UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| (Mark One) | ||
☑ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
| For the fiscal year ended December 31, 2010 | ||
or | ||
o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
| For the transition period from to | ||
Commission file number: 0-19311
Biogen Idec Inc.
(Exact name of registrant as specified in its charter)
| Delaware (State or other jurisdiction of incorporation or organization) | 33-0112644 (I.R.S. Employer Identification No.) | |
| 133 Boston Post Road, Weston, Massachusetts (Address of principal executive offices) | 02493 (Zip code) |
(781) 464-2000
(Registrant's telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class | Name of Each Exchange on Which Registered | |
Common Stock, $0.0005 par value | The Nasdaq Global Select Market |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☑ No o
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes o No ☑
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☑ No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files): Yes ☑ No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer ☑ | Accelerated filer o | Non-accelerated filer o | Smaller reporting company o | |||
| (Do not check if a smaller reporting company) | ||||||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No ☑
The aggregate market value of the registrant's common stock held by non-affiliates of the registrant (without admitting that any person whose shares are not included in such calculation is an affiliate) computed by reference to the price at which the common stock was last sold as of the last business day of the registrant's most recently completed second fiscal quarter was $11,688,813,825.
As of January 31, 2011, the registrant had 240,911,883 shares of common stock, $0.0005 par value, outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the definitive proxy statement for our 2011 Annual Meeting of Stockholders are incorporated by reference into Part III of this report.
BIOGEN
IDEC INC.
ANNUAL
REPORT ON FORM 10-K
For the
Year Ended December 31, 2010
TABLE OF CONTENTS
| Page | ||||||||
PART I | ||||||||
Item 1. | Business | 1 | ||||||
Item 1A. | Risk Factors | 21 | ||||||
Item 1B. | Unresolved Staff Comments | 31 | ||||||
Item 2. | Properties | 31 | ||||||
Item 3. | Legal Proceedings | 32 | ||||||
Item 4. | [Reserved] | 32 | ||||||
| PART II | ||||||||
Item 5. | Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | 33 | ||||||
Item 6. | Selected Consolidated Financial Data | 35 | ||||||
Item 7. | Management's Discussion and Analysis of Financial Condition and Results of Operations | 37 | ||||||
Item 7A. | Quantitative and Qualitative Disclosures About Market Risk | 70 | ||||||
Item 8. | Consolidated Financial Statements and Supplementary Data | 71 | ||||||
Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure | 71 | ||||||
Item 9A. | Controls and Procedures | 71 | ||||||
Item 9B. | Other Information | 72 | ||||||
| PART III | ||||||||
Item 10. | Directors, Executive Officers and Corporate Governance | 73 | ||||||
Item 11. | Executive Compensation | 73 | ||||||
Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 73 | ||||||
Item 13. | Certain Relationships and Related Transactions, and Director Independence | 73 | ||||||
Item 14. | Principal Accountant Fees and Services | 73 | ||||||
| PART IV | ||||||||
Item 15. | Exhibits, Financial Statement Schedules | 74 | ||||||
Signatures | 75 | |||||||
Consolidated Financial Statements | F-1 | |||||||
Exhibit Index | A-1 | |||||||
| EX-10.5 | ||||||||
| EX-10.6 | ||||||||
| EX-10.43 | ||||||||
| EX-10.55 | ||||||||
| EX-21 | ||||||||
| EX-23 | ||||||||
| EX-31.1 | ||||||||
| EX-31.2 | ||||||||
| EX-32.1 | ||||||||
| EX-101 INSTANCE DOCUMENT | ||||||||
| EX-101 SCHEMA DOCUMENT | ||||||||
| EX-101 CALCULATION LINKBASE DOCUMENT | ||||||||
| EX-101 LABELS LINKBASE DOCUMENT | ||||||||
| EX-101 PRESENTATION LINKBASE DOCUMENT | ||||||||
| EX-101 DEFINITION LINKBASE DOCUMENT | ||||||||
NOTE REGARDING FORWARD-LOOKING STATEMENTS
In addition to historical information, this report contains forward-looking statements that are based on our current beliefs and expectations. These forward-looking statements may be accompanied by such words as "anticipate," "believe," "estimate," "expect," "forecast," "intend," "may," "plan," "project," "target," "will" and other words and terms of similar meaning. Reference is made in particular to forward-looking statements regarding:
| • | the anticipated amount, mix and timing of future product sales, corporate partner revenue, foreign earnings, royalty revenues or obligations, milestone payments, expenses, liabilities, charges, contractual obligations, cash expenditures, share-based compensation, currency hedges, tax benefits and effective tax rate, and amortization of intangible assets; | |
| • | the growth trends for TYSABRI and our ability to improve the benefit-risk profile of TYSABRI; | |
| • | the assumed remaining life of the core technology relating to AVONEX and expected lifetime revenue of AVONEX; | |
| • | the incidence, timing, outcome and impact of litigation, proceedings related to patents and other intellectual property rights, tax audits and assessments and other legal proceedings; | |
| • | the timing and impact of accounting standards; | |
| • | the design, costs, development and timing of, and therapeutic area and indications targeted by, programs in our clinical pipeline; | |
| • | the timing and outcome of regulatory filings and communications with regulatory authorities; | |
| • | the impact and interpretation of healthcare reform and other measures designed to reduce healthcare costs; | |
| • | the impact of the global macroeconomic environment and the deterioration of the credit and economic conditions in certain countries in Europe; | |
| • | our ability to finance our operations and business initiatives and obtain funding for such activities; | |
| • | our reliance on third-parties for certain aspects of our business; | |
| • | opportunistic return of cash to shareholders; | |
| • | the structure, strategy, financial and operational impact, and timing of our framework for growth; | |
| • | the status, use, location and financial impact of our manufacturing facilities and other properties; and | |
| • | the drivers for growing our business, including our plans to pursue external business development and research opportunities, and the impact of competition. |
These forward-looking statements involve risks and uncertainties that could cause actual results to differ materially from those reflected in such forward-looking statements, including those discussed in the " Risk Factors " section of this report and elsewhere in this report. You should not place undue reliance on these statements. Forward-looking statements speak only as of the date of this report. We do not undertake any obligation to publicly update any forward-looking statements.
NOTE REGARDING COMPANY AND PRODUCT REFERENCES
Throughout this report, "Biogen Idec," the "Company," "we," "us" and "our" refer to Biogen Idec Inc. and its consolidated subsidiaries. References to "RITUXAN" refer to both RITUXAN (the trade name for rituximab in the U.S., Canada and Japan) and MabThera (the trade name for rituximab outside the U.S., Canada and Japan), and "ANGIOMAX" refers to both ANGIOMAX (the trade name for bivalirudin in the U.S., Canada and Latin America) and ANGIOX (the trade name for bivalirudin in Europe).
NOTE REGARDING TRADEMARKS
AVONEX ® , RITUXAN ® and ADENTRI ® are registered trademarks of Biogen Idec. FUMADERM tm is a common law trademark of Biogen Idec Inc. TYSABRI ® and TOUCH ® are registered trademarks of Elan Pharmaceuticals, Inc. The following are trademarks of the respective companies listed: ACTEMRA ® - Chugai Seiyaku Kabushiki Kaisha; AMEVIVE ® - Astellas US LLC; AMPYRA ® and FAMPYRA ® - Acorda Therapeutics, Inc.; ANGIOMAX ® and ANGIOX ® - The Medicines Company; ARZERRA tm - Glaxo Group Limited; BETASERON ® and BETAFERON ® - Bayer Schering Pharma AG; CAMPATH ® and LEMTRADA ® - Genzyme Corporation; CIMZIA ® - UCB Pharma, S.A.; COPAXONE ® - Teva Pharmaceutical Industries Limited; ENBREL ® - Immunex Corporation; EXTAVIA ® and GILENYA ® - Novartis AG; HUMIRA ® - Abbott Biotechnology Ltd.; ONCOVIN tm - Eli Lilly and Company; ORENCIA ® - Bristol-Myers Squibb Company; REBIF ® - Ares Trading S.A.; REMICADE ® - Centocor Ortho Biotech Inc.; SIMPONI tm - Johnson & Johnson; TREANDA ® - Cephalon, Inc.; and ZEVALIN ® - RIT Oncology, LLC
PART I
| Item 1. | Business |
Overview
Biogen Idec is a global biotechnology company focused on discovering, developing, manufacturing and marketing products for the treatment of neurological disorders and other serious diseases. Patients worldwide benefit from our significant products used for the treatment of multiple sclerosis, non-Hodgkin's lymphoma, rheumatoid arthritis, Crohn's disease, chronic lymphocytic leukemia and psoriasis.
