, contract manufacturers and distributors must comply with these regulations as applicable. In the United States, the FDA requires that all suppliers of pharmaceutical bulk material and all manufacturers of pharmaceuticals for sale in or from the United States achieve and maintain compliance with the FDA's cGMP regulations and guidelines. Our failure to comply, or failure of our third-party manufacturers to comply with applicable regulations could result in sanctions being imposed on them or us, including fines, injunctions, civil penalties, disgorgement, suspension or withdrawal of approvals, license revocation, seizures or recalls of products, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supplies of our products. In addition, before any product batch produced by our manufacturers can be shipped, it must conform to release specifications pre-approved by regulators for the content of the pharmaceutical product. If the operations of one or more of our manufacturers were to become unavailable for any reason, any required FDA review and approval of the operations of an alternative supplier could cause a delay in the manufacture of our products.
If our outside manufacturers do not meet our requirements for quality, quantity or timeliness, or do not achieve and maintain compliance with all applicable regulations, our ability to continue supplying such products at a level that meets demand could be adversely affected.
We have contracted with specialty distributors, to distribute THALOMID ® , REVLIMID ® and VIDAZA ® in the United States. If our distributors fail to perform and we cannot secure a replacement distributor within a reasonable period of time, we may experience adverse effects to our business and results of operations.
We are continuing to establish marketing and distribution capabilities in international markets with respect to our products. At the same time, we are in the process of obtaining necessary governmental and regulatory approvals to sell our products in certain countries. If we have not successfully completed and implemented adequate marketing and distribution support services upon our receipt of such approvals, our ability to effectively launch our products in these countries would be severely restricted.
29
The consolidation of drug wholesalers and other wholesaler actions could increase competitive and pricing pressures on pharmaceutical manufacturers, including us.
We sell our pharmaceutical products in the United States primarily through wholesale distributors and contracted pharmacies. These wholesale customers comprise a significant part of the distribution network for pharmaceutical products in the United States. This distribution network is continuing to undergo significant consolidation. As a result, a smaller number of large wholesale distributors control a significant share of the market. We expect that consolidation of drug wholesalers will increase competitive and pricing pressures on pharmaceutical manufacturers, including us. In addition, wholesalers may apply pricing pressure through fee-for-service arrangements, and their purchases may exceed customer demand, resulting in reduced wholesaler purchases in later quarters. We cannot assure you that we can manage these pressures or that wholesaler purchases will not decrease as a result of this potential excess buying.
Risks from the improper conduct of employees, agents or contractors or collaborators could adversely affect our business or reputation.
We cannot ensure that our compliance controls, policies and procedures will in every instance protect us from acts committed by our employees, agents, contractors or collaborators that would violate the laws or regulations of the jurisdictions in which we operate, including without limitation, employment, foreign corrupt practices, environmental, competition and privacy laws. Such improper actions could subject us to civil or criminal investigations, monetary and injunctive penalties and could adversely impact our ability to conduct business, results of operations and reputation.
We may face significant challenges in effectively integrating entities and businesses that we acquire and we may not realize the benefits that we anticipate from any such acquisition.
Achieving the anticipated benefits of our acquisition of entities will depend in part upon whether we can integrate our businesses in an efficient and effective manner. Our integration of these entities involves a number of risks, including, but not limited to:
| • | demands on management related to the increase in our size after the acquisition; | |
| • | the diversion of management's attention from the management of daily operations to the integration of operations; | |
| • | failure of the acquired entity to meet or exceed our expected returns; | |
| • | higher integration costs than anticipated; | |
| • | failure to achieve expected synergies and costs savings; | |
| • | difficulties in the assimilation and retention of employees; | |
| • | difficulties in the assimilation of different cultures and practices, as well as in the assimilation of broad and geographically dispersed personnel and operations; and | |
| • | difficulties in the integration of departments, systems, including accounting systems, technologies, books and records, and procedures, as well as in maintaining uniform standards, controls (including internal control over financial reporting required by Section 404 of the Sarbanes-Oxley Act of 2002) and related procedures and policies. |
30
If we cannot successfully integrate acquired businesses, we may experience material negative consequences to our business, financial condition or results of operations.
Our failure to attract and retain key managerial, technical, scientific, selling and marketing personnel could adversely affect our business.
The success of our business depends, in large part, on our continued ability to (i) attract and retain highly qualified management, scientific, manufacturing and sales and marketing personnel, (ii) successfully integrate large numbers of new employees into our corporate culture and (iii) develop and maintain important relationships with leading research and medical institutions and key distributors. Competition for these types of personnel and relationships is intense.
Among other benefits, we use share-based compensation to attract and retain personnel. Share-based compensation accounting rules require us to recognize all share-based compensation costs as expenses. These or other factors could reduce the number of shares and options management and our board of directors grants under our incentive plan. We cannot be sure that we will be able to attract or retain skilled personnel or maintain key relationships, or that the costs of retaining such personnel or maintaining such relationships will not materially increase.
We could be subject to significant liability as a result of risks associated with using hazardous materials in our business.
We use certain hazardous materials in our research, development, manufacturing and general business activities. While we believe we are currently in substantial compliance with the federal, state and local laws and regulations governing the use of these materials, we cannot be certain that accidental injury or contamination will not occur. If an accident or environmental discharge occurs, or if we discover contamination caused by prior operations, including by prior owners and operators of properties we acquire, we could be liable for cleanup obligations, damages and fines. This could result in substantial liabilities that could exceed our insurance coverage and financial resources. Additionally, the cost of compliance with environmental and safety laws and regulations may increase in the future, requiring us to expend more financial resources either in compliance or in purchasing supplemental insurance coverage.
Changes in our effective income tax rate could adversely affect our results of operations.
We are subject to income taxes in both the United States and various foreign jurisdictions, and our domestic and international tax liabilities are dependent upon the distribution of income among these different jurisdictions. Various factors may have favorable or unfavorable effects on our effective income tax rate. These factors include, but are not limited to, interpretations of existing tax laws, the accounting for stock options and other share-based compensation, changes in tax laws and rates, future levels of research and development spending, changes in accounting standards, changes in the mix of earnings in the various tax jurisdictions in which we operate, the outcome of examinations by the Internal Revenue Service and other jurisdictions, the accuracy of our estimates for unrecognized tax benefits and realization of deferred tax assets, and changes in overall levels of pre-tax earnings. The impact on our income tax provision resulting from the above-mentioned factors may be significant and could have an impact on our results of operations.
Currency fluctuations and changes in exchange rates could increase our costs and may cause our profitability to decline.
We collect and pay a substantial portion of our sales and expenditures in currencies other than the U.S. dollar. Therefore, fluctuations in foreign currency exchange rates affect our operating results.
31
We utilize foreign currency forward contracts to manage foreign currency risk, but not to engage in currency speculation. We use these forward contracts to hedge certain forecasted transactions and balance sheet exposures denominated in foreign currencies. We use derivative instruments, including those not designated as part of a hedging transaction, to manage our exposure to movements in foreign exchange rates. The use of these derivative instruments mitigates the exposure of these risks with the intent to reduce our risk or cost but may not fully offset any change in operating results that result from fluctuations in foreign currencies. Any significant foreign exchange rate fluctuations could adversely affect our financial condition and results of operations.
We may experience an adverse market reaction if we are unable to meet our financial reporting obligations.
As we continue to expand at a rapid pace, the development of new and/or improved automated systems will remain an ongoing priority. During this expansion period, our internal control over financial reporting may not prevent or detect misstatements in our financial reporting. Such misstatements may result in litigation and/or negative publicity and possibly cause an adverse market reaction that may negatively impact our growth plans and the value of our common stock.
The decline of global economic conditions could adversely affect our results of operations.
Sales of our products are dependent, in large part, on reimbursement from government health administration authorities, private health insurers, distribution partners and other organizations. As a result of the current global credit and financial market conditions, these organizations may be unable to satisfy their reimbursement obligations or may delay payment. In addition, U.S. federal and state health authorities may reduce Medicare and Medicaid reimbursements, and private insurers may increase their scrutiny of claims. A reduction in the availability or extent of reimbursement could negatively affect our product sales, revenue and cash flows.
Due to the recent tightening of global credit, there may be a disruption or delay in the performance of our third-party contractors, suppliers or collaborators. We rely on third parties for several important aspects of our business, including portions of our product manufacturing, royalty revenue, clinical development of future collaboration products, conduct of clinical trials and raw materials. If such third parties are unable to satisfy their commitments to us, our business could be adversely affected.
The price of our common stock may fluctuate significantly and you may lose some or all of your investment in us.
The market for our shares of common stock may be subject to disruptions that could cause volatility in its price. In general, the current global economic crisis has caused substantial market volatility and instability. Any such disruptions or continuing volatility may adversely affect the value of our common stock. In addition to current global economic instability in general, the following key factors may have an adverse impact on the market price of our common stock:
| • | results of our clinical trials or adverse events associated with our marketed products; |
| • | fluctuations in our commercial and operating results; |
| • | announcements of technical or product developments by us or our competitors; |
| • | market conditions for pharmaceutical and biotechnology stocks in particular; |
32
| • | stock market conditions generally; |
| • | changes in governmental regulations and laws, including, without limitation, changes in tax laws, health care legislation, environmental laws, competition laws, and patent laws; |
| • | new accounting pronouncements or regulatory rulings; |
| • | public announcements regarding medical advances in the treatment of the disease states that we are targeting; |
| • | patent or proprietary rights developments; |
| • | changes in pricing and third-party reimbursement policies for our products; |
| • | the outcome of litigation involving our products or processes related to production and formulation of those products or uses of those products; |
| • | other litigation or governmental investigations; |
| • | competition; and |
| • | investor reaction to announcements regarding business or product acquisitions. |
In addition, our operations may be materially affected by conditions in the global markets and economic conditions throughout the world, including the current global economic and market instability. The global market and economic climate may continue to deteriorate because of many factors beyond our control, including continued economic instability and market volatility, rising interest rates or inflation, terrorism or political uncertainty. In the event of a continued or future market downturn in general and/or the biotechnology sector in particular, the market price of our common stock may be adversely affected.
A breakdown or breach of our information technology systems could subject us to liability or interrupt the operation of our business.
We rely upon our information technology systems and infrastructure for our business. The size and complexity of our computer systems make them potentially vulnerable to breakdown, malicious intrusion and random attack. Likewise, data privacy breaches by employees and others who access our systems may pose a risk that sensitive data may be exposed to unauthorized persons or to the public. While we believe that we have taken appropriate security measures to protect our data and information technology systems, there can be no assurance that our efforts will prevent breakdowns or breaches in our systems that could adversely affect our business.
We have certain charter and by-law provisions that may deter a third-party from acquiring us and may impede the stockholders' ability to remove and replace our management or board of directors.
Our board of directors has the authority to issue, at any time, without further stockholder approval, up to 5,000,000 shares of preferred stock, and to determine the price, rights, privileges and preferences of those shares. An issuance of preferred stock could discourage a third-party from acquiring a majority of our outstanding voting stock. Additionally, our board of directors has adopted certain amendments to our by-laws intended to strengthen the board's position in the event of a hostile takeover attempt. These provisions could impede the stockholders' ability to remove and replace our management and/or board of directors. Furthermore, we are subject to the provisions of Section 203 of the Delaware General Corporation Law, an anti-takeover law, which may also dissuade a potential acquirer of our common stock.
33
AVAILABLE INFORMATION
Our current reports on Form 8-K, quarterly reports on Form 10-Q and Annual Reports on Form 10-K are electronically filed with or furnished to the SEC, and all such reports and amendments to such reports filed have been and will be made available, free of charge, through our website ( http://www.celgene.com ) as soon as reasonably practicable after such filing. Such reports will remain available on our website for at least 12 months. The contents of our website are not incorporated by reference into this Annual Report. The public may read and copy any materials filed by us with the SEC at the SEC's Public Reference Room at 100 F Street, NW, Washington, D.C. 20549.
The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains an Internet site (http://www.sec.gov) that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 2. PROPERTIES
Our corporate headquarters, which is located in Summit, New Jersey on approximately 45 acres of land, was purchased in 2004 and consists of several buildings, which house our administrative, sales, marketing and research functions.
Our international headquarters is located in Boudry, Switzerland and includes a drug product manufacturing facility to perform formulation, encapsulation, packaging, warehousing and distribution. We operate an API manufacturing facility located in Zofingen, Switzerland which has the capability to produce multiple drug substances. The facility is being used to produce REVLIMID ® and THALOMID ® API to supply global markets and may also be used to produce drug substance for our future drugs and drug candidates.
We occupy the following facilities, located in the United States, under operating lease arrangements that have remaining lease terms greater than one year. Under these lease arrangements, we also are required to reimburse the lessors for real estate taxes, insurance, utilities, maintenance and other operating costs. All leases are with unaffiliated parties.
| • | 78,000 square feet of office space in Basking Ridge, New Jersey with a term ending in September 2011 at an annual cost of $1.4 million. | ||
| • | 73,500 square feet of laboratory and office space in Warren, New Jersey. The two leases for this facility extend through May 2012 and July 2010, respectively, and contain five-year renewal options. Annual rent for these facilities is approximately $1.1 million. | ||
| • | 23,500 square feet of office space in Warren, New Jersey with a term ending in September 2010 at an annual cost of $0.5 million. |
34
| • | 20,800 square feet of office and laboratory space in Cedar Knolls, New Jersey. The lease for this facility has a term ending in October 2010 with renewal options for additional five-year terms. Annual rent for this facility is approximately $0.3 million and is subject to specified annual rental increases. | ||
| • | 78,200 square feet of laboratory and office space in San Diego, California. The lease for this facility has a term ending in August 2012 with one five-year renewal option. Annual rent for this facility is approximately $2.2 million and is subject to specified annual rental increases. | ||
| • | 55,900 square feet of office and research space in San Francisco, California with a term ending in September 2016 at an annual cost of $2.4 million. | ||
| • | 27,700 square feet of office space in Overland Park, Kansas with a term ending in May 2011 at an annual cost of $0.4 million. |
We also lease a number of offices under various lease agreements in Europe, Canada and Asia/Pacific. The minimum annual rents may be subject to specified annual rent increases. At December 31, 2009, the non-cancelable lease terms for these operating leases expire at various dates between 2010 and 2017 and in some cases include renewal options. The total amount of rent expense recorded for leased facilities in 2009 was $20.5 million.
ITEM 3. LEGAL PROCEEDINGS
We and certain of our subsidiaries are involved in various patent, commercial and other claims; government investigations; and other legal proceedings that arise from time to time in the ordinary course of our business.
Patent proceedings include challenges to scope, validity or enforceability of our patents relating to our various products or processes. Although we believe we have substantial defenses to these challenges with respect to all our material patents, there can be no assurance as to the outcome of these matters, and a loss in any of these cases could result in a loss of patent protection for the drug at issue, which could lead to a significant loss of sales of that drug and could materially affect future results of operations.
THALOMID ®
Barr Laboratories, Inc., or Barr, a generic drug manufacturer located in Pomona, New York, filed an ANDA for the treatment of ENL in the manner described in our label and seeking permission from the FDA to market a generic version of 50mg, 100mg and 200mg THALOMID ® . Barr has notified us that it merged with Teva, and Barr is now Barr Pharmaceuticals, LLC, a wholly-owned subsidiary of Teva. Under the federal Hatch-Waxman Act of 1984, any generic manufacturer may file an ANDA with a certification (a "Paragraph IV certification") challenging the validity or infringement of a patent listed in the FDA's Orange Book four years after the pioneer company obtains approval of its NDA. On or after December 5, 2006, Barr mailed notices of Paragraph IV certifications alleging that the following patents listed for THALOMID ® in the Orange Book are invalid, unenforceable, and/or not infringed: U.S. Patent Nos. 6,045,501 ("the '501 patent"), 6,315,720 ("the '720 patent"), 6,561,976 ("the '976 patent"), 6,561,977 ("the '977 patent"), 6,755,784 ("the '784 patent"), 6,869,399 ("the '399 patent"), 6,908,432 ("the '432 patent"), and 7,141,018 ("the ‘018 patent"). The ‘501, ‘976, and ‘432 patents do not expire until August 28, 2018, while the remaining patents do not expire until October 23, 2020. On January 18, 2007, we filed an infringement action in the U.S. District Court of New Jersey against Barr. By bringing suit, we are entitled to a 30-month stay, from the date of our receipt of the Paragraph IV certification, against the FDA's approval of a generic applicant's application to market a generic version of THALOMID ® . In June 2007, U.S. Patent No. 7,230,012, or ‘012 patent, was issued to us claiming formulations of thalidomide and was then timely listed in the Orange Book. Barr sent us a supplemental Paragraph IV certification against the ‘012 patent and alleged that the claims of the ‘012 patent, directed to formulations which encompass THALOMID ® , were invalid. On August 23, 2007, we filed an infringement action in the U.S. District Court of New Jersey with respect to the ‘012 patent. On or after October 4, 2007, Barr filed a second supplemental notice of Paragraph IV certifications relating to the 150mg dosage strength of THALOMID ® alleging that the '501 patent, '720 patent, '976 patent, '977 patent, '784 patent, '399 patent, '432 patent and the '018 patent are invalid, unenforceable, and/or not infringed. On November 14, 2007, we filed an infringement action in the U.S. District Court of New Jersey against Barr which entitled us to a second 30-month stay, expiring in November 2010. All three actions have subsequently been consolidated. We intend to enforce our patent rights. If the ANDA is approved by the FDA, and Barr is successful in challenging our patents listed in the Orange Book for THALOMID ® , Barr would be permitted to sell a generic thalidomide product. If we are unsuccessful in the suits and the FDA were to approve a comprehensive education and risk-management distribution program for a generic version of thalidomide, sales of THALOMID ® could be significantly reduced in the United States by the entrance of a generic thalidomide product, consequently reducing our revenue.
35
In July 2008, we and our co-plaintiff Children's Medical Center Corp., or CMCC, asserted two Orange-Book listed patents (U.S. Patent Nos. 5,629,327 and 6,235,756) relating to uses of thalidomide for the treatment of various cancers, including multiple myeloma. We filed the action in response to Notices of Paragraph IV certification in connection with Barr's ANDA seeking approval to market generic versions for our THALOMID ® capsules. Because both of those patents were listed in the Orange Book when Barr originally filed its ANDA (Barr originally failed to certify under Paragraph IV against either patent), a second 30-month stay applies, and Barr's ANDA may not receive final approval until November 2010. Barr has asserted counterclaims seeking declarations of noninfringement, invalidity, and unenforceability. In December 2008, we and CMCC asserted a third Orange-Book patent relating to uses of thalidomide for the treatment of various cancers, including multiple myeloma. We filed the action in response to Notices of Paragraph IV certification in connection with Barr's ANDA seeking approval to market generic versions for our THALOMID ® capsules. Barr has asserted counterclaims seeking declarations of noninfringement and invalidity. All of the above thalidomide actions have been consolidated.
The parties have completed the bulk of fact discovery, and general fact discovery is now closed. The parties expect the Court to resolve Barr's motion in February 2010. No schedule has been set for claim construction or expert discovery. No trial date has been set.