Marketed Products
We have four therapeutic products on the market, which are summarized in the tables below.
| Product Revenues | ||||||||||||||
| to Biogen Idec (in millions) | ||||||||||||||
| Product | Indications | 2010 | 2009 | 2008 | ||||||||||
| AVONEX (interferon beta-1a) | Multiple sclerosis | $ | 2,518.4 | $ | 2,322.9 | $ | 2,202.6 | |||||||
| ||||||||||||||
| TYSABRI (natalizumab) | Multiple sclerosis Crohn's disease | 900.2 | 776.0 | 588.6 | ||||||||||
| ||||||||||||||
| FUMADERM (dimethylfumarate and monoethylfumarate salts) | Psoriasis | 51.2 | 49.6 | 43.4 | ||||||||||
| Unconsolidated Joint Business | ||||||||||||||
| Revenues to Biogen Idec (in millions) | ||||||||||||||
| Product | Indications | 2010 | 2009 | 2008 | ||||||||||
| RITUXAN (rituximab) | Non-Hodgkin's lymphoma Rheumatoid arthritis Chronic lymphocytic leukemia | $ | 1,077.2 | $ | 1,094.9 | $ | 1,128.2 | |||||||
| ||||||||||||||
Additional financial information about our product revenues, other revenues and geographic areas in which we operate is set forth in our consolidated financial statements, Note 24, Segment Information to our consolidated financial statements, and Item 6. Selected Consolidated Financial Data included in this report.
Research and Development
We devote significant resources to research and development programs and external business development opportunities. We have incurred significant expenditures related to conducting clinical studies to develop new pharmaceutical products and explore the utility of our existing products in treating disorders beyond those currently approved in their labels.
In 2010, 2009 and 2008, our research and development costs totaled $1,248.6 million, $1,283.1 million, and $1,072.1 million, respectively. In addition, we incurred charges associated with acquired in process research and development as follows: $245.0 million in 2010 of which $145.0 million was attributed to noncontrolling interests; none in 2009; and $25.0 million in 2008.
CEO Appointment
On July 15, 2010, George A. Scangos, Ph.D. began serving as our Chief Executive Officer and member of our Board of Directors. Dr. Scangos succeeded James C. Mullen, who retired as our President and Chief Executive Officer on June 8, 2010.
1
Framework for Growth
On November 3, 2010, we announced a number of strategic, operational and organizational initiatives designed to provide a framework for the future growth of our business, which are summarized as follows:
| • | We intend to focus our business on neurology and leverage our strengths in biologics research, development and manufacturing to pursue select biological therapies where there is a significant unmet need and where the drug candidate has the potential to be highly differentiated. Accordingly, during the fourth quarter of 2010, we began to reallocate resources within our research and development organization to maximize our investment in our highest-potential programs. As a result, we have terminated or are in the process of discontinuing certain research and development programs, including substantially all of our oncology programs (which we are looking to spin out or out-license), our cardiovascular programs and selected neurology and immunology programs. In addition, we have substantially reduced our small molecule discovery activities in favor of outsourcing these efforts. | |
| • | We are in the process of closing the San Diego, California facility and consolidating our Massachusetts facilities. | |
| • | We eliminated our RITUXAN oncology and rheumatology sales force and Genentech, Inc. (Genentech), a wholly-owned member of the Roche Group, has assumed sole responsibility for the U.S. sales and marketing efforts related to RITUXAN. | |
| • | We are in the process of completing a 13% reduction in our workforce and realigning our overall structure to become a more efficient and cost-effective organization. |
We expect these initiatives to be substantially completed by the end of 2011 and to result in total restructuring charges of approximately $110.0 million.