FOCALIN ® and FOCALIN XR ®
On August 19, 2004, we, together with our exclusive licensee Novartis, filed an infringement action in the U.S. District Court of New Jersey against Teva Pharmaceuticals USA, Inc., or Teva, in response to notices of Paragraph IV certifications made by Teva in connection with the filing of an ANDA for FOCALIN ® . The notification letters from Teva contend that U.S. Patent Nos. 5,908,850, or ‘850 patent, and 6,355,656, or ‘656 patent, are invalid. After the suit was filed, Novartis listed another patent, U.S. Patent No. 6,528,530, or ‘530 patent, in the Orange Book in association with the FOCALIN ® NDA. The original 2004 action asserted infringement of the ‘850 patent. Teva amended its answer during discovery to contend that the ‘850 patent was not infringed by the filing of its ANDA, and that the ‘850 patent is not enforceable due to an allegation of inequitable conduct. Fact discovery in the original 2004 action expired on February 28, 2006. At about the time of the filing of the ‘850 patent infringement action, reexamination proceedings for the ‘656 patent were initiated in the U.S. PTO. On September 28, 2006, the U.S. PTO issued a Notice of Intent to Issue Ex Parte Reexamination Certificate, and on March 27, 2007, the Reexamination Certificate for the ‘656 patent issued. On December 21, 2006, we and Novartis filed an action in the U.S. District Court of New Jersey against Teva for infringement of the ‘656 patent. Teva filed an amended answer and counterclaim on March 23, 2007. The amended counterclaim seeks a declaratory judgment of patent invalidity, noninfringement, and unenforceability. The statutory 30-month stay, to which Paragraph IV certifications (including those below) are entitled to, expired on January 9, 2007, and Teva proceeded to market with a generic version of FOCALIN ® . Plaintiffs' complaints included a request for an injunction against future sales of Teva's generic products, as well as a claim for money damages for actual sales. This action has been resolved pursuant to a confidential settlement agreement dated December 9, 2009. Pursuant to the settlement agreement, the parties sought (and the Court allowed) a 60-day stay of the litigation, in order to allow for review of the settlement agreement by the Federal Trade Commission and Department of Justice. The case was dismissed on February 1, 2010.
36
On September 14, 2007, we, together with our exclusive licensee Novartis, filed an infringement action in the U.S. District Court for the District of New Jersey against Teva Pharmaceuticals USA, Inc. in response to a notice of a Paragraph IV certification made by Teva in connection with the filing of an ANDA for FOCALIN XR ® . The notification letter from Teva contends that claims in U.S. Patent Nos. 5,908,850 and 6,528,530 are invalid, unenforceable, and not infringed by the proposed Teva products, and it contends that U.S. Patent Nos. 5,837,284 and 6,635,284 are invalid and not infringed by the proposed Teva products. We and Novartis asserted each of these patents and additionally asserted U.S. Patent No. 6,355,656 in our complaint against Teva. Subsequently, plaintiffs added claims for infringement of U.S. Patent No. 7,431,944. This action has been resolved pursuant to a confidential settlement agreement dated December 9, 2009. Pursuant to the settlement agreement, the parties sought (and the Court allowed) a 60-day stay of the litigation, in order to allow for review of the settlement agreement by the Federal Trade Commission and Department of Justice. The case was dismissed on February 1, 2010.
On October 5, 2007, we, together with our exclusive licensee Novartis, filed an infringement action in the U.S. District Court for the District of New Jersey against IntelliPharmaCeutics Corp., or IPC, in response to a notice of a Paragraph IV certification made by IPC in connection with the filing of an ANDA for FOCALIN XR ® . The notification letter from IPC contends that claims in U.S. Patent Nos. 5,908,850, 5,837,284, and 6,635,284 are not infringed by the proposed IPC products. The notification letter also contends that claims in U.S. Patent Nos. 5,908,850, 6,355,656, 6,528,530, 5,837,284, and 6,635,284 are invalid, and that claims in U.S. Patent Nos. 5,908,850, 6,355,656 and 6,528,530 are unenforceable. In our complaint against IPC, we and Novartis asserted U.S. Patent Nos. 5,908,850, 6,355,656, 6,528,530, 5,837,284, and 6,635,284. IPC filed an answer and counterclaim on November 20, 2007. The counterclaim seeks a declaratory judgment of patent invalidity, non infringement, and unenforceability with respect to Patent Nos. 5,908,850, 6,355,656, and 6,528,530, and it seeks a declaratory judgment of patent invalidity and non infringement with respect to Patent Nos. 5,837,284 and 6,635,284. We and Novartis subsequently added claims against IPC for infringement of United States patent No. 7,431,944. Fact discovery has expired and claim construction briefing has been completed. Expert discovery has yet to be completed. On October 23, 2009, the court administratively struck the pleadings relating to claim construction, in order to afford the parties a chance to determine whether a settlement can be reached. If we are unsuccessful in proving infringement or defending our patents, Novartis' sales of FOCALIN XR ® could be significantly reduced in the United States by the entrance of a generic FOCALIN XR ® product, consequently reducing our revenue from royalties associated with these sales. If settlement cannot be reached, the claim construction and other litigation proceedings will move forward.
On November 8, 2007, we, together with our exclusive licensee Novartis, filed an infringement action in the U.S. District Court for the District of New Jersey against Actavis South Atlantic LLC and Abrika Pharmaceuticals, Inc. (collectively, "Actavis") in response to a notice of a Paragraph IV certification made by Actavis in connection with the filing of an ANDA for FOCALIN XR ® . The notification letter from Actavis contends that claims in U.S. Patent Nos. 5,908,850, 6,355,656, 5,837,284, and 6,635,284 are not infringed by the proposed Actavis products, and it contends that claims in U.S. Patent Nos. 5,908,850, 6,355,656, 6,528,530, 5,837,284 and 6,635,284 are invalid. In our complaint against Actavis, we and Novartis asserted U.S. Patent Nos. 5,908,850, 6,355,656, 6,528,530, 5,837,284, and 6,635,284. Actavis filed an answer and counterclaim, seeking a declaratory judgment of patent invalidity, non-infringement, and unenforceability with respect to the patents-in-suit. Plaintiffs subsequently added claims against Actavis for infringement of U.S. Patent No. 7,431,944. Fact discovery has expired and claim construction briefing has been completed. Expert discovery has yet to be completed. No trial date has been set. On October 23, 2009, the court administratively struck the pleadings relating to claim construction, in order to afford the parties a chance to determine whether a settlement can be reached. If we are unsuccessful in proving infringement or defending our patents, Novartis' sales of FOCALIN XR ® could be significantly reduced in the United States by the entrance of a generic FOCALIN XR ® product, consequently reducing our revenue from royalties associated with these sales. If settlement cannot be reached, the claim construction and other litigation proceedings will move forward.
37
On November 16, 2007, we, together with our exclusive licensee Novartis, filed an infringement action in the U.S. District Court for the District of New Jersey against Barr and Barr Pharmaceuticals, Inc. in response to a notice of a Paragraph IV certification made by Barr in connection with the filing of an ANDA for FOCALIN XR ® . The notification letter from Barr contends that claims in U.S. Patent Nos. 5,908,850, 6,355,656, 5,837,284, and 6,635,284 are not infringed by the proposed Barr products, and it contends that claims in U.S. Patent Nos. 5,908,850, 6,355,656, 6,528,530, 5,837,284 and 6,635,284 are invalid. In our complaint against Barr, we and Novartis asserted U.S. Patent Nos. 5,908,850, 6,355,656, 6,528,530, 5,837,284, and 6,635,284. We and Novartis subsequently added claims against Barr for infringement of U.S. Patent No. 7,431,944. Fact discovery has expired, claim construction briefing has been completed, and no trial date has been set. This action has been resolved pursuant to a confidential settlement agreement dated December 9, 2009. Pursuant to the settlement agreement, the parties sought (and the Court allowed) a 60-day stay of the litigation, in order to allow for review of the settlement agreement by the Federal Trade Commission and Department of Justice. The case was dismissed on February 1, 2010.
On December 5, 2008, we, together with our exclusive licensee Novartis, filed an infringement action in the United States District Court for the District of New Jersey against KV Pharmaceutical Company ("KV") in response to two notices of Paragraph IV certification made by KV in connection with its filing of an ANDA for generic versions of the FOCALIN XR ® products. In our complaint against KV, we and Novartis asserted U.S. Patent Nos. 5,908,850, 6,355,656, 6,528,530, 5,837,284, 6,635,284, and 7,431,944. KV filed an answer and counterclaim on January 20, 2009, seeking a declaratory judgment of patent invalidity, non-infringement and unenforceability with respect to the patents-in-suit. Fact discovery is complete or substantially complete, and claim construction briefing has been completed. Expert discovery has yet to be completed. No trial date has been set. On October 23, 2009, the court administratively struck the pleadings relating to claim construction, in order to afford the parties a chance to determine whether a settlement can be reached. If we are unsuccessful in proving infringement or defending our patents, Novartis' sales of FOCALIN XR ® could be significantly reduced in the United States by the entrance of a generic FOCALIN XR ® product, consequently reducing our revenue from royalties associated with these sales. If settlement cannot be reached, the claim construction and other litigation proceedings will move forward.
RITALIN LA ®
On December 4, 2006, we, together with our exclusive licensee Novartis, filed an infringement action in the U.S. District Court for the District of New Jersey against Abrika Pharmaceuticals, Inc. and Abrika Pharmaceuticals, LLP, (collectively, "Abrika Pharmaceuticals") in response to a notice of a Paragraph IV certification made by Abrika Pharmaceuticals in connection with the filing of an ANDA for RITALIN LA ® 20 mg, 30 mg, and 40 mg generic products. The notification letter from Abrika Pharmaceuticals contends that claims in U.S. Patent Nos. 5,837,284 and 6,635,284 are invalid and are not infringed by the proposed Abrika Pharmaceuticals products. In our complaint against Abrika Pharmaceuticals, we and Novartis asserted U.S. Patent Nos. 5,837,284 and 6,635,284. Abrika Pharmaceuticals filed an answer and counterclaim in the New Jersey court on June 1, 2007. The counterclaim seeks a declaratory judgment of patent invalidity, noninfringement, and unenforceability with respect to the patents-in-suit. On September 26, 2007, Abrika Pharmaceuticals sent a Paragraph IV certification to us and Novartis in connection with the filing of an ANDA supplement with respect to Abrika Pharmaceuticals' proposed generic 10 mg RITALIN LA ® product. We and Novartis filed an amended complaint against Abrika Pharmaceuticals on November 5, 2007 that includes infringement allegations directed to Abrika Pharmaceuticals' proposed generic 10 mg RITALIN LA ® product. Abrika Pharmaceuticals filed an answer and counterclaim to the amended complaint on December 5, 2007. The counterclaim seeks a declaratory judgment of patent invalidity, noninfringement, and unenforceability with respect to the patents-in-suit. If we are unsuccessful in proving infringement or defending our patents, Novartis' sales of RITALIN LA ® could be significantly reduced in the United States by the entrance of a generic RITALIN LA ® product, consequently reducing our revenue from royalties associated with these sales. Fact discovery has expired and claim construction briefing has been completed. Expert discovery will commence after the court has construed the claims of the patents-in-suit. No trial date has been set.
38
On October 4, 2007, we, together with our exclusive licensee Novartis, filed an infringement action in the U.S. District Court for the District of New Jersey against KV Pharmaceutical Company ("KV") in response to a notice of a Paragraph IV certification made by KV in connection with the filing of an ANDA for RITALIN LA ® . The notification letter from KV contends that claims in U.S. Patent Nos. 5,837,284 and 6,635,284 are not infringed by the proposed KV products. In our complaint against KV, we and Novartis asserted United States Patent Nos. 5,837,284 and 6,635,284. KV filed an answer and counterclaim on November 26, 2007. The counterclaim seeks a declaratory judgment of patent invalidity, noninfringement, and unenforceability with respect to the patents-in-suit. No pretrial or trial dates have been set. If we are unsuccessful in proving infringement or defending our patents, Novartis' sales of RITALIN LA ® could be significantly reduced in the United States by the entrance of a generic RITALIN LA ® product, consequently reducing our revenue from royalties associated with these sales. KV's counterclaims also include antitrust allegations, which have been severed and stayed from the rest of the case for a separate trial (if necessary). Fact discovery has expired and claim construction briefing has been completed. Expert discovery will commence after the court has construed the claims of the patents-in-suit. No trial date has been set. On October 23, 2009, the court administratively struck the pleadings relating to claim construction, in order to afford the parties a chance to determine whether a settlement can be reached. If settlement cannot be reached, the claim construction and other litigation proceedings will move forward.
On October 31, 2007, we, together with our exclusive licensee Novartis, filed an infringement action in the U.S. District Court for the District of New Jersey against Barr and Barr Pharmaceuticals, Inc. (collectively, "Barr"), in response to a notice of a Paragraph IV certification made by Barr in connection with the filing of an ANDA for RITALIN LA ® . The notification letter from Barr contends that claims in U.S. Patent Nos. 5,837,284 and 6,635,284 are invalid and not infringed by the proposed Barr products. In our complaint against Barr, we and Novartis asserted United States Patent Nos. 5,837,284 and 6,635,284. If we are unsuccessful in proving infringement or defending our patents, Novartis' sales of RITALIN LA ® could be significantly reduced in the United States by the entrance of a generic RITALIN LA ® product, consequently reducing our revenue from royalties associated with these sales. Fact discovery has expired and claim construction briefing has been completed. Expert discovery will commence after the court has construed the claims of the patents-in-suit. No trial date has been set. Barr has notified us that it merged with Teva, and Barr is now Barr Pharmaceuticals, LLC, a wholly-owned subsidiary of Teva. On October 23, 2009, the court administratively struck the pleadings relating to claim construction, in order to afford the parties a chance to determine whether a settlement can be reached. If settlement cannot be reached, the claim construction and other litigation proceedings will move forward.
ITEM 4. SUBMISSION OF MATTERS TO A VOTE OF SECURITY HOLDERS
None.
39
PART II
ITEM 5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
(a) MARKET INFORMATION
Our common stock is traded on the NASDAQ Global Select Market under the symbol "CELG." The following table sets forth, for the periods indicated, the intra-day high and low prices per share of common stock on the NASDAQ Global Select Market:
| High | Low | |||||||
| ||||||||
2009 | ||||||||
Fourth Quarter | $ | 57.79 | $ | 49.74 | ||||
Third Quarter | 58.31 | 45.27 | ||||||
Second Quarter | 48.77 | 36.90 | ||||||
First Quarter | 56.60 | 39.32 | ||||||
| ||||||||
2008 | ||||||||
Fourth Quarter | $ | 66.50 | $ | 45.44 | ||||
Third Quarter | 77.39 | 56.00 | ||||||
Second Quarter | 65.90 | 56.88 | ||||||
First Quarter | 62.20 | 46.07 | ||||||
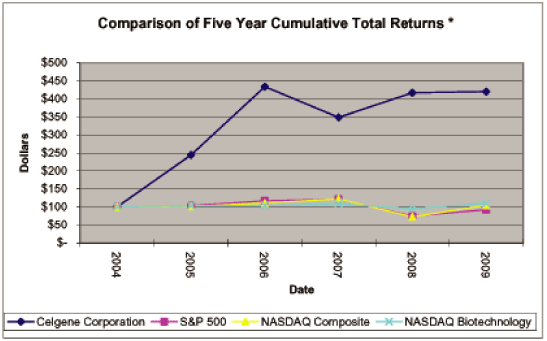
40
| Cumulative Total Return | ||||||||||||||||||||||||
| 12/04 | 12/05 | 12/06 | 12/07 | 12/08 | 12/09 | |||||||||||||||||||
| ||||||||||||||||||||||||
Celgene Corporation | $ | 100.00 | $ | 244.34 | $ | 433.86 | $ | 348.49 | $ | 416.89 | $ | 419.91 | ||||||||||||
S&P 500 | 100.00 | 103.00 | 117.03 | 121.16 | 74.53 | 92.01 | ||||||||||||||||||
NASDAQ Composite | 100.00 | 101.37 | 111.03 | 121.92 | 72.49 | 104.31 | ||||||||||||||||||
NASDAQ Biotechnology | 100.00 | 102.84 | 103.89 | 108.65 | 94.93 | 109.77 | ||||||||||||||||||
| * | $100 Invested on 12/31/04 in Stock or Index - Including Reinvestment of Dividends, Fiscal Year Ending December 31. |
(b) HOLDERS
The closing sales price per share of common stock on the NASDAQ Global Select Market on February 5, 2010 was $55.16. As of January 31, 2010, there were approximately 313,505 holders of record of our common stock.
(c) DIVIDEND POLICY
We have never declared or paid any cash dividends on our common stock. We currently intend to retain any future earnings for funding growth and, therefore, do not anticipate paying any cash dividends on our common stock in the foreseeable future.
(d) EQUITY COMPENSATION PLAN INFORMATION
We incorporate information regarding the securities authorized for issuance under our equity compensation plans into this section by reference from the section entitled "Equity Compensation Plan Information" in the proxy statement for our 2010 Annual Meeting of Stockholders.
(e) REPURCHASE OF EQUITY SECURITIES
In April 2009, our Board of Directors approved a $500.0 million common share repurchase program. As of December 31, 2009 an aggregate 4,314,625 common shares were repurchased under the program at an average price of $48.55 per common share and total cost of $209.5 million.
41
ITEM 6. SELECTED FINANCIAL DATA
The following Selected Consolidated Financial Data should be read in conjunction with our Consolidated Financial Statements and the related Notes thereto, Management's Discussion and Analysis of Financial Condition and Results of Operations and other financial information included elsewhere in this Annual Report. The data set forth below with respect to our Consolidated Statements of Operations for the years ended December 31, 2009, 2008 and 2007 and the Consolidated Balance Sheet data as of December 31, 2009 and 2008 are derived from our Consolidated Financial Statements which are included elsewhere in this Annual Report and are qualified by reference to such Consolidated Financial Statements and related Notes thereto. The data set forth below with respect to our Consolidated Statements of Operations for the years ended December 31, 2006 and 2005 and the Consolidated Balance Sheet data as of December 31, 2007, 2006 and 2005 are derived from our Consolidated Financial Statements, which are not included elsewhere in this Annual Report.
| Years ended December 31, | ||||||||||||||||||||
| In thousands, except per share data | 2009 | 2008 | 2007 | 2006 | 2005 | |||||||||||||||
| ||||||||||||||||||||
Consolidated Statements of Operations Data: | ||||||||||||||||||||
Total revenue | $ | 2,689,893 | $ | 2,254,781 | $ | 1,405,820 | $ | 898,873 | $ | 536,941 | ||||||||||
Costs and operating expenses | 1,848,367 | 3,718,999 | 980,699 | 724,182 | 453,357 | |||||||||||||||
| ||||||||||||||||||||
Operating income (loss) | 841,526 | (1,464,218 | ) | 425,121 | 174,691 | 83,584 | ||||||||||||||
| ||||||||||||||||||||
Interest and investment income, net | 76,785 | 84,835 | 109,813 | 40,352 | 24,557 | |||||||||||||||
Equity in losses of affiliated companies | 1,103 | 9,727 | 4,488 | 8,233 | 6,923 | |||||||||||||||
Interest expense | 1,966 | 4,437 | 11,127 | 9,417 | 9,497 | |||||||||||||||
Other income (expense), net | 60,461 | 24,722 | (2,350 | ) | 5,502 | (7,509 | ) | |||||||||||||
| ||||||||||||||||||||
Income (loss) before tax | 975,703 | (1,368,825 | ) | 516,969 | 202,895 | 84,212 | ||||||||||||||
Income tax provision | 198,956 | 164,828 | 290,536 | 133,914 | 20,556 | |||||||||||||||
| ||||||||||||||||||||
Net income (loss) | $ | 776,747 | $ | (1,533,653 | ) | $ | 226,433 | $ | 68,981 | $ | 63,656 | |||||||||
| ||||||||||||||||||||
| Years ended December 31, | ||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||||||
| ||||||||||||||||||||
Net income (loss) per common share (1): | ||||||||||||||||||||
Basic | $ | 1.69 | $ | (3.46 | ) | $ | 0.59 | $ | 0.20 | $ | 0.19 | |||||||||
Diluted | $ | 1.66 | $ | (3.46 | ) | $ | 0.54 | $ | 0.18 | $ | 0.18 | |||||||||
| ||||||||||||||||||||
Weighted average shares (1): | ||||||||||||||||||||
Basic | 459,304 | 442,620 | 383,225 | 352,217 | 335,512 | |||||||||||||||
Diluted | 467,354 | 442,620 | 431,858 | 407,181 | 390,585 | |||||||||||||||
| (1) | Amounts have been adjusted for the two-for-one stock split effected in February 2006. |
| As of December 31, | ||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||||||
| ||||||||||||||||||||
Consolidated Balance Sheets Data: | ||||||||||||||||||||
Cash, cash equivalents and marketable securities | $ | 2,996,752 | $ | 2,222,091 | $ | 2,738,918 | $ | 1,982,220 | $ | 724,260 | ||||||||||
Total assets | 5,389,311 | 4,445,270 | 3,611,284 | 2,735,791 | 1,258,313 | |||||||||||||||
Convertible notes | - | - | 196,555 | 399,889 | 399,984 | |||||||||||||||
(Accumulated deficit) retained earnings | (632,246 | ) | (1,408,993 | ) | 124,660 | (101,773 | ) | (170,754 | ) | |||||||||||
Stockholders' equity | 4,394,606 | 3,491,328 | 2,843,944 | 1,976,177 | 635,775 | |||||||||||||||
42
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Executive Summary
Celgene Corporation and its subsidiaries (collectively "we" or "our") is a global integrated biopharmaceutical company primarily engaged in the discovery, development and commercialization of innovative therapies designed to treat cancer and immune-inflammatory related diseases.
Our primary commercial stage products include REVLIMID ® , THALOMID ® (inclusive of Thalidomide Celgene TM and Thalidomide Pharmion TM , subsequent to the acquisition of Pharmion Corporation, or Pharmion) and VIDAZA ® . ALKERAN ® was licensed from GlaxoSmithKline, or GSK, and sold under our label through March 31, 2009, the conclusion date of the ALKERAN ® license with GSK. REVLIMID ® is an oral immunomodulatory drug marketed in the United States, Europe and Asia / Pacific for patients with multiple myeloma who have received at least one prior therapy and in the United States, Canada and certain countries in Latin America for the treatment of transfusion-dependent anemia due to low- or intermediate-1-risk myelodysplastic syndromes, or MDS, associated with a deletion 5q cytogenetic abnormality with or without additional cytogenetic abnormalities. THALOMID ® is marketed for patients with newly diagnosed multiple myeloma and for the acute treatment of the cutaneous manifestations of moderate to severe erythema nodosum leprosum, or ENL, an inflammatory complication of leprosy. VIDAZA ® is a pyrimidine nucleoside analog that has been shown to reverse the effects of DNA hypermethylation and promote subsequent gene re-expression. VIDAZA ® is licensed from Pfizer, and is marketed in the United States for the treatment of all subtypes of MDS and was granted orphan drug designation for the treatment of MDS through May 2011. In the third quarter of 2009, the National Comprehensive Cancer Network, or NCCN, upgraded VIDAZA ® to a Category 1 recommended treatment for patients with intermediate-2 and high-risk MDS. In Europe, VIDAZA ® is marketed for the treatment of certain qualified adult patients and was granted orphan drug designation for the treatment of MDS and acute myeloid leukemia, or AML, in the European Union, or EU, expiring December 2018.
We continue to invest substantially in research and development, and the drug candidates in our pipeline are at various stages of preclinical and clinical development. These candidates include our IMiDs ® compounds, which are a class of compounds proprietary to us and having certain immunomodulatory and other biologically important properties, in addition to our leading oral anti-inflammatory agents and cell products. We believe that continued acceptance of our primary commercial stage products, depth of our product pipeline, regulatory approvals of both new products and expanded use of existing products provide the catalysts for future growth.
For the year ended December 31, 2009, we reported revenue of $2.690 billion, net income of $776.7 million and diluted earnings per share of $1.66. Revenue increased by $435.1 million in 2009 compared to 2008 primarily due to our continued expansion into international markets and revenue growth of REVLIMID ® and VIDAZA ® , which more than offset decreases in revenues from THALOMID ® and ALKERAN ® . The decrease in THALOMID ® was primarily due to lower unit volumes in the United States resulting from the increased use of REVLIMID ® , while the decrease in ALKERAN ® was due to the March 31, 2009 conclusion of the ALKERAN ® license with GSK. Net income and earnings per share for 2009 reflect the earnings contributions from higher REVLIMID ® and VIDAZA ® revenues, partly offset by increased spending for new product launches, recurring research and development activities and the expansion of our international operations. The year ended December 31, 2008 included a $1.740 billion charge for acquired in-process research and development, or IPR&D, related to the Pharmion acquisition in March 2008.
43
Factors Affecting Future Results
Future operating results will depend on many factors, including demand for our existing products, regulatory approvals of our products and product candidates, the timing and market acceptance of new products launched by us or competing companies, the timing of research and development milestones, challenges to our intellectual property and our ability to control costs. See "Risk Factors" contained in Part I, Item 1A of this Annual Report on Form 10-K.
Results of Operations -
Fiscal Years Ended December 31, 2009, 2008 and 2007
Total Revenue: Total revenue and related percentage changes for the years ended December 31, 2009, 2008 and 2007 were as follows:
| % Change | ||||||||||||||||||||
| 2009 | 2008 | |||||||||||||||||||
| versus | versus | |||||||||||||||||||
| In thousands $ | 2009 | 2008 | 2007 | 2008 | 2007 | |||||||||||||||
| ||||||||||||||||||||
Net product sales: | ||||||||||||||||||||
REVLIMID ® | $ | 1,706,437 | $ | 1,324,671 | $ | 773,877 | 28.8 | % | 71.2 | % | ||||||||||
THALOMID ® | 436,906 | 504,713 | 447,089 | (13.4 | )% | 12.9 | % | |||||||||||||
VIDAZA ® | 387,219 | 206,692 | - | 87.3 | % | N/A | ||||||||||||||
ALKERAN ® | 20,111 | 81,734 | 73,551 | (75.4 | )% | 11.1 | % | |||||||||||||
Other | 16,681 | 19,868 | 5,924 | (16.0 | )% | 235.4 | % | |||||||||||||
| ||||||||||||||||||||
Total net product sales | $ | 2,567,354 | $ | 2,137,678 | $ | 1,300,441 | 20.1 | % | 64.4 | % | ||||||||||
Collaborative agreements and other revenue | 13,743 | 14,945 | 20,109 | (8.0 | )% | (25.7 | )% | |||||||||||||
Royalty revenue | 108,796 | 102,158 | 85,270 | 6.5 | % | 19.8 | % | |||||||||||||
| ||||||||||||||||||||
Total revenue | $ | 2,689,893 | $ | 2,254,781 | $ | 1,405,820 | 19.3 | % | 60.4 | % | ||||||||||
| ||||||||||||||||||||
2009 compared to 2008: Total revenue increased by $435.1 million, or 19.3%, in 2009 compared to 2008. The revenue increase in the United States was $132.5 million, or 8.3% and the increase in international markets was $302.6 million, or 46.3%.
2008 compared to 2007: Total revenue increased by $849.0 million, or 60.4%, in 2008 compared to 2007. The revenue increase in the United States was $379.8 million, or 31.6% and the increase in international markets was $469.2 million, or 230.2%.
Net Product Sales:
2009 compared to 2008: Net product sales increased by $429.7 million, or 20.1% to $2.567 billion in 2009 compared to 2008. The increase was comprised of net volume increases of $428.0 million and price increases of $61.5 million, partly offset by a decrease due to the impact of foreign exchange of $59.8 million.
REVLIMID ® net sales increased by $381.8 million, or 28.8% to $1.706 billion in 2009 compared to 2008 primarily due to increased unit sales in both U.S. and international markets. Increased market penetration and the increase in duration of therapy and number of patients using REVLIMID ® in multiple myeloma contributed to U.S. growth. The growth in international markets reflects the expansion of our commercial activities in over 65 countries and product reimbursement approvals.
44
THALOMID ® net sales decreased by $67.8 million, or 13.4%, to $436.9 million in 2009 compared to 2008. The decrease was primarily due to lower unit volumes in the United States resulting from the increased use of REVLIMID ® , partially offset by higher pricing and volume increases in international markets.
VIDAZA ® net sales increased by $180.5 million, or 87.3%, to $387.2 million in 2009 compared to 2008 primarily due to the December 2008 full marketing authorization granted by the European Commission, or EC, for the treatment of adult patients who are not eligible for haematopoietic stem cell transplantation with Intermediate-2 and high-risk MDS according to the International Prognostic System Score, or IPSS, or chronic myelomonocytic leukaemia, or CMML, with 10-29 percent marrow blasts without myeloproliferative disorder, or AML with 20-30 percent blasts and multi-lineage dysplasia, according to World Health Organization, or WHO, classification of VIDAZA ® . In addition, sales for 2008 only included sales subsequent to the March 7, 2008 acquisition of Pharmion.
ALKERAN ® net sales decreased by $61.6 million to $20.1 million in 2009 compared to 2008. This product was licensed from GSK and sold under our label through March 31, 2009, the conclusion date of the ALKERAN ® license with GSK.
2008 compared to 2007: Net product sales increased by $837.2 million, or 64.4% to $2.138 billion in 2008 compared to 2007. The increase was comprised of net volume increases of $742.8 million, as well as price increases of $93.0 million, and the favorable impact of foreign exchange of $1.4 million.
REVLIMID ® net sales increased by $550.8 million, or 71.2%, to $1.325 billion in 2008 compared to 2007 primarily due to increased sales in the United States and continued expansion in international markets. Increased market penetration and the increase in duration of patients using REVLIMID ® in multiple myeloma accounted for most of the U.S. growth. International sales growth primarily reflects the impact of the June 2007 EC's approval for the use of REVLIMID ® for treatment in combination with dexamethasone of patients with multiple myeloma who have received at least one prior therapy and continued expansion in international markets.
THALOMID ® net sales increased by $57.6 million, or 12.9%, to $504.7 million in 2008 compared to 2007 primarily due to the 2008 inclusion of international sales, resulting from the acquisition of Pharmion. In addition, U.S. price increases were offset by lower sales volumes.
VIDAZA ® net sales of $206.7 million represented sales recorded subsequent to the March 7, 2008 Pharmion acquisition in both the United States and international markets.
ALKERAN â net sales increased by $8.2 million, or 11.1%, to $81.7 million in 2008 compared to 2007 primarily due to an increase in unit sales of the injectable form.
Gross to Net Sales Accruals: We record gross to net sales accruals for sales returns and allowances, sales discounts, government rebates, and chargebacks and distributor service fees.
THALOMID ® is distributed in the United States under our S.T.E.P.S. ® program which we developed and is a proprietary comprehensive education and risk-management distribution program with the objective of providing for the safe and appropriate distribution and use of THALOMID ® . Internationally, THALOMID ® is also distributed under mandatory risk-management distribution programs tailored to meet local competent authorities' specifications to help ensure the safe and appropriate distribution and use of THALOMID ® . These programs may vary by country and, depending upon the country and the design of the risk-management program, the product may be sold through hospitals or retail pharmacies. REVLIMID ® is distributed in the United States primarily through contracted pharmacies under the RevAssist ® program, which is a proprietary risk-management distribution program tailored specifically to help ensure the safe and appropriate distribution and use of REVLIMID ® . Internationally, REVLIMID ® is also distributed under mandatory risk-management distribution programs tailored to meet local competent authorities' specifications to help ensure the safe and appropriate distribution and use of REVLIMID ® . These programs may vary by country and, depending upon the country and the design of the risk-management program, the product may be sold through hospitals or retail pharmacies. VIDAZA ® is distributed through the more traditional pharmaceutical industry supply chain. VIDAZA ® is not subjected to the same risk-management distribution programs as THALOMID ® and REVLIMID ® .
45
We base our sales returns allowance on estimated on-hand retail/hospital inventories, measured end-customer demand as reported by third-party sources, actual returns history and other factors, such as the trend experience for lots where product is still being returned or inventory centralization and rationalization initiatives conducted by major pharmacy chains, as applicable. If the historical data we use to calculate these estimates does not properly reflect future returns, then a change in the allowance would be made in the period in which such a determination is made and revenues in that period could be materially affected. Under this methodology, we track actual returns by individual production lots. Returns on closed lots, that is, lots no longer eligible for return credits, are analyzed to determine historical returns experience. Returns on open lots, that is, lots still eligible for return credits, are monitored and compared with historical return trend rates. Any changes from the historical trend rates are considered in determining the current sales return allowance. THALOMID ® is drop-shipped directly to the prescribing pharmacy and, as a result, wholesalers do not stock the product. REVLIMID ® is distributed primarily through hospitals and contracted pharmacies, lending itself to tighter controls of inventory quantities within the supply channel and, thus, resulting in lower returns activity to date.
Sales discount accruals are based on payment terms extended to customers.
Government rebate accruals are based on estimated payments due to governmental agencies for purchases made by third parties under various governmental programs. U.S. Medicaid rebate accruals are based on historical payment data and estimates of future Medicaid beneficiary utilization applied to the Medicaid unit rebate formula established by the Center for Medicaid and Medicare Services. Certain international markets have government-sponsored programs that require rebates to be paid based on program specific rules and accordingly, the rebate accruals are determined primarily on estimated eligible sales.
Chargebacks accruals are based on the differentials between product acquisition prices paid by wholesalers and lower government contract pricing paid by eligible customers covered under federally qualified programs. Distributor service fee accruals are based on contractual fees to be paid to the wholesale distributor for services provided. On January 28, 2008, the Fiscal Year 2008 National Defense Authorization Act was enacted, which expands TRICARE to include prescription drugs dispensed by TRICARE retail network pharmacies. TRICARE is a health care program of the U.S. Department of Defense Military Health System that provides civilian health benefits for military personnel, military retirees and their dependents. TRICARE rebate accruals reflect this program expansion and are based on estimated Department of Defense eligible sales multiplied by the TRICARE rebate formula.
See Critical Accounting Estimates and Significant Accounting Policies for further discussion of gross to net sales accruals.
46
Gross to net sales accruals and the balance in the related allowance accounts for the years ended December 31, 2009, 2008 and 2007 were as follows:
| Returns | Chargebacks | |||||||||||||||||||
| and | Government | and Dist. | ||||||||||||||||||
| In thousands $ | Allowances | Discounts | Rebates | Service Fees | Total | |||||||||||||||
| ||||||||||||||||||||
Balance at December 31, 2006 | $ | 9,480 | $ | 2,296 | $ | 7,468 | $ | 10,633 | $ | 29,877 | ||||||||||
Allowances for sales during 2007 | 22,303 | 27,999 | 28,420 | 72,982 | 151,704 | |||||||||||||||
Allowances for sales during prior periods | 17,498 | - | - | (2,776 | ) | 14,722 | ||||||||||||||
Credits/deductions issued for prior year sales | (26,979 | ) | (2,206 | ) | (7,071 | ) | (6,725 | ) | (42,981 | ) | ||||||||||
Credits/deductions issued for sales during 2007 | (5,568 | ) | (25,194 | ) | (19,615 | ) | (65,275 | ) | (115,652 | ) | ||||||||||
| ||||||||||||||||||||
Balance at December 31, 2007 | $ | 16,734 | $ | 2,895 | $ | 9,202 | $ | 8,839 | $ | 37,670 | ||||||||||
Pharmion balance at March 7, 2008 | 926 | 283 | 1,266 | 2,037 | 4,512 | |||||||||||||||
Allowances for sales during 2008 | 20,624 | 36,024 | 35,456 | 100,258 | 192,362 | |||||||||||||||
Credits/deductions issued for prior year sales | (17,066 | ) | (2,428 | ) | (7,951 | ) | (4,127 | ) | (31,572 | ) | ||||||||||
Credits/deductions issued for sales during 2008 | (3,419 | ) | (33,115 | ) | (27,163 | ) | (83,621 | ) | (147,318 | ) | ||||||||||
| ||||||||||||||||||||
Balance at December 31, 2008 | $ | 17,799 | $ | 3,659 | $ | 10,810 | $ | 23,386 | $ | 55,654 | ||||||||||
Allowances for sales during 2009 | 14,742 | 37,315 | 48,082 | 88,807 | 188,946 | |||||||||||||||
Credits/deductions issued for prior year sales | (13,168 | ) | (2,306 | ) | (11,042 | ) | (10,333 | ) | (36,849 | ) | ||||||||||
Credits/deductions issued for sales during 2009 | (12,013 | ) | (35,070 | ) | (29,739 | ) | (72,619 | ) | (149,441 | ) | ||||||||||
| ||||||||||||||||||||
Balance at December 31, 2009 | $ | 7,360 | $ | 3,598 | $ | 18,111 | $ | 29,241 | $ | 58,310 | ||||||||||
| ||||||||||||||||||||
2009 compared to 2008 : Returns and allowances decreased by $5.9 million in 2009 compared to 2008 primarily due to the completion of an inventory centralization and rationalization initiative conducted by a major pharmacy chain during 2009, decreased revenue from products with a higher return rate history in 2009 compared to 2008 and a decrease in ALKERAN ® returns due to the March 31, 2009 conclusion of the ALKERAN ® license with GSK. In addition, 2008 includes an increase in THALOMID ® returns resulting from the anticipated increase in the use of REVLIMID ® in multiple myeloma.
Discounts increased by $1.3 million in 2009 compared to 2008 primarily due to revenue increases in the United States and international markets, both of which offer different discount programs.
Government rebates increased by $12.6 million in 2009 compared to 2008 primarily due to increased sales levels of REVLIMID ® and VIDAZA ® in the United States and international markets, as well as reimbursement approvals in new markets.
Chargebacks and distributor service fees decreased by $11.5 million in 2009 compared to 2008 primarily due to reduced revenue from products with a higher chargeback history in 2009 compared to 2008 and a decrease in ALKERAN ® chargebacks, partially offset by an increase in international distributor service fees due to certain programs commenced in 2009.
2008 compared to 2007 : Returns and allowances decreased by $19.2 million in 2008 compared 2007 primarily due to reduced THALOMID ® inventory in the sales channel resulting from the 2007 THALOMID ® inventory centralization and rationalization at several major pharmacy chains, which also resulted in additional returns during 2007. In addition, 2007 includes an increase in THALOMID ® returns resulting from the anticipated increase in use of REVLIMID ® in multiple myeloma.
Discounts increased by $8.0 million in 2008 compared to 2007 primarily due to increased sales of REVLIMID â as well as the inclusion of former Pharmion products, which resulted in additional discounts taken.
47
Government rebates increased by $7.0 million in 2008 compared to 2007 primarily due to increased international government rebates resulting from our global expansion, as well as the inclusion of former Pharmion products.
Chargebacks and distributor service fees increased by $30.1 million in 2008 compared to 2007 primarily due to the new TRICARE rebate program, as well as the inclusion of former Pharmion products.
Collaborative Agreements and Other Revenue:
2009 compared to 2008: Revenues from collaborative agreements and other sources decreased by $1.2 million to $13.7 million in 2009 compared to 2008. The decrease was primarily due to the elimination of license fees and amortization of deferred revenues related to Pharmion subsequent to the March 7, 2008 acquisition and was partly offset by an increase in milestone payments received in 2009.
2008 compared to 2007: Revenues from collaborative agreements and other sources totaled $14.9 million and $20.1 million for 2008 and 2007, respectively. The $5.2 million decrease in 2008 compared to 2007 was primarily due to the elimination of license fees and amortization of deferred revenues related to Pharmion.
Royalty Revenue:
2009 compared to 2008: Royalty revenue increased by $6.6 million to $108.8 million in 2009 compared to 2008 due to the 2009 inclusion of $9.0 million in residual ALKERAN ® payments earned by us based upon GSK's ALKERAN ® revenues subsequent to the conclusion of the ALKERAN ® license with GSK. Royalty revenue related to Novartis' sales of RITALIN ® decreased by $2.1 million from 2008.
2008 compared to 2007: Royalty revenue totaled $102.2 million in 2008, representing an increase of $16.9 million compared to 2007. The increase was primarily due to amounts received from Novartis on sales of FOCALIN XR ® , partly due to patients transitioning from FOCALIN ® to FOCALIN XR ® . We sell FOCALIN ® to Novartis and receive royalties on sales of Novartis' FOCALIN XR ® .