Business Development
| • | In December 2010, we completed our acquisition of 100% of the stock of Panima Pharmaceuticals AG (Panima), an affiliate of Neurimmune AG. The purchase price is comprised of a $32.5 million cash payment, plus contingent consideration in the form of development milestones of up to $395.0 million in cash. Panima is involved in the discovery of antibodies designed to treat neurological disorders. For a more detailed description of this transaction, please read Note 2, Acquisitions to our consolidated financial statements included in this report. | |
| • | In October 2010, we amended our collaboration agreement with Genentech with regard to the development of ocrelizumab and agreed to terms for the development of GA101. Under the terms of the amended agreement, Genentech is responsible for the further development and commercialization of ocrelizumab and funding future costs. We will receive tiered royalties between 13.5% and 24% on U.S. sales of ocrelizumab. Commercialization of ocrelizumab will not impact our percentage of the co-promotion profits for RITUXAN. In addition, we will pay 35% of the development and commercialization expenses of GA101 and will receive between 35% and 39% of the profits of GA101 based upon the achievement of certain sales milestones. Commercialization of GA101 will impact our percentage of the co-promotion profits for RITUXAN. This amendment did not have an impact on our share of the co-promotion operating profits of RITUXAN in 2010. For a more detailed description of this collaboration, please read Note 19, Collaborations to our consolidated financial statements included in this report. | |
| • | In August 2010, we entered into a license agreement with Knopp Neurosciences, Inc. (Knopp), for the development, manufacture and commercialization of dexpramipexole, an orally administered small molecule in clinical development for the treatment of amyotrophic lateral sclerosis (ALS). Under the terms of the license agreement we made a $26.4 million upfront payment and agreed to pay Knopp up to an additional $265.0 million in development and sales-based milestone payments, as well as royalties on future commercial sales. For a more detailed description of this transaction, please read Note 18, Investments in Variable Interest Entities to our consolidated financial statements included in this report. |
2
Available Information
We were formed as a California corporation in 1985 and became a Delaware corporation in 1997. In 2003, we acquired Biogen, Inc. and changed our corporate name from IDEC Pharmaceuticals Corporation to Biogen Idec Inc. Our principal executive offices are located at 133 Boston Post Road, Weston, MA 02493 and our telephone number is (781) 464-2000. Our website address is www.biogenidec.com. We make available free of charge through the Investors section of our website our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and all amendments to those reports as soon as reasonably practicable after such material is electronically filed with or furnished to the Securities and Exchange Commission (SEC). We include our website address in this report only as an inactive textual reference and do not intend it to be an active link to our website. The contents of our website are not incorporated into this filing.
Marketed Products
Our marketed products address the following diseases: multiple sclerosis (MS); non-Hodgkin's lymphoma (NHL); rheumatoid arthritis (RA); Crohn's disease (CD); chronic lymphocytic leukemia (CLL); and psoriasis. In addition, we are exploring the expansion of our marketed products into other diseases through ongoing development efforts. The approved indications for, and ongoing development of, our marketed products are summarized in the table below.
| Development or Marketing | ||||||
| Product | Approved Indication | Development Program | Collaborators | |||
| AVONEX (1) (interferon beta-1a) | Relapsing MS | None | ||||
| ||||||
| TYSABRI (2) (natalizumab) | Relapsing MS | Elan Pharmaceuticals | ||||
| ||||||
| CD | Elan Pharmaceuticals | |||||
| ||||||
| RITUXAN (3) (rituximab) | NHL | Genentech (Roche Group) | ||||
| RA | Genentech (Roche Group) | |||||
| ||||||
| CLL | Genentech (Roche Group) | |||||
| ||||||
| ANCA-associated vasculitis in registration | Genentech (Roche Group) (Our rights are limited to U.S.) | |||||
| ||||||
| FUMADERM (4) (dimethylfurnarate and monoethylfumarate salts) | Severe psoriasis | None | ||||
| ||||||
| (1) | AVONEX is indicated for the treatment of patients with relapsing forms of MS to slow the accumulation of physical disability and decrease the frequency of clinical exacerbations. | |
| (2) | TYSABRI is indicated for the treatment of (1) relapsing forms of MS as a monotherapy to delay the accumulation of physical disability and reduce the frequency of clinical exacerbations and (2) in the U.S., moderately to severely active CD with evidence of inflammation in adult patients who have had an inadequate response to or inability to tolerate conventional CD therapies and TNF inhibitors. | |
| (3) | RITUXAN is indicated for the treatment of (1)(a) relapsed or refractory, low-grade or follicular, CD20-positive, B-cell NHL as a single agent, (b) previously untreated follicular, CD20-positive, B-cell NHL in combination with first line chemotherapy and, in patients achieving a complete or partial response to RITUXAN in combination with chemotherapy, as a single-agent maintenance therapy, (c) non-progressing (including stable disease), low-grade, CD20-positive, B-cell NHL, as a single agent, after first-line CVP chemotherapy, and (d) previously untreated diffuse large B-cell, CD20-positive NHL in combination with cyclophosphamide, doxorubicin, ONCOVIN and prednisone or other anthracycline-based chemotherapy regimens, (2) CD20-positive CLL in combination with fludarabine and cyclophosphamide, and (3) moderately- to severely-active RA, in combination with methotrexate, in adult patients who have had an inadequate response to one or more tumor necrosis factor (TNF) antagonist therapies. | |
| (4) | FUMADERM is only approved in Germany and is indicated for the treatment of adult patients with moderate to severe plaque psoriasis for whom topical therapy is ineffective. |
3
AVONEX
AVONEX is a leading therapeutic for relapsing forms of MS. MS is a progressive neurological disease in which the body loses the ability to transmit messages along nerve cells, leading to a loss of muscle control, paralysis and, in some cases, death. Patients with active relapsing MS experience an uneven pattern of disease progression characterized by periods of stability that are interrupted by flare-ups of the disease after which the patient returns to a new baseline of functioning. AVONEX is a recombinant form of the interferon beta protein produced in the body in response to viral infection.