Cost of Goods Sold (excluding amortization of acquired intangible assets): Cost of goods sold and related percentages for the years ended December 31, 2009, 2008 and 2007 were as follows:
| In thousands $ | 2009 | 2008 | 2007 | |||||||||
| ||||||||||||
Cost of goods sold (excluding amortization of acquired intangible assets) | $ | 216,289 | $ | 258,267 | $ | 130,211 | ||||||
Increase (decrease) from prior year | $ | (41,978 | ) | $ | 128,056 | $ | 4,452 | |||||
Percent increase (decrease) from prior year | (16.3 | )% | 98.3 | % | 3.5 | % | ||||||
Percent of net product sales | 8.4 | % | 12.1 | % | 10.0 | % | ||||||
2009 compared to 2008 : Cost of goods sold (excluding amortization of acquired intangible assets) decreased by $42.0 million to $216.3 million in 2009 compared to 2008 partly due to the March 31, 2009 conclusion date of the ALKERAN ® license with GSK, reducing cost of goods sold by approximately $39.0 million compared to 2008. In addition, costs related to THALOMID â decreased as a result of lower unit volumes. Finally, 2008 included a $24.6 million inventory step-up adjustment related to the March 7, 2008 acquisition of Pharmion compared to an adjustment of $0.4 million included in 2009. The impact of these reductions was partly offset by higher costs related to increased unit volumes for REVLIMID â and VIDAZA â . As a percent of net product sales, cost of goods sold (excluding amortization of acquired intangible assets) decreased to 8.4% in 2009 from 12.1% in 2008 primarily due to lower ALKERAN ® sales, which carried a higher cost to sales ratio relative to our other products, and the decrease in the inventory step-up adjustment.
48
2008 compared to 2007 : Cost of goods sold increased by $128.1 million in 2008 compared to 2007 primarily due to the inclusion of costs related to VIDAZA â and THALOMID â , which were obtained in the Pharmion acquisition. Also included in 2008 is $24.6 million of the $25.0 million of inventory step-up cost related to the acquisition date fair value of former Pharmion inventories. Cost of sales also increased due to an increase in material costs for ALKERAN â for injection and an increase in unit volume for REVLIMID â , resulting in higher royalties. As a percent of net product sales, cost of goods sold increased to 12.1% in the 2008 from 10.0% in 2007 primarily due to the inclusion of higher costs for VIDAZA â and ALKERAN â and the $24.6 million of inventory step-up cost.
Research and Development: Research and development expenses and related percentages for the years ended December 31, 2009, 2008 and 2007 were as follows:
| In thousands $ | 2009 | 2008 | 2007 | |||||||||
| ||||||||||||
Research and development | $ | 794,848 | $ | 931,218 | $ | 400,456 | ||||||
Increase (decrease) from prior year | $ | (136,370 | ) | $ | 530,762 | $ | 140,500 | |||||
Percent increase (decrease) from prior year | (14.6 | )% | 132.5 | % | 54.0 | % | ||||||
Percent of total revenue | 29.5 | % | 41.3 | % | 28.5 | % | ||||||
2009 compared to 2008 : Research and development expenses decreased by $136.4 million in 2009 compared to 2008 primarily due to a $303.1 million charge included in 2008 for a royalty obligation payment to Pfizer that related to the unapproved forms of VIDAZA â partly offset by 2009 spending increases related to drug discovery and clinical research and development in support of multiple programs across a broad range of diseases. Included in 2009 were upfront payments of $30.0 million and $4.5 million to GlobeImmune, Inc. and Array BioPharma, Inc., respectively, related to research and development collaboration agreements. Included in 2008 was an upfront payment of $45.0 million made to Acceleron Pharma, Inc. related to a research and development collaboration agreement.
The following table provides an additional breakdown of research and development expenses:
| Increase | ||||||||||||
| In thousands $ | 2009 | 2008 | (Decrease) | |||||||||
| ||||||||||||
Human pharmaceutical clinical programs | $ | 371,189 | $ | 288,222 | $ | 82,967 | ||||||
Other pharmaceutical programs | 323,702 | 549,841 | (226,139 | ) | ||||||||
Biopharmaceutical discovery and development | 85,208 | 77,293 | 7,915 | |||||||||
Placental stem cell and biomaterials | 14,749 | 15,862 | (1,113 | ) | ||||||||
| ||||||||||||
Total | $ | 794,848 | $ | 931,218 | $ | (136,370 | ) | |||||
| ||||||||||||
Other pharmaceutical programs for 2009 includes $34.5 million for the GlobeImmune, Inc. and Array BioPharma, Inc., or Array, research and development collaboration agreements noted above in addition to spending for toxicology, analytical research and development, quality and regulatory affairs. Other pharmaceutical programs for 2008 includes the $303.1 million VIDAZA â royalty obligation payment, $45.0 million for the Acceleron Pharma, Inc., or Acceleron, research and development collaboration agreement noted above, in addition to spending for toxicology, analytical research and development, quality and regulatory affairs.
49
Research and development expenditures support ongoing clinical progress in multiple proprietary development programs for REVLIMID â and other IMiDs â compounds; VIDAZA â ; amrubicin, our lead compound for small cell lung cancer; apremilast (CC-10004), our lead anti-inflammatory compound that inhibits PDE-4, which results in the inhibition of multiple proinflammatory mediators such as TNF- a and which is currently being evaluated in Phase II clinical trials in the treatment of psoriasis and psoriatic arthritis; pomalidomide, which is currently being evaluated in Phase I and II clinical trials; CC-11050, for which Phase II clinical trials are planned; our kinase and ligase inhibitor programs; as well as the placental stem cell program. In June 2009, we filed a New Drug Application, or NDA, with the Japanese Ministry of Health, Labour and Welfare, or MHLW, for REVLIMID â in combination with dexamethasone for the treatment of patients with multiple myeloma who have received at least one prior therapy. REVLIMID â had previously been granted orphan drug status by the MHLW in Japan for this same indication.
Research and development expense may continue to grow as earlier stage compounds are moved through the preclinical and clinical stages. Due to the significant risk factors and uncertainties inherent in preclinical tests and clinical trials associated with each of our research and development projects, the cost to complete such projects can vary. The data obtained from these tests and trials may be susceptible to varying interpretation that could delay, limit or prevent a project's advancement through the various stages of clinical development, which would significantly impact the costs incurred to bring a project to completion.
For information about the commercial and development status and target diseases of our drug compounds, refer to the product overview table contained in Part I, Item I, "Business," of this Annual Report on Form 10-K.
2008 compared to 2007 : Research and development expenses increased by $530.8 million in 2008 compared to 2007, primarily due to a $303.1 million charge for the October 3, 2008 royalty obligation payment to Pfizer that related to the unapproved forms of VIDAZA â . Clinical program spending increased by $147.4 million in support of ongoing multiple proprietary development programs. Regulatory spending increased by $20.2 million primarily due to the expansion of REVLIMID â in international markets and costs related to apremilast. Also included in 2008 was $45.0 million in upfront payments made to Acceleron related to a research and development collaboration arrangement. The increase was partly offset by the 2007 inclusion of a combined $41.1 million in upfront payments for collaborative research and development arrangements for early stage compounds with Array and PTC Therapeutics.
Selling, General and Administrative: Selling, general and administrative expenses and related percentages for the years ended December 31, 2009, 2008 and 2007 were as follows:
| In thousands $ | 2009 | 2008 | 2007 | |||||||||
| ||||||||||||
Selling, general and administrative | $ | 753,827 | $ | 685,547 | $ | 440,962 | ||||||
Increase from prior year | $ | 68,280 | $ | 244,585 | $ | 111,213 | ||||||
Percent increase from prior year | 10.0 | % | 55.5 | % | 33.7 | % | ||||||
Percent of total revenue | 28.0 | % | 30.4 | % | 31.4 | % | ||||||
2009 compared to 2008: Selling, general and administrative expenses increased by $68.3 million to $753.8 million in 2009 compared to 2008, primarily reflecting increases in marketing and sales related expenses of $75.1 million, which were partly offset by a $6.7 million reduction in bad debt expense and other customer account charges. Marketing and sales related expenses in 2009 included product launch activities for REVLIMID â , VIDAZA â and THALOMID ® in Europe, Canada and Australia, in addition to VIDAZA â relaunch expenses in the United States upon receipt of an expanded FDA approval to reflect new overall survival data. The increase in expense also reflects the continued expansion of our international commercial activities.
50
2008 compared to 2007: Selling, general and administrative expenses increased by $244.6 million in 2008 compared to 2007, primarily reflecting an increase in marketing and sales related expenses of $167.5 million, general and administrative expenses of $63.8 million and an increase in donations to non-profit foundations that assist patients with their co-payments of $13.3 million. The increase reflects marketing and sales expenses related to product launch activities for REVLIMID â and THALOMID ® in Europe, Canada and Australia, in addition to the activities related to the relaunch of VIDAZA â in the United States and new launches in Europe.
Amortization of Acquired Intangible Assets: Amortization of acquired intangible assets decreased by $20.6 million to $83.4 million in 2009 compared to 2008 primarily due to several intangible assets obtained in the Pharmion acquisition in March 2008 becoming fully amortized during the fourth quarter of 2008 and third quarter of 2009.
Acquired In-Process Research and Development: The $1.74 billion IPR&D charge in 2008 represents the fair value of compounds under development by Pharmion at the date of acquisition that had not yet achieved regulatory approval for marketing in certain markets or had not yet been completed and have no alternative future use. These intangibles primarily related to development and approval initiatives for VIDAZA â IV in the EU market, the oral form of azacitidine in the U.S. and EU markets and THALOMID ® in the EU market.
Interest and investment income, net: The following table provides a summary of interest and investment income, net for the years ended December 31, 2009, 2008 and 2007:
| In thousands $ | 2009 | 2008 | 2007 | |||||||||
| ||||||||||||
Interest and investment income, net | $ | 76,785 | $ | 84,835 | $ | 109,813 | ||||||
Increase (decrease) from prior year | $ | (8,050 | ) | $ | (24,978 | ) | $ | 69,461 | ||||
Percentage increase (decrease) from prior year | (9.5 | )% | (22.7 | )% | 172.1 | % | ||||||
Interest and investment income decreased by $8.1 million to $76.8 million in 2009 compared to 2008 primarily due to reduced yields on invested balances, partly offset by higher invested balances.
Interest and investment income decreased by $25.0 million to $84.8 million in 2008 compared to 2007 primarily due to lower average cash, cash equivalents and marketable securities balances resulting from the March 2008 cash payment of $746.8 million related to the Pharmion acquisition and the October 3, 2008 payment of $425.0 million to Pfizer where we prepaid our royalty obligation under the June 7, 2001 5-azacytidine license in full, in addition to reduced yields on invested balances. Interest and investment income, net included other-than-temporary impairment losses on marketable securities available for sale totaling $2.4 million in 2008 and $5.5 million in 2007.
51
Equity in losses of affiliated companies: Under the equity method of accounting, we recorded losses of $1.1 million, $9.7 million and $4.5 million in 2009, 2008 and 2007, respectively. Included in 2008 were impairment losses of $6.0 million which were based on an evaluation of several factors, including an other-than-temporary decrease in fair value of an equity investment below our cost.
Interest expense : Interest expense was $2.0 million, $4.4 million and $11.1 million in 2009, 2008 and 2007, respectively. The $2.4 million decrease in expense in 2009 compared to 2008 and the $6.7 million expense decrease in 2008 compared to 2007 were primarily due to the June 2008 completion of convertible debt conversions related to our $400 million convertible notes issued on June 3, 2003 and the completion of amortization of their debt issuance costs.
Other income (expense), net: Other income (expense), net for the years ended December 31, 2009, 2008 and 2007 were as follows:
| In thousands $ | 2009 | 2008 | 2007 | |||||||||
| ||||||||||||
Other income (expense), net | $ | 60,461 | $ | 24,722 | $ | (2,350 | ) | |||||
Increase (decrease) in income from prior year | $ | 35,739 | $ | 27,072 | $ | (7,852 | ) | |||||
Other income increased by $35.7 million to $60.5 million in 2009 compared to 2008 primarily due to transaction exchange gains and net gains on foreign currency forward contracts that have not been designated as hedges entered into in order to offset net foreign exchange gains and losses. In addition, 2008 included an impairment loss of $4.1 million.
Other income increased by $27.1 million to $24.7 million in 2008 compared to 2007 primarily due to favorable foreign exchange rates, which was partly offset by an other-than-temporary impairment loss recorded on an equity investment. The $2.4 million expense in 2007 included expenses related to a termination benefit resulting from the modification of certain outstanding stock options of a terminated employee and was partly offset by foreign exchange gains.
Income tax provision :
2009 compared to 2008: The income tax provision increased by $34.2 million to $199.0 million in 2009 compared to 2008. The 2009 effective tax rate of 20.4% reflects the impact from our low tax Swiss manufacturing operations and our overall global mix of income. The income tax provision included a $17.0 million net tax benefit, which was primarily the result of filing 2008 income tax returns with certain items being more favorable than originally estimated, reduction in a valuation allowance related to capital loss carryforwards and the settlement of tax examinations, partially offset by an increase in unrecognized tax benefits related to certain ongoing income tax audits.
2008 compared to 2007: The income tax provision decreased by $125.7 million to $164.8 million in 2008 compared to 2007. The effective tax rate of negative 12% reflects non-deductible IPR&D charges incurred in connection with the acquisition of Pharmion. The effective tax rate, excluding the impact of IPR&D and the expense related to the prepayment of our royalty obligation for unapproved products, was 24.8%, which reflects the benefit of our low tax Swiss manufacturing operations and our overall global mix of income.
52
Net income (loss): Net income (loss) and per common share amounts for the years ended December 31, 2009, 2008 and 2007 were as follows:
| In thousands $, except per share amounts | 2009 | 2008 | 2007 | |||||||||
| ||||||||||||
Net income (loss) | $ | 776,747 | $ | (1,533,653 | ) | $ | 226,433 | |||||
| ||||||||||||
Per common share amounts: | ||||||||||||
Basic | $ | 1.69 | $ | (3.46 | ) | $ | 0.59 | |||||
Diluted (1) | $ | 1.66 | $ | (3.46 | ) | $ | 0.54 | |||||
| ||||||||||||
Weighted average shares: | ||||||||||||
Basic | 459,304 | 442,620 | 383,225 | |||||||||
Diluted | 467,354 | 442,620 | 431,858 | |||||||||
| (1) | In computing diluted earnings per share for 2007, the numerator has been adjusted to add back the after-tax amount of interest expense recognized in the year on our convertible debt. No adjustment to the numerator or denominator was made in 2008 due to the anti-dilutive effect of any potential common stock as a result of our net loss. As of their maturity date, June 1, 2008, substantially all of our convertible notes were converted into shares of common stock. |
2009 compared to 2008: Net income for 2009 reflects the earnings impact from higher sales of REVLIMID ® and VIDAZA ® , which was partly due to sales increases in the United States and our continued expansion into new international markets and the granting of full marketing authorization by the EC of VIDAZA ® for specified treatment of adult patients. The earnings generated from increased sales were partly offset by increased R&D spending, the costs related to new product launches and our ongoing expansion of international operations. Net loss for 2008 included $1.74 billion in IPR&D charges related to our acquisition of Pharmion and a $303.1 million charge for the October 2008 royalty obligation payment to Pfizer related to unapproved forms of VIDAZA ® .
2008 compared to 2007: Net income decreased by $1.76 billion in 2008 compared to 2007 primarily due to $1.74 billion in IPR&D charges and $102.3 million in acquired intangibles amortization related to the acquisition of Pharmion in March 2008, in addition to a $303.1 million charge for the October 2008 royalty obligation payment to Pfizer related to the unapproved forms of VIDAZA ® . These costs were partly offset by an increase in net revenues provided by REVLIMID ® and VIDAZA ® .
Liquidity and Capital Resources
Cash flows from operating, investing and financing activities for the years ended December 31, 2009, 2008 and 2007 were as follows:
| Increase (Decrease) | ||||||||||||||||||||
| 2009 | 2008 | |||||||||||||||||||
| versus | versus | |||||||||||||||||||
| In thousands $ | 2009 | 2008 | 2007 | 2008 | 2007 | |||||||||||||||
| ||||||||||||||||||||
Net cash provided by operating activities | $ | 909,855 | $ | 182,187 | $ | 477,500 | $ | 727,668 | $ | (295,313 | ) | |||||||||
Net cash used in investing activities | $ | (856,078 | ) | $ | (522,246 | ) | $ | (990,186 | ) | $ | (333,832 | ) | $ | 467,940 | ||||||
Net cash provided by (used in) financing activities | $ | (61,872 | ) | $ | 281,629 | $ | 287,695 | $ | (343,501 | ) | $ | (6,066 | ) | |||||||
53
Operating Activities: Net cash provided by operating activities in 2009 increased by $727.7 million to $909.9 million as compared to 2008. The increase in net cash provided by operating activities was primarily attributable to:
| • | higher net income, |
| • | timing of receipts and payments in the ordinary course of business and |
| • | the October 3, 2008 prepayment of our royalty obligation under the June 7, 2001 5-azacytidine license in full for $425.0 million. |
Also see discussion of cash, cash equivalents, marketable securities and working capital below.
Investing Activities: Net cash used in investing activities in 2009 increased by $333.8 million to $856.1 million as compared to 2008. The increase in net cash used in investing activities was primarily attributable to net purchases of marketable securities available for sale of $749.3 million in 2009 compared to net proceeds from net sales of marketable securities available for sale of $312.1 million in 2008, partly offset by the $746.8 million of cash paid to acquire Pharmion in 2008.
Capital expenditures made in 2009, 2008 and 2007 related primarily to the expansion of our manufacturing capabilities, upgrades to our facilities, as well as spending on computer and laboratory equipment to accommodate our business growth. In 2009, capital expenditures included the cost of developing an enhanced risk management system and in 2008, capital expenditures included the cost of implementing the Oracle Enterprise Business Suite, or EBS. In 2007, capital expenditures included the cost of building our international headquarters in Boudry, Switzerland and computer equipment. For 2010, we are forecasting capital expenditures in the range of approximately $140 million to $150 million compared to approximately $93.4 million in 2009, and we expect to fund this with our operating cash flows.
Financing Activities: Net cash used in financing activities was $61.9 million in 2009 compared to net cash provided by financing activities of $281.6 million in 2008. The increase in net cash used in financing activities compared to net cash provided by financing activities was primarily attributable to:
| • | purchase of $209 million of treasury shares in 2009 |
| • | a decrease in the proceeds from the exercise of common stock options and warrants in 2009 and |
| • | a decrease in the tax benefit from share-based compensation arrangements in 2009. |
Cash, cash equivalents, marketable securities and working capital : Working capital and cash, cash equivalents and marketable securities for the years ended December 31, 2009 and 2008 were as follows:
| 2009 | ||||||||||||
| In thousands $ | 2009 | 2008 | Increase | |||||||||
| ||||||||||||
Cash, cash equivalents and marketable securities | $ | 2,996,752 | $ | 2,222,091 | $ | 774,661 | ||||||
Working capital (1) | $ | 3,302,109 | $ | 2,299,122 | $ | 1,002,987 | ||||||
| (1) | Includes cash, cash equivalents and marketable securities, accounts receivable, net of allowances, inventory and other current assets, less accounts payable, accrued expenses, income taxes payable and other current liabilities. |
Cash, Cash Equivalents and Marketable Securities Available for Sale: We invest our excess cash primarily in money market funds, U.S. Treasury fixed rate securities, U.S. government-sponsored agency fixed rate securities, U.S. government-sponsored agency mortgage-backed fixed rate securities, Federal Deposit Insurance Corporation, or FDIC, guaranteed fixed rate corporate debt, non-U.S. government issued securities and non-U.S. government guaranteed securities. All liquid investments with maturities of three months or less from the date of purchase are classified as cash equivalents and all investments with maturities of greater than three months from the date of purchase are classified as marketable securities available for sale. We determine the appropriate classification of our investments in marketable debt and equity securities at the time of purchase. The increase in cash, cash equivalents and marketable securities available for sale at the end of 2009 compared to 2008 was primarily due to increased cash generated from operations, which more than offset the cash paid out under our share repurchase program announced in April 2009 and capital expenditures.
54
Accounts Receivable, Net : Accounts receivable, net increased by $126.4 million to $438.6 million in 2009 compared to 2008 primarily due to increased sales of REVLIMID ® and VIDAZA ® . Days of sales outstanding, or DSO, in 2009 amounted to 56 days compared to 42 days in 2008. The DSO increase was primarily due to increased international sales for which the collection period is longer than for U.S. sales. We expect this trend to continue as our international sales continue to expand.
Inventory : Inventory balances increased by $0.5 million to $100.7 million in 2009 compared to 2008. The increase reflected higher levels of REVLIMID ® and VIDAZA ® inventories, which were mostly offset by the elimination of ALKERAN ® inventories resulting from the conclusion of the GSK supply agreement and reductions in THALOMID ® due to lower sales volumes.
Other Current Assets : Other current assets increased by $68.5 million to $258.9 million in 2009 compared to 2008 primarily due to an increase in the fair value of foreign currency forward derivative contracts and an increase in prepaid expenses, primarily sales, use and value added taxes.
Accounts Payable, Accrued Expenses and Other Current Liabilities : Accounts payable, accrued expenses and other current liabilities decreased by $28.7 million to $446.0 million in 2009 compared to 2008. The decrease was primarily due to the impact of changes in the fair value of foreign currency forward derivative contracts, which was partly offset by an increase in clinical trial accruals and accrued payroll related expenses, resulting from our expanded business activities.
Income Taxes Payable (Current and Non-Current) : Income taxes payable increased $59.5 million in 2009 compared to 2008 primarily from the current provision for income taxes of $225.9 million partially offset by tax payments of $60.0 million and a tax benefit on stock option exercises of $103.4 million.
We expect continued growth in our expenditures, particularly those related to research and product development, clinical trials, regulatory approvals, international expansion, commercialization of products and capital investments. However, we anticipate that existing cash, cash equivalents and marketable securities available for sale, combined with cash received from expected net product sales and royalty agreements, will provide sufficient capital resources to fund our operations for the foreseeable future.
Contractual Obligations
The following table sets forth our contractual obligations as of December 31, 2009:
| Payment Due By Period | ||||||||||||||||||||
| Less than | More than | |||||||||||||||||||
| In thousands $ | 1 Year | 1 to 3 Years | 3 to 5 Years | 5 Years | Total | |||||||||||||||
| ||||||||||||||||||||
Operating leases | $ | 26,578 | $ | 37,207 | $ | 15,815 | $ | 19,541 | $ | 99,141 | ||||||||||
Manufacturing facility note payable | 3,964 | 7,832 | 7,736 | 7,736 | 27,268 | |||||||||||||||
Other contract commitments | 97,121 | 60,424 | - | - | 157,545 | |||||||||||||||
| ||||||||||||||||||||
Total | $ | 127,663 | $ | 105,463 | $ | 23,551 | $ | 27,277 | $ | 283,954 | ||||||||||
| ||||||||||||||||||||
Operating leases: We lease office and research facilities under various operating lease agreements in the United States and various international markets. The non-cancelable lease terms for the operating leases expire at various dates between 2010 and 2018 and include renewal options. In general, we are also required to reimburse the lessors for real estate taxes, insurance, utilities, maintenance and other operating costs associated with the leases. For more information on the major facilities that we occupy under lease arrangements refer to Part I, Item 2, "Properties" of this Annual Report on Form 10-K.
55
Manufacturing Facility Note Payable : In December 2006, we purchased an API manufacturing facility and certain other assets and liabilities from Siegfried located in Zofingen, Switzerland. At December 31, 2009, the fair value of our note payable to Siegfried approximated the carrying value of the note of $25.0 million.
Other Contract Commitments : Other contract commitments include $146.2 million in contractual obligations related to product supply contracts. In addition, we have committed to invest $20.0 million in an investment fund over a ten-year period, which is callable at any time. On December 31, 2009, our remaining investment commitment was $10.5 million. For more information refer to Note 19 of the Notes to the Consolidated Financial Statements included in this Annual Report on Form 10-K.
Income Taxes Payable: We have provided a liability for unrecognized tax benefits related to various federal, state and foreign income tax matters of $450.2 million at December 31, 2009 of which $27.8 million is classified as current. The remaining balance of $422.4 million is classified as non-current because the timing of the settlement of these amounts is not reasonably estimable as of December 31, 2009. We do not expect a settlement of the unrecognized tax benefits classified as non-current within the next 12 months.
Collaboration Arrangements: We have entered into certain research and development collaboration arrangements with third parties that include the funding of certain development, manufacturing and commercialization efforts with the potential for future milestone and royalty payments upon the achievement of pre-established developmental, regulatory and /or commercial targets. See Note 17 to the Consolidated Financial Statements included in this Annual Report on 10-K for a description of our collaboration agreements. Our obligation to fund these efforts is contingent upon continued involvement in the programs, the successful development of research compounds that we choose to license and/or the lack of any adverse events which could cause the discontinuance of the programs.
The table of contractual obligations in this Annual Report on Form 10-K does not include potential milestone payments totaling approximately $3.750 billion, which are either contingent on the achievement of various research, development and regulatory approval milestones (approximately $2.220 billion) or are sales-based milestones (approximately $1.530 billion). Research, development and regulatory approval milestones depend primarily upon future favorable clinical developments and regulatory agency actions, neither of which may ever occur. Sales-based milestones are contingent on generating certain levels of future sales of products. Since the achievement and timing of these milestones is neither determinable nor reasonably estimable, such contingencies have not been included in the contractual obligations table or recorded on our consolidated balance sheets.
New Accounting Principles
In June 2009, the Financial Accounting Standards Board, or FASB, established the FASB Accounting Standards Codification TM , or ASC, as the source of authoritative accounting principles recognized by the FASB to be applied by nongovernmental entities in preparation of financial statements in conformity with generally accepted accounting principles in the United States. All other accounting literature not included in the ASC is now nonauthoritative. The ASC was effective for financial statements issued for interim and annual periods ending after September 15, 2009 and its adoption did not have any impact on our consolidated financial statements. The ASC is updated through the FASB's issuance of Accounting Standard Updates, or ASUs. Summarized below are recently issued accounting pronouncements as described under the new ASC structure.
In September 2006, the FASB issued ASC No. 825, "Fair Value Measurements," or ASC 825, which establishes a framework for measuring fair value and expands disclosures about fair value measurements. The FASB partially deferred the effective date of ASC 825 for non-financial assets and liabilities that are recognized or disclosed at fair value in the financial statements on a nonrecurring basis to fiscal years beginning after November 15, 2008. Our adoption of ASC 825 related to non-financial assets beginning January 1, 2009 did not have any impact on our consolidated financial statements.
56
In December 2007, the FASB ratified ASC No. 808, "Accounting for Collaborative Arrangements Related to the Development and Commercialization of Intellectual Property," or ASC 808, which provides guidance for ASC No. 730, "Research and Development," related to how the parties to a collaborative agreement should account for costs incurred and revenue generated on sales to third parties, how sharing payments pursuant to a collaboration agreement should be presented in the income statement and certain related disclosure requirements. The guidance for ASC 808 was effective for us beginning January 1, 2009 on a retrospective basis and did not have any impact on our consolidated financial statements.
In December 2007, the FASB issued ASC No. 805, "Business Combinations," or ASC 805, which requires an acquirer to recognize the assets acquired, the liabilities assumed and any noncontrolling interest in the acquiree at the acquisition date, measured at their fair values as of that date, with limited exceptions. This Statement also requires the capitalization of research and development assets acquired in a business combination at their acquisition date fair values, separately from goodwill. In addition, ASC 805 requires that any post-acquisition adjustments to deferred tax asset valuation allowances and liabilities related to uncertain tax positions be recognized in current period income tax expense. ASC 805 was effective for us beginning January 1, 2009 and we accounted for post-acquisition tax-related adjustments for pre-2009 business combinations and will account for future business combinations and certain other developments from past combinations in accordance with its provisions.
In December 2007, the FASB issued an amendment to ASC No. 810, "Noncontrolling Interests in Consolidated Financial Statements," which changes the accounting for and reporting of noncontrolling interests (formerly known as minority interests) in consolidated financial statements. The amendment was effective for us beginning January 1, 2009 and did not have any impact on our consolidated financial statements.
In March 2008, the FASB issued an amendment to ASC No. 815, "Disclosures about Derivative Instruments and Hedging Activities," which is intended to improve financial reporting about derivative instruments and hedging activities by requiring enhanced disclosures to enable investors to better understand their effects on an entity's financial position, financial performance and cash flows. The amendment was effective for us beginning January 1, 2009 and the expanded disclosures are included in Note 6.
In April 2008, the FASB issued an amendment to ASC No. 350, "Determination of the Useful Life of Intangible Assets," which amends the factors that should be considered in developing renewal or extension assumptions used to determine the useful life of a recognized intangible asset. The amendment was effective for us beginning January 1, 2009 and did not have any impact on our consolidated financial statements.
In May 2008, the FASB issued an amendment to ASC No. 470 "Accounting for Convertible Debt Instruments That May Be Settled in Cash upon Conversion (Including Partial Cash Settlement)," which requires separate accounting for the debt and equity components of convertible debt issuances that have a cash settlement feature permitting settlement partially or fully in cash upon conversion. A component of such debt issuances that is representative of the approximate fair value of the conversion feature at inception should be bifurcated and recorded to equity, with the resulting debt discount amortized to interest expense in a manner that reflects the issuer's nonconvertible, unsecured debt borrowing rate. The requirements for separate accounting must be applied retrospectively to previously issued convertible debt issuances as well as prospectively to newly issued convertible debt issuances, negatively affecting both net income and earnings per share, in financial statements issued for fiscal years beginning after December 15, 2008. Since our past convertible debt issuance did not include a cash settlement feature, the amendment did not have any impact on our consolidated financial statements.
57
In June 2008, the FASB issued ASC No. 260, "Determining Whether Instruments Granted in Share-Based Payment Transactions Are Participating Securities," or ASC 260. The ASC addresses whether instruments granted in share-based payment transactions are participating securities prior to vesting and therefore need to be included in the earnings allocation in calculating earnings per share under the two-class method and requires companies to treat unvested share-based payment awards that have non-forfeitable rights to dividends or dividend equivalents as a separate class of securities in calculating earnings per share. ASC 260 was effective for us beginning January 1, 2009. Since our past share-based payment awards did not include non-forfeitable rights to dividends or dividend equivalents, the adoption of ASC 260 did not have any impact on our consolidated financial statements.
In November 2008, the FASB ratified ASC No. 323, "Equity Method Investment Accounting Considerations," or ASC 323, which clarifies the accounting for certain transactions and impairment considerations involving equity method investments. ASC 323 was effective for us beginning January 1, 2009 and did not have any impact on our consolidated financial statements.
In November 2008, the FASB ratified an amendment to ASC No. 350, "Accounting for Defensive Intangible Assets," which clarifies the accounting for certain separately identifiable intangible assets which an acquirer does not intend to actively use but intends to hold to prevent its competitors from obtaining access to them. The amendment requires an acquirer in a business combination to account for a defensive intangible asset as a separate unit of accounting, which should be amortized to expense over the period the asset diminishes in value. The amendment was effective for us beginning January 1, 2009 and we will account for defensive intangible assets acquired in future business combinations in accordance with its provisions.
In April 2009, the FASB issued an amendment to ASC No. 820, "Determining Fair Value When the Volume and Level of Activity for the Asset or Liability Have Significantly Decreased and Identifying Transactions That Are Not Orderly," or ASC 820. This amendment provides additional guidance for estimating fair value in accordance with ASC 820 when the volume and level of activity for the asset or liability have significantly decreased and also includes guidance on identifying circumstances that indicate a transaction is not orderly for fair value measurements. This amendment shall be applied prospectively with retrospective application not permitted. This amendment was effective for interim and annual periods ending after June 15, 2009. The adoption did not have any impact on our consolidated financial statements.
In April 2009, the FASB issued an amendment to ASC 320, "Recognition and Presentation of Other-Than-Temporary Impairments." This amendment was issued to make the other-than-temporary impairments guidance more operational and to improve the presentation of other-than-temporary impairments in the financial statements. This amendment replaces the existing requirement that the entity's management assert it has both the intent and ability to hold an impaired debt security until recovery with a requirement that management assert it does not have the intent to sell the security, and it is more likely than not it will not have to sell the security before recovery of its cost basis. This amendment provides increased disclosure about the credit and noncredit components of impaired debt securities that are not expected to be sold and also requires increased and more frequent disclosures regarding expected cash flows, credit losses and an aging of securities with unrealized losses. Although this amendment does not result in a change in the carrying amount of debt securities, it does require that the portion of an other-than-temporary impairment not related to a credit loss for a held-to-maturity security be recognized in a new category of other comprehensive income and be amortized over the remaining life of the debt security as an increase in the carrying value of the security. This amendment was effective for interim and annual periods ending after June 15, 2009. The adoption of this amendment did not have any impact on our consolidated financial statements.
58
In April 2009, the FASB issued an amendment to ASC 825, "Interim Disclosures About Fair Value of Financial Instruments," to require disclosures about fair value of financial instruments not measured on the balance sheet at fair value in interim financial statements as well as in annual financial statements. Prior to this amendment, fair values for these assets and liabilities were only disclosed annually. This amendment applies to all financial instruments within the scope of ASC 825 and requires all entities to disclose the method(s) and significant assumptions used to estimate the fair value of financial instruments. This amendment was effective for interim periods ending after June 15, 2009. This amendment does not require disclosures for earlier periods presented for comparative purposes at initial adoption. In periods after initial adoption, this amendment requires comparative disclosures only. The adoption did not have any impact on our consolidated financial statements.
In April 2009, the FASB issued an amendment to ASC No. 805, "Accounting for Assets Acquired and Liabilities Assumed in a Business Combination That Arise from Contingencies." This amendment clarifies application issues associated with initial recognition and measurement, subsequent measurement and accounting, and disclosure of assets and liabilities arising from contingencies in a business combination. This amendment was effective for us beginning January 1, 2009 and we will account for assets or liabilities arising from contingencies acquired in future business combinations in accordance with its provisions.
In May 2009, the FASB issued ASC No. 855, "Subsequent Events," or ASC 855, which established general standards of accounting for and disclosure of events that occur after the balance sheet date but before financial statements are issued. It sets forth the period after the balance sheet date during which management of a reporting entity should evaluate events or transactions that occur for potential recognition or disclosure in the financial statements, the circumstances under which an entity should recognize events or transactions occurring after the balance sheet date in its financial statements and the disclosures that an entity should make about events or transactions that occurred after the balance sheet date. ASC 855 was effective for financial statements issued for interim and annual periods ending after June 15, 2009.
In June 2009, the FASB issued an amendment to ASC No. 860, "Accounting for Transfers of Financial Assets," which eliminates the concept of a "qualifying special-purpose entity," changes the requirements for derecognizing financial assets and requires additional disclosures. This amendment clarifies the determination whether a transferor and all of the entities included in the transferor's financial statements being presented have surrendered control over transferred financial assets. It also enhances information reported to users of financial statements by providing greater transparency about transfers of financial assets and a company's continuing involvement in transferred financial assets. This amendment will be effective for our fiscal year beginning January 1, 2010. We are currently evaluating the impact, if any, that the adoption of this amendment will have on our consolidated financial statements.
In June 2009, the FASB issued an amendment to ASC 810, "Consolidation of Variable Interest Entities," which changes how a company determines when an entity that is insufficiently capitalized or is not controlled through voting (or similar rights) should be consolidated. The determination of whether a company is required to consolidate an entity is based on, among other things, an entity's purpose and design and a company's ability to direct the activities of the entity that most significantly impact the entity's economic performance. This amendment requires ongoing reassessments of whether an enterprise is the primary beneficiary of a variable interest entity and will require a company to provide additional disclosures about its involvement with variable interest entities, any significant changes in risk exposure due to that involvement and how its involvement with a variable interest entity affects the company's financial statements. This amendment will be effective for our fiscal year beginning January 1, 2010. We are currently evaluating the impact, if any, that the adoption of this amendment will have on our consolidated financial statements.
59
In August 2009, the FASB issued ASU No. 2009-05, "Measuring Liabilities at Fair Value," or ASU 2009-05, which amends ASC 820 to provide clarification of a circumstances in which a quoted price in an active market for an identical liability is not available. A reporting entity is required to measure fair value using one or more of the following methods: 1) a valuation technique that uses a) the quoted price of the identical liability when traded as an asset or b) quoted prices for similar liabilities (or similar liabilities when traded as assets) and/or 2) a valuation technique that is consistent with the principles of ASC 820. ASU 2009-05 also clarifies that when estimating the fair value of a liability, a reporting entity is not required to adjust to include inputs relating to the existence of transfer restrictions on that liability. The adoption of this ASU did not have an impact on our consolidated financial statements.
In September 2009, the FASB issued ASU No. 2009-12, "Fair Value Measurements and Disclosure," or ASU 2009-12, which provides additional guidance on using the net asset value per share, provided by an investee, when estimating the fair value of an alternate investment that does not have a readily determinable fair value and enhances the disclosures concerning these investments. ASU 2009-12 was effective for our interim and annual periods ending after December 15, 2009.
In October 2009, the FASB issued ASU No. 2009-13, "Multiple-Deliverable Revenue Arrangements," or ASU 2009-13, which amends existing revenue recognition accounting pronouncements that are currently within the scope of ASC 605. This guidance eliminates the requirement to establish the fair value of undelivered products and services and instead provides for separate revenue recognition based upon management's estimate of the selling price for an undelivered item when there is no other means to determine the fair value of that undelivered item. ASU 2009-13 is effective for us prospectively for revenue arrangements entered into or materially modified beginning January 1, 2011. We are currently evaluating the impact, if any, that the adoption of this amendment will have on our consolidated financial statements.
Critical Accounting Estimates and Significant Accounting Policies
A critical accounting policy is one that is both important to the portrayal of our financial condition and results of operation and requires management's most difficult, subjective or complex judgments, often as a result of the need to make estimates about the effect of matters that are inherently uncertain. While our significant accounting policies are more fully described in Note 1 of the Notes to the Consolidated Financial Statements included in this Annual Report, we believe the following accounting estimates and policies to be critical:
Revenue Recognition: Revenue from the sale of products is recognized when title and risk of loss of the product is transferred to the customer. Provisions for discounts, early payments, rebates, sales returns and distributor chargebacks under terms customary in the industry are provided for in the same period the related sales are recorded. We record estimated reductions to revenue for volume-based discounts and rebates at the time of the initial sale. The estimated reductions to revenue for such volume-based discounts and rebates are based on the sales terms, historical experience and trend analysis.
We recognize revenue from royalties based on licensees' sales of our products or products using our technologies. Royalties are recognized as earned in accordance with the contract terms when royalties from licensees can be reasonably estimated and collectibility is reasonably assured. If royalties cannot be reasonably estimated or collectibility of a royalty amount is not reasonably assured, royalties are recognized as revenue when the cash is received.
Gross to Net Sales Accruals: We record gross to net sales accruals for sales returns and allowances, sales discounts, government rebates, and chargebacks and distributor service fees.