TYSABRI
TYSABRI is a treatment for MS with powerful efficacy. TYSABRI is a monoclonal antibody that was initially approved by the U.S. Food and Drug Administration (FDA) in November 2004 to treat relapsing MS. In February 2005, in consultation with the FDA, we and our collaborator Elan Corporation plc (Elan) voluntarily suspended the marketing and commercial distribution of TYSABRI based on reports of cases of progressive multifocal leukoencephalopathy (PML) in patients treated with TYSABRI in clinical studies. PML is an opportunistic viral infection of the brain that often leads to death or severe disability. In July 2006, TYSABRI was reintroduced in the U.S., and introduced in the European Union (E.U.), as a monotherapy treatment for relapsing MS. TYSABRI is also approved in the U.S. to treat CD, which is an inflammatory disease of the intestines.
Because of the risk of PML, TYSABRI has a boxed warning and is marketed under risk management or minimization plans approved by local regulatory authorities. In the U.S., TYSABRI was reintroduced under the TOUCH Prescribing Program, a restricted distribution program designed to assess and minimize the risk of PML, minimize death and disability due to PML, and promote informed risk-benefit decisions regarding TYSABRI use.
Based upon data available to us through the TOUCH prescribing program and other third-party sources, we estimate that as of December 31, 2010 approximately 56,600 patients were on commercial and clinical TYSABRI therapy worldwide. We continue to monitor the growth of TYSABRI unit sales, which may be adversely impacted by the significant safety warnings in the prescribing information. We continue to research and develop protocols that may reduce risk and improve outcomes of PML in patients. Our efforts have included working to identify patient or viral characteristics which contribute to the risk of developing PML, including the presence of asymptomatic JC virus infection with an assay to detect an immune response against the JC virus.
We have initiated the five year renewal process for TYSABRI's marketing authorization in the E.U. This marketing authorization review by E.U. regulators, in addition to ongoing label discussions with U.S. regulators, includes assessment of the criteria for confirming PML diagnosis, the number of PML cases, the incidence of PML in TYSABRI patients, the risk factors for PML, as well as an overall assessment of TYSABRI's benefit-risk profile. Our interactions with E.U. and U.S. regulators could result in modifications to the respective labels or other restrictions for TYSABRI. Upon completion of the assessment of the TYSABRI renewal in the E.U. the marketing authorization is expected to be valid for either an unlimited period or for an additional five year term.
We collaborate with Elan on the development and commercialization of TYSABRI. For a more detailed description of this collaboration, please read Note 19, Collaborations to our consolidated financial statements included in this report.