60
THALOMID ® is distributed in the United States under our S.T.E.P.S. ® program which we developed and is a proprietary comprehensive education and risk-management distribution program with the objective of providing for the safe and appropriate distribution and use of THALOMID ® . Internationally, THALOMID ® is also distributed under mandatory risk-management distribution programs tailored to meet local competent authorities' specifications to help ensure the safe and appropriate distribution and use of THALOMID ® . These programs may vary by country and, depending upon the country and the design of the risk-management program, the product may be sold through hospitals or retail pharmacies. REVLIMID ® is distributed in the United States primarily through contracted pharmacies under the RevAssist ® program, which is a proprietary risk-management distribution program tailored specifically to help ensure the safe and appropriate distribution and use of REVLIMID ® . Internationally, REVLIMID ® is also distributed under mandatory risk-management distribution programs tailored to meet local competent authorities' specifications to help ensure the safe and appropriate distribution and use of REVLIMID ® . These programs may vary by country and, depending upon the country and the design of the risk-management program, the product may be sold through hospitals or retail pharmacies. VIDAZA ® is distributed through the more traditional pharmaceutical industry supply chain. VIDAZA ® is not subjected to the same risk-management distribution programs as THALOMID ® and REVLIMID ® .
We base our sales returns allowance on estimated on-hand retail/hospital inventories, measured end-customer demand as reported by third-party sources, actual returns history and other factors, such as the trend experience for lots where product is still being returned or inventory centralization and rationalization initiatives conducted by major pharmacy chains, as applicable. If the historical data we use to calculate these estimates does not properly reflect future returns, then a change in the allowance would be made in the period in which such a determination is made and revenues in that period could be materially affected. Under this methodology, we track actual returns by individual production lots. Returns on closed lots, that is, lots no longer eligible for return credits, are analyzed to determine historical returns experience. Returns on open lots, that is, lots still eligible for return credits, are monitored and compared with historical return trend rates. Any changes from the historical trend rates are considered in determining the current sales return allowance. THALOMID ® is drop-shipped directly to the prescribing pharmacy and, as a result, wholesalers do not stock the product. REVLIMID ® is distributed primarily through hospitals and contracted pharmacies, lending itself to tighter controls of inventory quantities within the supply channel and, thus, resulting in lower returns activity to date.
Sales discount accruals are based on payment terms extended to customers.
Government rebate accruals are based on estimated payments due to governmental agencies for purchases made by third parties under various governmental programs. U.S. Medicaid rebate accruals are based on historical payment data and estimates of future Medicaid beneficiary utilization applied to the Medicaid unit rebate amount formula established by the Center for Medicaid and Medicare Services. Certain international markets have government-sponsored programs that require rebates to be paid and accordingly the rebate accruals are determined primarily on estimated eligible sales.
Chargebacks are based on the differentials between product acquisition prices paid by wholesalers and lower government contract pricing paid by eligible customers covered under federally qualified programs. Distributor service fee accruals are based on contractual fees to be paid to the wholesale distributor for services provided. On January 28, 2008, the Fiscal Year 2008 National Defense Authorization Act was enacted, which expands TRICARE to include prescription drugs dispensed by TRICARE retail network pharmacies. TRICARE is a health care program of the U.S. Department of Defense Military Health System that provides civilian health benefits for military personnel, military retirees and their dependents. TRICARE rebate accruals reflect this program expansion and are based on estimated Department of Defense eligible sales multiplied by the TRICARE rebate formula.
Income Taxes : We utilize the asset and liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on the difference between the financial statement carrying amounts and tax bases of assets and liabilities using enacted tax rates in effect for years in which the temporary differences are expected to reverse. We provide a valuation allowance when it is more likely than not that deferred tax assets will not be realized.
We account for interest and penalties related to uncertain tax positions as part of our provision for income taxes. These unrecognized tax benefits relate primarily to issues common among multinational corporations in our industry. We apply a variety of methodologies in making these estimates which include studies performed by independent economists, advice from industry and subject experts, evaluation of public actions taken by the IRS and other taxing authorities, as well as our own industry experience. We provide estimates for unrecognized tax benefits. If our estimates are not representative of actual outcomes, our results of operations could be materially impacted.
61
We periodically evaluate the likelihood of the realization of deferred tax assets, and reduce the carrying amount of these deferred tax assets by a valuation allowance to the extent we believe a portion will not be realized. We consider many factors when assessing the likelihood of future realization of deferred tax assets, including our recent cumulative earnings experience by taxing jurisdiction, expectations of future taxable income, carryforward periods available to us for tax reporting purposes, various income tax strategies and other relevant factors. Significant judgment is required in making this assessment and, to the extent future expectations change, we would have to assess the recoverability of our deferred tax assets at that time. At December 31, 2009, it was more likely than not that we would realize our deferred tax assets, net of valuation allowances.
Share-Based Compensation : The cost of share-based compensation is recognized in the Consolidated Statements of Operations based on the fair value of all awards granted, using the Black-Scholes method of valuation. The fair value of each award is determined and the compensation cost is recognized over the service period required to obtain full vesting. Compensation cost to be recognized reflects an estimate of the number of awards expected to vest after taking into consideration an estimate of award forfeitures based on actual experience.
Other-Than-Temporary Impairments of Available-For-Sale Marketable Securities: A decline in the market value of any available-for-sale marketable security below its cost that is deemed to be other-than-temporary results in a reduction in carrying amount to fair value. The impairment is charged to operations and a new cost basis for the security established. The determination of whether an available-for-sale marketable security is other-than-temporarily impaired requires significant judgment and requires consideration of available quantitative and qualitative evidence in evaluating the potential impairment. Factors evaluated to determine whether the investment is other-than-temporarily impaired include: significant deterioration in the issuer's earnings performance, credit rating, asset quality, business prospects of the issuer, adverse changes in the general market conditions in which the issuer operates, length of time that the fair value has been below our cost, our expected future cash flows from the security, our intent not to sell and an evaluation as to whether it is more likely than not that we will not have to sell before recovery of our cost basis. Assumptions associated with these factors are subject to future market and economic conditions, which could differ from our assessment.
Derivatives and Hedging Activities: All derivative instruments are recognized on the balance sheet at their fair value. Changes in the fair value of derivative instruments are recorded each period in current earnings or other comprehensive income (loss), depending on whether a derivative instrument is designated as part of a hedging transaction and, if it is, the type of hedging transaction. For a derivative to qualify as a hedge at inception and throughout the hedged period, we formally document the nature and relationships between the hedging instruments and hedged item. We assess, both at inception and on an on-going basis, whether the derivative instruments that are used in cash flow hedging transactions are highly effective in offsetting the changes in cash flows of hedged items. We assess hedge effectiveness on a quarterly basis and record the gain or loss related to the ineffective portion of derivative instruments, if any, to current earnings. If we determine that a forecasted transaction is no longer probable of occurring, we discontinue hedge accounting and any related unrealized gain or loss on the derivative instrument is recognized in current earnings. We use derivative instruments, including those not designated as part of a hedging transaction, to manage our exposure to movements in foreign exchange rates. The use of these derivative instruments modifies the exposure of these risks with the intent to reduce our risk or cost. We do not use derivative instruments for speculative trading purposes and are not a party to leveraged derivatives.
62
Investment in Affiliated Companies: We apply the equity method of accounting to our investments in common stock of affiliated companies and certain investment funds, which primarily invest in companies conducting business in life sciences such as biotechnology, pharmaceuticals, medical technology, medical devices, diagnostics and health and wellness.
Equity investments are reviewed on a regular basis for possible impairment. If an investment's fair value is determined to be less than its net carrying value and the decline is determined to be other-than-temporary, the investment is written down to its fair value. Such an evaluation is judgmental and dependent on specific facts and circumstances. Factors considered in determining whether an other-than-temporary decline in value has occurred include: market value or exit price of the investment based on either market-quoted prices or future rounds of financing by the investee; length of time that the market value was below its cost basis; financial condition and business prospects of the investee; our intent and ability to retain the investment for a sufficient period of time to allow for recovery in market value of the investment; issues that raise concerns about the investee's ability to continue as a going concern; and any other information that we may be aware of related to the investment.
Accounting for Long-Term Incentive Plans : We have established a Long-Term Incentive Plan, or LTIP, designed to provide key officers and executives with performance-based incentive opportunities contingent upon achievement of pre-established corporate performance objectives covering a three-year period. We currently have three three-year performance cycles running concurrently ending December 31, 2010, 2011 and 2012. Performance measures for each LTIP are based on the following components in the last year of the three-year cycle: 25% on non-GAAP earnings per share, 25% on non-GAAP net income and 50% on total non-GAAP revenue, as defined.
Payouts may be in the range of 0% to 200% of the participant's salary for the plans. Awards are payable in cash or, at our discretion, in our common stock based upon our stock price at the payout date. We accrue the long-term incentive liability over each three-year cycle. Prior to the end of a three-year cycle, the accrual is based on an estimate of our level of achievement during the cycle. Upon a change in control, participants will be entitled to an immediate payment equal to their target award, or an award based on actual performance, if higher, through the date of the change in control.
Accruals recorded for the LTIP entail making certain assumptions concerning future non-GAAP earnings per share, non-GAAP net income and non-GAAP revenues, as defined; the actual results of which could be materially different than the assumptions used. Accruals for the LTIP are reviewed on a regular basis and revised accordingly so that the liability recorded reflects updated estimates of future payouts. In estimating the accruals, management considers actual results to date for the performance period, expected results for the remainder of the performance period, operating trends, product development, pricing and competition.
63
Valuation of acquired intangible assets and acquired in-process research and development: We have acquired intangible assets primarily through business combinations. When identifiable intangible assets, including in-process research and development, are acquired we determine the fair values of these assets as of the acquisition date. Discounted cash flow models are typically used in these valuations, and the models require the use of significant estimates and assumptions including but not limited to:
| • | projecting regulatory approvals, |
| • | estimating future cash flows from product sales resulting from completed products and in-process projects and |
| • | developing appropriate discount rates and probability rates. |
Goodwill and Other Intangible Assets: Goodwill represents the excess of purchase price over fair value of net assets acquired in a business combination accounted for by the purchase method of accounting and is not amortized, but subject to impairment testing at least annually or when a triggering event occurs that could indicate a potential impairment. We test our goodwill annually for impairment each November 30. Intangible assets with definite useful lives are amortized to their estimated residual values over their estimated useful lives and reviewed for impairment if certain events occur. We currently have no intangible assets with indefinite useful lives.
64
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
The following discussion provides forward-looking quantitative and qualitative information about our potential exposure to market risk. Market risk represents the potential loss arising from adverse changes in the value of financial instruments. The risk of loss is assessed based on the likelihood of adverse changes in fair values, cash flows or future earnings.
We have established guidelines relative to the diversification and maturities of investments to maintain safety and liquidity. These guidelines are reviewed periodically and may be modified depending on market conditions. Although investments may be subject to credit risk, our investment policy specifies credit quality standards for our investments and limits the amount of credit exposure from any single issue, issuer or type of investment. At December 31, 2009, our market risk sensitive instruments consisted of marketable securities available for sale, our manufacturing facility note payable and certain foreign currency forward contracts.
Marketable Securities Available for Sale: At December 31, 2009, our marketable securities available for sale consisted primarily of U.S. Treasury fixed rate securities, U.S. government-sponsored agency fixed rate securities, U.S. government-sponsored agency mortgage-backed fixed rate securities, FDIC guaranteed fixed rate corporate debt, non-U.S. government issued fixed rate securities, non-U.S. government guaranteed fixed rate securities and a marketable equity security. U.S. government-sponsored agency securities include general unsecured obligations of the issuing agency, including issues from the Federal Home Loan Bank, or FHLB, Federal National Mortgage Association, or Fannie Mae, and Federal Home Loan Mortgage Corporation, or Freddie Mac. U.S. government-sponsored agency mortgage-backed securities include fixed rate asset-backed securities issued by Fannie Mae, Freddie Mac and Government National Mortgage Association, or Ginnie Mae. Federal Deposit Insurance Corporation, or FDIC, guaranteed corporate debt includes obligations of bank holding companies that meet certain criteria set forth under the Federal Temporary Liquidity Guarantee Program, or TLGP, and is unconditionally guaranteed by the FDIC.
Fannie Mae, Freddie Mac, FHLB and Ginnie Mae are regulated by the Federal Housing Finance Agency, or FHFA. Working with the Congress and the Office of the President, the U.S. Treasury and the Federal Reserve have pledged to continue to provide capital and liquidity to these U.S. government-sponsored agencies. We have not recorded any impairment against our holdings in these securities due to the support of the U.S. government of these agencies.
Non-U.S. government issued securities consist of direct obligations of highly-rated governments of nations other then the United States. Non-U.S. government guaranteed securities consist of obligations of agencies and other entities that are explicitly guaranteed by highly-rated governments of nations other then the United States. We have not recorded impairments against our holdings in these securities due to the support of the governments of these agencies and entities.
Marketable securities available for sale are carried at fair value, held for an unspecified period of time and are intended for use in meeting our ongoing liquidity needs. Unrealized gains and losses on available-for-sale securities, which are deemed to be temporary, are reported as a separate component of stockholders' equity, net of tax. The cost of debt securities is adjusted for amortization of premiums and accretion of discounts to maturity. The amortization, along with realized gains and losses and other than temporary impairment charges, is included in interest and investment income, net.
65
As of December 31, 2009, the principal amounts, fair values and related weighted average interest rates of our investments in debt securities classified as marketable securities available-for-sale were as follows:
| Less than | More Than | |||||||||||||||||||
| In thousands $ | 1 Year | 1 to 3 Years | 3 to 5 Years | 5 Years | Total | |||||||||||||||
| ||||||||||||||||||||
Principal amount | $ | 514,553 | $ | 1,234,906 | $ | 103,003 | $ | 9,777 | $ | 1,862,239 | ||||||||||
Fair value | $ | 524,766 | $ | 1,252,237 | $ | 106,628 | $ | 10,437 | $ | 1,894,068 | ||||||||||
Average interest rate | 1.1 | % | 1.6 | % | 2.5 | % | 2.9 | % | 1.5 | % | ||||||||||
Note Payable: In December 2006, we purchased an API manufacturing facility and certain other assets and liabilities from Siegfried. At December 31, 2009, the fair value of our note payable to Siegfried approximated the carrying value of the note of $25.0 million (See Note 11 of the Notes to the Consolidated Financial Statements included in this Annual Report). Assuming other factors are held constant, an increase in interest rates generally will result in a decrease in the fair value of the note. The note is denominated in Swiss francs and its fair value will also be affected by changes in the U.S. dollar / Swiss franc exchange rate. The carrying value of the note reflects the U.S. dollar / Swiss franc exchange rate and Swiss interest rates.
Foreign Currency Forward Contracts: We use foreign currency forward contracts to hedge specific forecasted transactions denominated in foreign currencies and to reduce exposures to foreign currency fluctuations of certain assets and liabilities denominated in foreign currencies.
We enter into foreign currency forward contracts to protect against changes in anticipated foreign currency cash flows resulting from changes in foreign currency exchange rates, primarily associated with non-functional currency denominated revenues and expenses of foreign subsidiaries. The foreign currency forward hedging contracts outstanding at December 31, 2009 and 2008 had settlement dates within 24 months. These foreign currency forward contracts are designated as cash flow hedges under ASC 815 and, accordingly, to the extent effective, any unrealized gains or losses on them are reported in other comprehensive income (loss), or OCI, and reclassified to operations in the same periods during which the underlying hedged transactions affect operations. Any ineffectiveness on these foreign currency forward contracts is reported in other income, net. Foreign currency forward contracts entered into to hedge forecasted revenue and expenses were as follows:
| Notional Amount | ||||||||
| Foreign Currency | 2009 | 2008 | ||||||
| ||||||||
Euro | $ | 1,107,340 | $ | 704,198 | ||||
| ||||||||
The notional settlement amounts of the foreign currency forward contracts outstanding as of December 31, 2009 and 2008 were $1.107 billion and $704.2 million, respectively. We consider the impact of our own and the counterparties' credit risk on the fair value of the contracts as well as the ability of each party to execute its obligations under the contract. As of December 31, 2009 and 2008, credit risk did not materially change the fair value of our foreign currency forward contracts.
66
We recognized reductions in net product sales for certain effective cash flow hedge instruments of $36.4 million for 2009 and $0.4 million for 2008. These settlements were recorded in the same period as the related forecasted sales occurred. We recognized an increase in research and development expenses for the settlement of certain effective cash flow hedge instruments of $0.6 million for 2009 and a decrease in research and development expenses for the settlement of certain effective cash flow hedge instruments of $4.0 million for 2008. These settlements were recorded in the same period as the related forecasted research and development expenses occurred. We recognized an increase in other income, net for the settlement of certain effective cash flow hedge instruments of $11.6 million for the year ended December 31, 2008. These settlements were recorded in the same period as the related forecasted expenses occurred. Changes in time value, which we excluded from the hedge effectiveness assessment, were included in other income, net.
We also enter into foreign currency forward contracts to reduce exposures to foreign currency fluctuations of certain recognized assets and liabilities denominated in foreign currencies. These foreign currency forward contracts have not been designated as hedges under ASC 815 and, accordingly, any changes in their fair value are recognized in other income, net in the current period. The aggregate notional amount of the foreign currency forward non-designated hedging contracts outstanding at December 31, 2009 and 2008 were $483.2 million and $56.6 million, respectively.
Although not predictive in nature, we believe a hypothetical 10% threshold reflects a reasonably possible near-term change in foreign currency rates. Assuming that the December 31, 2009 exchange rates were to change by a hypothetical 10%, the fair value of the foreign currency forward contracts would change by approximately $154.1 million. However, since the contracts either hedge specific forecasted intercompany transactions denominated in foreign currencies or hedge assets and liabilities denominated in currencies other than the entities' functional currencies, any change in the fair value of the contract would be either reported in other comprehensive income (loss) and reclassified to earnings in the same periods during which the underlying hedged transactions affect earnings or remeasured through earnings each period along with the underlying hedged item.
67
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
CELGENE CORPORATION AND SUBSIDIARIES
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| Page | ||||
Consolidated Financial Statements | ||||
| ||||
Report of Independent Registered Public Accounting Firm | 69 | |||
| ||||
Consolidated Balance Sheets as of December 31, 2009 and 2008 | 70 | |||
| ||||
Consolidated Statements of Operations - Years Ended December 31, 2009, 2008 and 2007 | 71 | |||
| ||||
Consolidated Statements of Cash Flows - Years Ended December 31, 2009, 2008 and 2007 | 72 | |||
Consolidated Statements of Stockholders' Equity - Years Ended December 31, 2009, 2008 and 2007 | 74 | |||
| ||||
Notes to Consolidated Financial Statements | 75 | |||
| ||||
Financial Statement Schedule | ||||
| ||||
Schedule II - Valuation and Qualifying Accounts | 130 | |||
| ||||
68
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Celgene Corporation:
We have audited the accompanying consolidated balance sheets of Celgene Corporation and subsidiaries (the Company) as of December 31, 2009 and 2008, and the related consolidated statements of operations, cash flows and stockholders' equity for each of the years in the three-year period ended December 31, 2009. In connection with our audits of the consolidated financial statements, we also have audited the consolidated financial statement schedule, "Schedule II - Valuation and Qualifying Accounts." These consolidated financial statements and consolidated financial statement schedule are the responsibility of the Company's management. Our responsibility is to express an opinion on these consolidated financial statements and consolidated financial statement schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Celgene Corporation and subsidiaries as of December 31, 2009 and 2008, and the results of their operations and their cash flows for each of the years in the three-year period ended December 31, 2009, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the related consolidated financial statement schedule, when considered in relation to the basic consolidated financial statements taken as a whole, presents fairly, in all material respects, the information set forth therein.
As discussed in the Notes to the consolidated financial statements, the Company has, as of January 1, 2009, changed its method of accounting for business combinations, as of January 1, 2008, changed its method of accounting for the measurement of the fair value of financial assets and liabilities, and, as of January 1, 2007, changed its method of recognizing and measuring the tax effects related to uncertain tax positions, each due to the adoption of new accounting requirements issued by the Financial Accounting Standards Board.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the effectiveness of the Company's internal control over financial reporting as of December 31, 2009, based on criteria established in Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated February 18, 2010 expressed an unqualified opinion on the effectiveness of the Company's internal control over financial reporting.
/s/ KPMG LLP
February 18, 2010
69
CELGENE CORPORATION AND SUBSIDIARIES
CONSOLIDATED BALANCE SHEETS
(Dollars in thousands, except per share amounts)
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| ||||||||
Assets | ||||||||
| ||||||||
Current assets: | ||||||||
Cash and cash equivalents | $ | 1,102,172 | $ | 1,092,386 | ||||
Marketable securities available for sale | 1,894,580 | 1,129,705 | ||||||
Accounts receivable, net of allowances of $10,787 and $9,391 at December 31, 2009 and 2008, respectively | 438,617 | 312,243 | ||||||
Inventory | 100,683 | 100,176 | ||||||
Deferred income taxes | 49,817 | 16,415 | ||||||
Other current assets | 258,935 | 190,441 | ||||||
| ||||||||
Total current assets | 3,844,804 | 2,841,366 | ||||||
Property, plant and equipment, net | 297,792 | 248,971 | ||||||
Investment in affiliated companies | 21,476 | 18,392 | ||||||
Intangible assets, net | 349,542 | 434,764 | ||||||
Goodwill | 578,116 | 588,822 | ||||||
Other assets | 297,581 | 312,955 | ||||||
| ||||||||
Total assets | $ | 5,389,311 | $ | 4,445,270 | ||||
| ||||||||
| ||||||||
Liabilities and Stockholders' Equity | ||||||||
| ||||||||
Current liabilities: | ||||||||
Accounts payable | $ | 36,629 | $ | 53,859 | ||||
Accrued expenses | 315,608 | 306,120 | ||||||
Income taxes payable | 46,874 | 51,162 | ||||||
Current portion of deferred revenue | 1,827 | 1,419 | ||||||
Other current liabilities | 93,767 | 114,688 | ||||||
| ||||||||
Total current liabilities | 494,705 | 527,248 | ||||||
| ||||||||
Deferred revenue, net of current portion | 6,527 | 3,127 | ||||||
Non-current income taxes payable | 422,358 | 358,578 | ||||||
Other non-current liabilities | 71,115 | 64,989 | ||||||
| ||||||||
Total liabilities | 994,705 | 953,942 | ||||||
| ||||||||
| ||||||||
Commitments and Contingencies | ||||||||
| ||||||||
Stockholders' equity: | ||||||||
| ||||||||
Preferred stock, $.01 par value per share, 5,000,000 shares authorized; none outstanding at December 31, 2009 and 2008 | - | - | ||||||
Common stock, $.01 par value per share, 575,000,000 shares authorized; issued 467,629,433 and 463,274,296 shares at December 31, 2009 and 2008, respectively | 4,676 | 4,633 | ||||||
Common stock in treasury, at cost; 8,337,961 and 4,144,667 shares at December 31, 2009 and 2008, respectively | (362,521 | ) | (157,165 | ) | ||||
Additional paid-in capital | 5,474,122 | 5,180,397 | ||||||
Accumulated deficit | (632,246 | ) | (1,408,993 | ) | ||||
Accumulated other comprehensive loss | (89,425 | ) | (127,544 | ) | ||||
| ||||||||
Total stockholders' equity | 4,394,606 | 3,491,328 | ||||||
| ||||||||
| ||||||||
Total liabilities and stockholders' equity | $ | 5,389,311 | $ | 4,445,270 | ||||
| ||||||||
See accompanying Notes to Consolidated Financial Statements
70
CELGENE CORPORATION AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF OPERATIONS
(Dollars in thousands, except per share amounts)
| Years Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
Revenue: | ||||||||||||
| ||||||||||||
Net product sales | $ | 2,567,354 | $ | 2,137,678 | $ | 1,300,441 | ||||||
Collaborative agreements and other revenue | 13,743 | 14,945 | 20,109 | |||||||||
Royalty revenue | 108,796 | 102,158 | 85,270 | |||||||||
| ||||||||||||
Total revenue | 2,689,893 | 2,254,781 | 1,405,820 | |||||||||
| ||||||||||||
| ||||||||||||
Expenses: | ||||||||||||
| ||||||||||||
Cost of goods sold (excluding amortization expense) | 216,289 | 258,267 | 130,211 | |||||||||
Research and development | 794,848 | 931,218 | 400,456 | |||||||||
Selling, general and administrative | 753,827 | 685,547 | 440,962 | |||||||||
Amortization of acquired intangible assets | 83,403 | 103,967 | 9,070 | |||||||||
Acquired in-process research and development | - | 1,740,000 | - | |||||||||
| ||||||||||||
Total expenses | 1,848,367 | 3,718,999 | 980,699 | |||||||||
| ||||||||||||
| ||||||||||||
Operating income (loss) | 841,526 | (1,464,218 | ) | 425,121 | ||||||||
| ||||||||||||
Other income and expense: | ||||||||||||
Interest and investment income, net | 76,785 | 84,835 | 109,813 | |||||||||
Equity in losses of affiliated companies | 1,103 | 9,727 | 4,488 | |||||||||
Interest expense | 1,966 | 4,437 | 11,127 | |||||||||
Other income (expense), net | 60,461 | 24,722 | (2,350 | ) | ||||||||
| ||||||||||||
Income (loss) before income taxes | 975,703 | (1,368,825 | ) | 516,969 | ||||||||
| ||||||||||||
Income tax provision | 198,956 | 164,828 | 290,536 | |||||||||
| ||||||||||||
| ||||||||||||
Net income (loss) | $ | 776,747 | $ | (1,533,653 | ) | $ | 226,433 | |||||
| ||||||||||||
| ||||||||||||
Net income (loss) per common share: | ||||||||||||
Basic | $ | 1.69 | $ | (3.46 | ) | $ | 0.59 | |||||
Diluted | $ | 1.66 | $ | (3.46 | ) | $ | 0.54 | |||||
| ||||||||||||
Weighted average shares (in thousands): | ||||||||||||
Basic | 459,304 | 442,620 | 383,225 | |||||||||
| ||||||||||||
| ||||||||||||
Diluted | 467,354 | 442,620 | 431,858 | |||||||||
| ||||||||||||
See accompanying Notes to Consolidated Financial Statements
71
CELGENE CORPORATION AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
(Dollars in thousands)
| Years Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
Cash flows from operating activities: | ||||||||||||
Net income (loss) | $ | 776,747 | $ | (1,533,653 | ) | $ | 226,433 | |||||
Adjustments to reconcile net income (loss) to net cash provided by operating activities: | ||||||||||||
Depreciation of long-term assets | 41,682 | 33,797 | 22,057 | |||||||||
Amortization of intangible assets | 84,386 | 104,365 | 9,478 | |||||||||
Allocation of pre-paid royalties | 36,045 | 10,739 | - | |||||||||
Provision for accounts receivable allowances | 2,664 | 6,232 | 9,489 | |||||||||
Deferred income taxes | (26,939 | ) | (104,588 | ) | (10,077 | ) | ||||||
Acquired in-process research and development | - | 1,740,000 | - | |||||||||
Share-based compensation cost | 145,929 | 106,578 | 58,825 | |||||||||
Equity in losses of affiliated companies | 518 | 8,884 | 3,578 | |||||||||
Share-based employee benefit plan expense | 11,515 | 8,314 | 5,365 | |||||||||
Unrealized change in value of foreign currency forward contracts | (9,738 | ) | 8,250 | (70 | ) | |||||||
Realized (gain) loss on marketable securities available for sale | (31,013 | ) | 1,206 | 6,232 | ||||||||
Other, net | 8,715 | 2,224 | (287 | ) | ||||||||
| ||||||||||||
Change in current assets and liabilities, excluding the effect of the 2008 acquisition: | ||||||||||||
Accounts receivable | (122,615 | ) | (107,685 | ) | (47,367 | ) | ||||||
Inventory | 1,540 | (25,867 | ) | (23,967 | ) | |||||||
Other operating assets | (53,847 | ) | (129,199 | ) | (19,933 | ) | ||||||
Accounts payable and other operating liabilities | 652 | (17,087 | ) | 83,729 | ||||||||
Income tax payable | 39,823 | 69,610 | 157,621 | |||||||||
Deferred revenue | 3,791 | 67 | (3,606 | ) | ||||||||
| ||||||||||||
Net cash provided by operating activities | 909,855 | 182,187 | 477,500 | |||||||||
| ||||||||||||
| ||||||||||||
Cash flows from investing activities: | ||||||||||||
Proceeds from sales of marketable securities available for sale | 2,258,376 | 1,148,116 | 1,654,354 | |||||||||
Purchases of marketable securities available for sale | (3,007,673 | ) | (835,967 | ) | (2,547,686 | ) | ||||||
Payments for acquisition of business, net of cash acquired | - | (746,779 | ) | - | ||||||||
Capital expenditures | (93,384 | ) | (77,379 | ) | (64,359 | ) | ||||||
Investment in affiliated companies | (3,603 | ) | (12,855 | ) | (1,621 | ) | ||||||
Purchases of investment securities | (13,127 | ) | (9,436 | ) | (23,356 | ) | ||||||
Other | 3,333 | 12,054 | (7,518 | ) | ||||||||
| ||||||||||||
Net cash used in investing activities | (856,078 | ) | (522,246 | ) | (990,186 | ) | ||||||
| ||||||||||||
| ||||||||||||
Cash flows from financing activities: | ||||||||||||
Purchase of treasury shares | (209,461 | ) | - | - | ||||||||
Net proceeds from exercise of common stock options and warrants | 49,751 | 128,583 | 144,703 | |||||||||
Excess tax benefit from share-based compensation arrangements | 97,838 | 153,046 | 142,992 | |||||||||
| ||||||||||||
Net cash provided by (used in) financing activities | (61,872 | ) | 281,629 | 287,695 | ||||||||
| ||||||||||||
| ||||||||||||
Effect of currency rate changes on cash and cash equivalents | 17,881 | (67,457 | ) | 3,849 | ||||||||
| ||||||||||||
| ||||||||||||
Net increase (decrease) in cash and cash equivalents | 9,786 | (125,887 | ) | (221,142 | ) | |||||||
| ||||||||||||
Cash and cash equivalents at beginning of year | 1,092,386 | 1,218,273 | 1,439,415 | |||||||||
| ||||||||||||
| ||||||||||||
Cash and cash equivalents at end of year | $ | 1,102,172 | $ | 1,092,386 | $ | 1,218,273 | ||||||
| ||||||||||||
See accompanying Notes to Consolidated Financial Statements
72
CELGENE CORPORATION AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS - (Continued)
(Dollars in thousands)
| Years Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| ||||||||||||
Supplemental schedule of non-cash investing and financing activity: | ||||||||||||
Change in net unrealized (gain) loss on marketable securities available for sale | $ | (3,326 | ) | $ | 87,349 | $ | (81,325 | ) | ||||
| ||||||||||||
| ||||||||||||
Matured shares tendered in connection with stock option exercises | $ | (2,014 | ) | $ | (7,646 | ) | $ | (6,457 | ) | |||
| ||||||||||||
| ||||||||||||
Conversion of convertible notes | $ | - | $ | 196,543 | $ | 203,334 | ||||||
| ||||||||||||
| ||||||||||||
Supplemental disclosure of cash flow information: | ||||||||||||
Interest paid on convertible notes | $ | - | $ | 1,640 | $ | 6,700 | ||||||
| ||||||||||||
| ||||||||||||
Income taxes paid | $ | 70,539 | $ | 29,319 | $ | - | ||||||
| ||||||||||||
See accompanying Notes to Consolidated Financial Statements
73
CELGENE CORPORATION AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY
(Dollars in thousands)
| Accumulated | ||||||||||||||||||||||||
| Other | ||||||||||||||||||||||||
| Additional | Retained | Comprehensive | ||||||||||||||||||||||
| Common | Treasury | Paid-in | Earnings | Income | ||||||||||||||||||||
| Years Ended December 31, 2009, 2008 and 2007 | Stock | Stock | Capital | (Deficit) | (Loss) | Total | ||||||||||||||||||
Balances at December 31, 2006 | 3,801 | (148,097 | ) | 2,209,889 | (101,773 | ) | 12,357 | 1,976,177 | ||||||||||||||||
| ||||||||||||||||||||||||
Net income | 226,433 | 226,433 | ||||||||||||||||||||||
Other comprehensive income: | ||||||||||||||||||||||||
Increase in unrealized gains on available for sale securities, net of $29,631 tax | 47,834 | 47,834 | ||||||||||||||||||||||
Reclassification of losses on available for sale securities included in net income, net of $3,860 tax | 6,232 | 6,232 | ||||||||||||||||||||||
Pension liability adjustment | (31 | ) | (31 | ) | ||||||||||||||||||||
Currency translation adjustments | 17,490 | 17,490 | ||||||||||||||||||||||
| ||||||||||||||||||||||||
Comprehensive income | $ | 297,958 | ||||||||||||||||||||||
Mature shares tendered related to option exercise | (6,457 | ) | (6,457 | ) | ||||||||||||||||||||
Costs related to 2006 secondary stock offering | (3 | ) | (3 | ) | ||||||||||||||||||||
Conversion of long-term convertible notes | 168 | 203,166 | 203,334 | |||||||||||||||||||||
Exercise of stock options and warrants | 103 | 146,763 | 146,866 | |||||||||||||||||||||
Issuance of common stock for employee benefit plans | 5,035 | 2,901 | 7,936 | |||||||||||||||||||||
Expense related to share-based compensation | 58,825 | 58,825 | ||||||||||||||||||||||
Income tax benefit upon exercise of stock options | 159,308 | 159,308 | ||||||||||||||||||||||
| ||||||||||||||||||||||||
Balances at December 31, 2007 | 4,072 | (149,519 | ) | 2,780,849 | 124,660 | 83,882 | 2,843,944 | |||||||||||||||||
| ||||||||||||||||||||||||
Net loss | (1,533,653 | ) | (1,533,653 | ) | ||||||||||||||||||||
Other comprehensive income: | ||||||||||||||||||||||||
Increase in unrealized gains on available for sale securities, net of $5,211 tax | 8,413 | 8,413 | ||||||||||||||||||||||
Reversal of unrealized gains on Pharmion investment, net of $38,904 tax | (62,806 | ) | (62,806 | ) | ||||||||||||||||||||
Reclassification of losses on available for sale securities included in net loss, net of $736 tax | 1,188 | 1,188 | ||||||||||||||||||||||
Unrealized losses on cash flow hedges | (50,117 | ) | (50,117 | ) | ||||||||||||||||||||
Pension liability adjustment | (3,290 | ) | (3,290 | ) | ||||||||||||||||||||
Net asset transfer of common control foreign subsidiaries | 4,337 | (4,337 | ) | - | ||||||||||||||||||||
Currency translation adjustments | (100,477 | ) | (100,477 | ) | ||||||||||||||||||||
| ||||||||||||||||||||||||
Comprehensive (loss) | $ | (1,740,742 | ) | |||||||||||||||||||||
Mature shares tendered related to option exercise | (7,646 | ) | 3,861 | (3,785 | ) | |||||||||||||||||||
Acquisition of Pharmion Corp. | 308 | 1,793,838 | 1,794,146 | |||||||||||||||||||||
Conversion of long-term convertible notes | 162 | 196,381 | 196,543 | |||||||||||||||||||||
Exercise of stock options and warrants | 90 | 128,439 | 128,529 | |||||||||||||||||||||
Issuance of common stock for employee benefit plans | 1 | 5,178 | 5,179 | |||||||||||||||||||||
Expense related to share-based compensation | 106,951 | 106,951 | ||||||||||||||||||||||
Income tax benefit upon exercise of stock options | 160,563 | 160,563 | ||||||||||||||||||||||
| ||||||||||||||||||||||||
Balances at December 31, 2008 | $ | 4,633 | $ | (157,165 | ) | $ | 5,180,397 | $ | (1,408,993 | ) | $ | (127,544 | ) | $ | 3,491,328 | |||||||||
| ||||||||||||||||||||||||
Net income | 776,747 | 776,747 | ||||||||||||||||||||||
Other comprehensive income: | ||||||||||||||||||||||||
Increase in unrealized gains on available for sale securities, net of $11,316 tax | 14,642 | 14,642 | ||||||||||||||||||||||
Reclassification of gains on available for sale securities included in net income, net of $20,675 tax | (31,013 | ) | (31,013 | ) | ||||||||||||||||||||
Unrealized gains on cash flow hedges | 55,479 | 55,479 | ||||||||||||||||||||||
Pension liability adjustment | 5,180 | 5,180 | ||||||||||||||||||||||
Net asset transfer of common control foreign subsidiaries | (3,198 | ) | 3,198 | - | ||||||||||||||||||||
Currency translation adjustments | (9,367 | ) | (9,367 | ) | ||||||||||||||||||||
| ||||||||||||||||||||||||
Comprehensive income | $ | 811,668 | ||||||||||||||||||||||
Mature shares tendered related to option exercise | (2,014 | ) | 1,213 | (801 | ) | |||||||||||||||||||
Exercise of stock options and warrants | 43 | (33 | ) | 50,491 | 50,501 | |||||||||||||||||||
Shares purchased under share repurchase program | (209,461 | ) | (209,461 | ) | ||||||||||||||||||||
Issuance of common stock for employee benefit plans | 6,152 | 2,784 | 8,936 | |||||||||||||||||||||
Expense related to share-based compensation | 143,659 | 143,659 | ||||||||||||||||||||||
Income tax benefit upon exercise of stock options | 98,776 | 98,776 | ||||||||||||||||||||||
| ||||||||||||||||||||||||
Balances at December 31, 2009 | $ | 4,676 | $ | (362,521 | ) | $ | 5,474,122 | $ | (632,246 | ) | $ | (89,425 | ) | $ | 4,394,606 | |||||||||
| ||||||||||||||||||||||||
See accompanying Notes to Consolidated Financial Statements
74
CELGENE CORPORATION AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Thousands of dollars, except per share amounts, unless otherwise indicated)
1. Nature of Business and Summary of Significant Accounting Policies
Nature of Business and Basis of Presentation: Celgene Corporation and its subsidiaries (collectively "Celgene" or the "Company") is a global integrated biopharmaceutical company primarily engaged in the discovery, development and commercialization of innovative therapies designed to treat cancer and immune-inflammatory diseases.
The Company's primary commercial stage products include REVLIMID ® , THALOMID ® (inclusive of Thalidomide Celgene TM and Thalidomide Pharmion TM , subsequent to the acquisition of Pharmion Corporation, or Pharmion), VIDAZA ® and FOCALIN ® . FOCALIN ® is sold exclusively to Novartis Pharma AG, or Novartis. The Company also derives revenues from a licensing agreement with Novartis, which entitles it to royalties on FOCALIN XR ® and the entire RITALIN ® family of drugs, and sales of bio-therapeutic products and services through the Company's Cellular Therapeutics subsidiary. ALKERAN ® was licensed from GlaxoSmithKline, or GSK, and sold under the Celgene label through March 31, 2009, the conclusion date of the ALKERAN ® license with GSK. For the ensuing two years, the Company will continue to earn residual payments based upon GSK's ALKERAN ® revenues.
The consolidated financial statements include the accounts of Celgene Corporation and its subsidiaries. Investments in limited partnerships and interests where the Company has an equity interest of 50% or less and does not otherwise have a controlling financial interest are accounted for by either the equity or cost method. Certain prior year amounts have been reclassified to conform to the current year's presentation.
The preparation of the consolidated financial statements requires management to make estimates and assumptions that affect reported amounts and disclosures. Actual results could differ from those estimates. The Company is subject to certain risks and uncertainties related to product development, regulatory approval, market acceptance, scope of patent and proprietary rights, intense competition, rapid technological change and product liability.
In January 2010, the Company acquired Gloucester Pharmaceuticals Inc., or Gloucester, a privately held pharmaceutical company, for $340.0 million in cash plus $300.0 million in contingent U.S. and international regulatory milestone payments. The acquisition is expected to advance the Company's leadership position in the development of disease-altering therapies through innovative approaches for patients with rare and debilitating blood cancers. Gloucester developed ISTODAX ® (romidepsin), which was approved in November 2009 by the U.S. Food and Drug Administration, or FDA, for the treatment of cutaneous T-cell lymphoma, or CTCL, in patients who have received at least one prior systemic therapy. Additionally, ISTODAX ® has received both orphan drug designation for the treatment of non-Hodgkin's T-cell lymphomas, which includes CTCL and peripheral T-cell lymphoma, or PTCL, and Fast Track status in PTCL from the FDA. The European Agency for the Evaluation of Medicinal Products (EMEA) has granted orphan status designation for ISTODAX ® for the treatment of both CTCL and PTCL. Due to the limitations on access to Gloucester information prior to the acquisition date and the limited time since the acquisition date, the initial accounting for the business combination is incomplete at this time. As a result, the Company is unable to provide the contingent consideration disclosures and amounts recognized as of the acquisition date for the major classes of assets acquired and liabilities assumed, including the information required for pre-acquisition contingencies and goodwill.
75
CELGENE CORPORATION AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
Financial Instruments: Certain financial instruments reflected in the Consolidated Balance Sheets, (e.g., cash, cash equivalents, accounts receivable, certain other assets, accounts payable and certain other liabilities) are recorded at cost, which approximates fair value due to their short-term nature. The fair values of financial instruments other than marketable securities are determined through a combination of management estimates and information obtained from third parties using the latest market data. The fair value of available-for-sale marketable securities is determined utilizing the valuation techniques appropriate to the type of security (See Note 5).
Derivative Instruments and Hedges: All derivative instruments are recognized on the balance sheet at their fair value. Changes in the fair value of derivative instruments are recorded each period in current earnings or other comprehensive income (loss), depending on whether a derivative instrument is designated as part of a hedging transaction and, if it is, the type of hedging transaction. For a derivative to qualify as a hedge at inception and throughout the hedged period, the Company formally documents the nature and relationships between the hedging instruments and hedged item. The Company assesses, both at inception and on an on-going basis, whether the derivative instruments that are used in cash flow hedging transactions are highly effective in offsetting the changes in cash flows of hedged items. The Company assesses hedge ineffectiveness on a quarterly basis and records the gain or loss related to the ineffective portion of derivative instruments, if any, to current earnings. If the Company determines that a forecasted transaction is no longer probable of occurring, it discontinues hedge accounting and any related unrealized gain or loss on the derivative instrument is recognized in current earnings. The Company uses derivative instruments, including those not designated as part of a hedging transaction, to manage its exposure to movements in foreign exchange rates. The use of these derivative instruments modifies the exposure of these risks with the intent to reduce the Company's risk or cost. The Company does not use derivative instruments for speculative trading purposes and is not a party to leveraged derivatives.
Cash, Cash Equivalents and Marketable Securities Available for Sale: The Company invests its excess cash primarily in money market funds, U.S. Treasury fixed rate securities, U.S. government-sponsored agency fixed rate securities, U.S. government-sponsored agency mortgage-backed fixed rate securities, Federal Deposit Insurance Corporation, or FDIC, guaranteed fixed rate corporate debt, non-U.S. government issued securities and non-U.S. government guaranteed securities. All liquid investments with maturities of three months or less from the date of purchase are classified as cash equivalents and all investments with maturities of greater than three months from date of purchase are classified as marketable securities available for sale. The Company determines the appropriate classification of its investments in marketable debt and equity securities at the time of purchase. Marketable securities available for sale are carried at fair value, held for an unspecified period of time and are intended for use in meeting the Company's ongoing liquidity needs. Unrealized gains and losses on available-for-sale securities, which are deemed to be temporary, are reported as a separate component of stockholders' equity, net of tax. The cost of debt securities is adjusted for amortization of premiums and accretion of discounts to maturity. The amortization, along with realized gains and losses and other than temporary impairment charges, is included in interest and investment income, net.
A decline in the market value of any available-for-sale security below its carrying value that is determined to be other-than-temporary would result in a charge to earnings and decrease in the security's carrying value down to its newly established fair value. Factors evaluated to determine if an investment is other-than-temporarily impaired include significant deterioration in earnings performance, credit rating, asset quality or business prospects of the issuer; adverse changes in the general market condition in which the issuer operates; the Company's intent not to sell and an evaluation as to whether it is more likely than not that the Company will not have to sell before recovery of its cost basis; and issues that raise concerns about the issuer's ability to continue as a going concern.
76
CELGENE CORPORATION AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
Concentration of Credit Risk: Cash, cash equivalents and marketable securities are financial instruments that potentially subject the Company to concentration of credit risk. The Company invests its excess cash primarily in money market funds, U.S. Treasury fixed rate securities, U.S. government-sponsored agency fixed rate securities, U.S. government-sponsored agency mortgage-backed fixed rate securities and FDIC guaranteed fixed rate corporate debt, non-U.S. government issued securities and non-U.S. government guaranteed securities (See Note 7). The Company may also invest in unrated or below investment grade securities, such as equity in private companies. The Company has established guidelines relative to diversification and maturities to maintain safety and liquidity. These guidelines are reviewed periodically and may be modified to take advantage of trends in yields and interest rates.
The Company sells its products in the United States primarily through wholesale distributors and contracted pharmacies. Therefore, wholesale distributors and large pharmacy chains account for a large portion of the Company's U.S. trade receivables and net product revenues (See Note 20). International sales are primarily made directly to hospitals, clinics and retail chains. The Company continuously monitors the creditworthiness of its customers and has internal policies regarding customer credit limits. The Company estimates an allowance for doubtful accounts primarily based on the credit worthiness of its customers, aging of receivable balances and general economic conditions.
Inventory: Inventories are recorded at the lower of cost or market, with cost determined on a first-in, first-out basis. The Company periodically reviews the composition of inventory in order to identify obsolete, slow-moving or otherwise non-saleable items. If non-saleable items are observed and there are no alternate uses for the inventory, the Company will record a write-down to net realizable value in the period that the decline in value is first recognized. Included in inventory are raw materials used in the production of preclinical and clinical products, which are charged to research and development expense when consumed.
Property, Plant and Equipment: Property, plant and equipment are stated at cost less accumulated depreciation. Depreciation of plant and equipment is recorded using the straight-line method. Leasehold improvements are depreciated over the lesser of the economic useful life of the asset or the remaining term of the lease, including anticipated renewal options. The estimated useful lives of plant and equipment are as follows:
Buildings | 40 years | |
Building and operating equipment | 15 years | |
Manufacturing machinery and equipment | 10 years | |
Other machinery and equipment | 5 years | |
Furniture and fixtures | 5 years | |
Computer equipment and software | 3-7 years |
Maintenance and repairs are charged to operations as incurred, while expenditures for improvements which extend the life of an asset are capitalized.
Investment in Affiliated Companies: The Company applies the equity method of accounting to its investments in common stock of affiliated companies and certain investment funds, which primarily invest in companies conducting business in life sciences such as biotechnology, pharmaceuticals, medical technology, medical devices, diagnostics and health and wellness.
Equity investments are reviewed on a regular basis for possible impairment. If an investment's fair value is determined to be less than its net carrying value and the decline is determined to be other-than-temporary, the investment is written down to its fair value. Such an evaluation is judgmental and dependent on specific facts and circumstances. Factors considered in determining whether an other-than-temporary decline in value has occurred include: market value or exit price of the investment based on either market-quoted prices or future rounds of financing by the investee; length of time that the market value was below its cost basis; financial condition and business prospects of the investee; the Company's intent and ability to retain the investment for a sufficient period of time to allow for recovery in market value of the investment; issues that raise concerns about the investee's ability to continue as a going concern; any other information that the Company may be aware of related to the investment.
77
CELGENE CORPORATION AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
Goodwill and Other Intangible Assets: Goodwill represents the excess of purchase price over fair value of net assets acquired in a business combination accounted for by the purchase method of accounting and is not amortized, but subject to impairment testing at least annually or when a triggering event occurs that could indicate a potential impairment. The Company tests its goodwill annually for impairment each November 30. Intangible assets with definite useful lives are amortized to their estimated residual values over their estimated useful lives and reviewed for impairment if certain events occur as described in "Impairment of Long-Lived Assets" below. The Company currently has no intangible assets with indefinite useful lives.
Impairment of Long-Lived Assets: Long-lived assets, such as property, plant and equipment are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable.
Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset or asset group to the estimated undiscounted future cash flows expected to be generated by the asset or asset group. If the carrying amount of the assets exceed their estimated future undiscounted net cash flows, an impairment charge is recognized by the amount by which the carrying amount of the assets exceed the fair value of the assets. Assets to be disposed of would be separately presented in the consolidated balance sheet and reported at the lower of their carrying amount or fair value less costs to sell, and are no longer depreciated. The assets and liabilities of a disposal group classified as held for sale would be presented separately in the appropriate asset and liability sections of the Consolidated Balance Sheet.
Foreign Currency Translation: Operations in non-U.S. entities are recorded in the functional currency of each entity. For financial reporting purposes, the functional currency of an entity is determined by a review of the source of an entity's most predominant cash flows. The results of operations for non-U.S. dollar functional currency entities are translated from functional currencies into U. S. dollars using the average currency rate during each period, which approximates the results that would be obtained using actual currency rates on the dates of individual transactions. Assets and liabilities are translated using currency rates at the end of the period. Adjustments resulting from translating the financial statements of the Company's foreign entities into the U.S. dollar are excluded from the determination of net income and are recorded as a component of other comprehensive income (loss). Transaction gains and losses are recorded in other income (expense), net in the Consolidated Statements of Operations. The Company had net foreign exchange gains of $54.5 million, $4.7 million, and $1.1 million in 2009, 2008, and 2007, respectively.
Research and Development Costs: Research and development costs are expensed as incurred. These include all internal costs, external costs related to services contracted by the Company. Upfront and milestone payments made to third parties in connection with research and development collaborations are expensed as incurred up to the point of regulatory approval. Milestone payments made to third parties subsequent to regulatory approval are capitalized and amortized over the remaining useful life of the related product.
78
CELGENE CORPORATION AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
Income Taxes: The Company utilizes the asset and liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on the difference between the financial statement carrying amounts and tax bases of assets and liabilities using enacted tax rates in effect for years in which the temporary differences are expected to reverse. A valuation allowance is provided when it is more likely than not that some portion or all of a deferred tax asset will not be realized. The Company recognizes the benefit of an uncertain tax position that it has taken or expects to take on income tax returns it files if such tax position is more likely than not to be sustained.
Revenue Recognition: Revenue from the sale of products is recognized when title and risk of loss of the product is transferred to the customer. Provisions for discounts, early payments, rebates, sales returns and distributor chargebacks under terms customary in the industry are provided for in the same period the related sales are recorded. The Company records estimated reductions to revenue for free goods and volume-based discounts at the time of the initial sale. The estimated reductions to revenue for such free goods and volume-based discounts are based on the sales terms, historical experience and trend analysis. The cost of free goods is included in Cost of Goods Sold (excluding amortization of acquired intangible assets).
The Company bases its sales returns allowance on estimated on-hand retail/hospital inventories, measured end-customer demand as reported by third-party sources, actual returns history and other factors, such as the trend experience for lots where product is still being returned or inventory centralization and rationalization initiatives conducted by major pharmacy chains. If the historical data used by the Company to calculate these estimates does not properly reflect future returns, then a change in the allowance would be made in the period in which such a determination is made and revenues in that period could be materially affected. Under this methodology, the Company tracks actual returns by individual production lots. Returns on closed lots, that is, lots no longer eligible for return credits, are analyzed to determine historical returns experience. Returns on open lots, that is, lots still eligible for return credits, are monitored and compared with historical return trend rates. Any changes from the historical trend rates are considered in determining the current sales return allowance.
The Company recognizes revenue from royalties based on licensees' sales of its products or products using its technologies. Royalties are recognized as earned in accordance with the contract terms when royalties from licensees can be reasonably estimated and collectibility is reasonably assured. If royalties cannot be reasonably estimated or collectibility of a royalty amount is not reasonably assured, royalties are recognized as revenue when the cash is received.
Share-Based Compensation: The cost of share-based compensation is recognized in the Consolidated Statements of Operations based on the fair value of all awards granted, using the Black-Scholes method of valuation. The fair value of each award is determined and the compensation cost is recognized over the service period required to obtain full vesting. Compensation cost to be recognized reflects an estimate of the number of awards expected to vest after taking into consideration an estimate of award forfeitures based on actual experience.
Earnings Per Share: Basic earnings per share is computed by dividing net income by the weighted-average number of common shares outstanding during the period. Diluted earnings per share is computed by dividing net income adjusted to add back the after-tax amount of interest recognized in the period associated with any convertible debt issuance that may be dilutive by the weighted-average number of common shares outstanding during the period increased to include all additional common shares that would have been outstanding as if the outstanding convertible debt was converted into shares of common stock and assuming potentially dilutive common shares, resulting from option exercises, restricted stock units, warrants and other incentives had been issued and any proceeds thereof used to repurchase common stock at the average market price during the period. The assumed proceeds used to repurchase common stock is the sum of the amount to be paid to the Company upon exercise of options, the amount of compensation cost attributed to future services and not yet recognized and, if applicable, the amount of excess income tax benefit that would be credited to paid-in capital upon exercise. As of their maturity date, June 1, 2008, substantially all of the Company's convertible notes were converted into shares of common stock.
79
CELGENE CORPORATION AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
Comprehensive Income: The components of comprehensive income (loss) consist of net income (loss), changes in pension liability, changes in net unrealized gains (losses) on marketable securities classified as available-for-sale, net unrealized gains (losses) related to cash flow hedges and changes in foreign currency translation adjustments.
A summary of accumulated other comprehensive income (loss), net of tax, is summarized as follows:
| Foreign | Accumulated | |||||||||||||||||||
| Net Unrealized | Net Unrealized | Currency | Other | |||||||||||||||||
| Pension | Gains (Losses) From | Gains (Losses) | Translation | Comprehensive | ||||||||||||||||
| Liability | Marketable Securities | From Hedges | Adjustment | Income (Loss) | ||||||||||||||||
Balance December 31, 2007 | $ | (31 | ) | $ | 69,788 | $ | - | $ | 14,125 | $ | 83,882 | |||||||||
Period Change | (3,290 | ) | (53,205 | ) | (50,117 | ) | (104,814 | ) | (211,426 | ) | ||||||||||
| ||||||||||||||||||||
Balance December 31, 2008 | (3,321 | ) | 16,583 | (50,117 | ) | (90,689 | ) | (127,544 | ) | |||||||||||
Period Change | 5,180 | (16,371 | ) | 55,479 | (6,169 | ) | 38,119 | |||||||||||||
| ||||||||||||||||||||
Balance December 31, 2009 | $ | 1,859 | $ | 212 | $ | 5,362 | $ | (96,858 | ) | $ | (89,425 | ) | ||||||||
| ||||||||||||||||||||
Capitalized Software Costs: The Company capitalizes software costs incurred in connection with developing or obtaining software. Capitalized software costs are included in property, plant and equipment, net and are amortized over their estimated useful life of three to seven years from the date the systems are ready for their intended use.
New Accounting Principles: In June 2009, the Financial Accounting Standards Board, or FASB, established the FASB Accounting Standards Codification TM , or ASC, as the source of authoritative accounting principles recognized by the FASB to be applied by nongovernmental entities in preparation of financial statements in conformity with generally accepted accounting principles in the United States. All other accounting literature not included in the ASC is now nonauthoritative. The ASC was effective for financial statements issued for interim and annual periods ending after September 15, 2009 and its adoption did not have any impact on the Company's consolidated financial statements. The ASC is updated through the FASB's issuance of Accounting Standard Updates, or ASUs. Summarized below are recently issued accounting pronouncements as described under the new ASC structure.
In December 2007, the FASB issued ASC No. 805, "Business Combinations," or ASC 805, which requires an acquirer to recognize the assets acquired, the liabilities assumed and any noncontrolling interest in the acquiree at the acquisition date, measured at their fair values as of that date, with limited exceptions. This Statement also requires the capitalization of research and development assets acquired in a business combination at their acquisition date fair values, separately from goodwill. In addition, ASC 805 requires that any post-acquisition adjustments to deferred tax asset valuation allowances and liabilities related to uncertain tax positions be recognized in current period income tax expense. ASC 805 was effective for the Company beginning January 1, 2009. The Company accounted for post-acquisition tax-related adjustments for pre-2009 business combinations and will account for future business combinations in accordance with its provisions.
In March 2008, the FASB issued an amendment to ASC No. 815, "Disclosures about Derivative Instruments and Hedging Activities," or ASC 815, which is intended to improve financial reporting about derivative instruments and hedging activities by requiring enhanced disclosures to enable investors to better understand their effects on an entity's financial position, financial performance and cash flows. The amendment was effective for the Company beginning January 1, 2009 and the expanded disclosures are included in Note 6.
80
CELGENE CORPORATION AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
In November 2008, the FASB ratified an amendment to ASC No. 350, "Accounting for Defensive Intangible Assets," which clarifies the accounting for certain separately identifiable intangible assets which an acquirer does not intend to actively use but intends to hold to prevent its competitors from obtaining access to them. The amendment requires an acquirer in a business combination to account for a defensive intangible asset as a separate unit of accounting, which should be amortized to expense over the period the asset diminishes in value. The amendment was effective for the Company beginning January 1, 2009 and the Company will account for defensive intangible assets acquired in future business combinations in accordance with its provisions.
In April 2009, the FASB issued an amendment to ASC No. 805, "Accounting for Assets Acquired and Liabilities Assumed in a Business Combination That Arise from Contingencies." This amendment clarifies application issues associated with initial recognition and measurement, subsequent measurement and accounting, and disclosure of assets and liabilities arising from contingencies in a business combination. This amendment was effective for the Company beginning January 1, 2009 and the Company will account for assets or liabilities arising from contingencies acquired in future business combinations in accordance with its provisions.
In June 2009, the FASB issued an amendment to ASC No. 860, "Accounting for Transfers of Financial Assets," which eliminates the concept of a "qualifying special-purpose entity," changes the requirements for derecognizing financial assets and requires additional disclosures. This amendment clarifies the determination whether a transferor and all of the entities included in the transferor's financial statements being presented have surrendered control over transferred financial assets. It also enhances information reported to users of financial statements by providing greater transparency about transfers of financial assets and a company's continuing involvement in transferred financial assets. This amendment will be effective for the Company's fiscal year beginning after January 1, 2010. The Company is currently evaluating the impact, if any, that the adoption of this amendment will have on its consolidated financial statements.
In June 2009, the FASB issued an amendment to ASC 810, "Consolidation of Variable Interest Entities," which changes how a company determines when an entity that is insufficiently capitalized or is not controlled through voting (or similar rights) should be consolidated. The determination of whether a company is required to consolidate an entity is based on, among other things, an entity's purpose and design and a company's ability to direct the activities of the entity that most significantly impact the entity's economic performance. This amendment requires ongoing reassessments of whether an enterprise is the primary beneficiary of a variable interest entity and will require a company to provide additional disclosures about its involvement with variable interest entities, any significant changes in risk exposure due to that involvement and how its involvement with a variable interest entity affects the company's financial statements. This amendment will be effective for the Company's fiscal year beginning January 1, 2010. The Company is currently evaluating the impact, if any, that the adoption of this amendment will have on its consolidated financial statements.
In September 2009, the FASB issued ASU No. 2009-12, "Fair Value Measurements and Disclosure," or ASU 2009-12, which provides additional guidance on using the net asset value per share, provided by an investee, when estimating the fair value of an alternate investment that does not have a readily determinable fair value and enhances the disclosures concerning these investments. ASU 2009-12 was effective for the Company's interim and annual periods ending after December 15, 2009.
81