2010 Developments
| • | In December 2010, we and Elan submitted a supplemental Biologics License Application (sBLA) to the FDA and a Type II Variation to the European Medicines Agency (EMA) to request review and approval to update the respective TYSABRI Prescribing Information and Summary of Product Characteristics. We are proposing updated product labeling to include anti-JC virus antibody status as one potential factor to help stratify the risk of PML in the TYSABRI-treated population. | |
| • | In November 2010, we updated the E.U. TYSABRI label to include information about the increased risk of PML in patients who have a history of prior treatment with immunosuppressant therapy. | |
| • | In July 2010, we updated the U.S. TYSABRI label to reflect that the risk of PML increases in patients with prior immunosuppressant use. |
4
| • | In May 2010, we updated the U.S. TYSABRI label to reflect that Immune Reconstitution Inflammatory Syndrome (IRIS) may occur in patients who developed PML and subsequently discontinued TYSABRI. | |
| • | In May 2010, we updated the E.U. TYSABRI label to reflect that the risk of PML increases after two years of therapy, with limited experience beyond three years, and there is a risk for the occurrence of IRIS in patients with TYSABRI induced PML following discontinuation or removal of TYSABRI by plasma exchange, a process that clears TYSABRI from patients' blood allowing the immune system to fight the infection. | |
| • | In March 2010, we began enrolling patients in a Phase 3 study, known as SURPASS, designed to evaluate switching patients with relapsing MS to TYSABRI from COPAXONE or REBIF. Although enrollment targets have not been met, we have stopped enrollment and will continue the study for currently enrolled patients. | |
| • | In March 2010, we began enrolling patients in two Phase 4 studies, known as STRATIFY-1 and STRATIFY-2, designed to evaluate the potential utility of a blood test that is designed to detect antibodies to the JC virus. |
RITUXAN
RITUXAN is a widely prescribed oncology therapeutic with over 2.4 million patient exposures across all indications. RITUXAN is a monoclonal antibody used to treat NHL, CLL and RA. NHL and CLL are cancers that affect lymphocytes, which are a type of white blood cell that help to fight infection. RA is a chronic disease that occurs when the immune system mistakenly attacks the body's joints, resulting in inflammation, pain and joint damage.
We collaborate with Genentech on the development and commercialization of RITUXAN. In October 2010, we amended our collaboration agreement with Genentech with regard to the development of ocrelizumab, a humanized anti-CD20 antibody, and agreed to terms for the development of GA101, a next-generation anti-CD20 antibody. For a more detailed description of this collaboration, please read Note 19, Collaborations to our consolidated financial statements included in this report.
2010 Developments
| • | In October 2010, we and Genentech filed a supplemental biologics license application with the FDA to expand the U.S. RITUXAN label for the treatment of antineutrophil cytoplasmic antibody (ANCA) associated vasculitis, a systemic inflammation of the blood vessels. | |
| • | In May 2010, we and Genentech announced that data from a Phase 3 study, known as PRIMA, showed that continuing RITUXAN for two years in patients who responded to initial treatment with RITUXAN plus chemotherapy doubled the likelihood of them living without their disease worsening compared to those who stopped treatment. The RITUXAN label has since been expanded to include maintenance treatment for patients with advanced follicular lymphoma who responded to initial treatment with RITUXAN plus chemotherapy. | |
| • | In March 2010, we and Genentech were issued a patent by the U.S. Patent and Trademark Office (PTO) related to a method of treating CLL using an anti-CD20 antibody. For information about legal proceedings related to this patent, please read Note 20, Litigation to our consolidated financial statements included in this report. | |
| • | In February 2010, the FDA approved RITUXAN for the treatment of CD20-positive CLL in combination with fludarabine and cyclophosphamide, expanding the label beyond the treatment of NHL and RA. |
FUMADERM
FUMADERM is approved for the treatment of severe psoriasis in Germany. Psoriasis is a skin disease in which cells build up on the skin surface and form scales and red patches.
5
Other Sources of Revenue
Our primary source of other revenue is derived from royalties received on sales by our licensees of other products covered under patents that we own. Our royalty revenues are dependent upon our licensees' sales of licensed products which could vary significantly due to competition, manufacturing, regulatory, safety or efficacy issues or other factors that are outside our control. In addition, the expiration or invalidation of any underlying patents could reduce or eliminate the royalty revenues derived from such patents. Royalties on sales of ANGIOMAX (bivalirudin) by The Medicines Company (TMC) represent our most significant source of other revenue. TMC markets ANGIOMAX primarily in the U.S. and the E.U. for use as an anticoagulant in patients undergoing percutaneous coronary intervention. For a description of this royalty arrangement and factors that could adversely affect this portion of our revenues, please read the subsection entitled " Other Revenue - Royalty Revenues " in the " Management's Discussion and Analysis of Financial Condition and Results of Operations " section of this report.
We have also sold or exclusively licensed to third parties rights to certain products previously included within our product line. Royalty or supply agreement revenues received based upon those products are recorded as corporate partner revenue. Amounts recorded as corporate partner revenue also include amounts earned upon delivery of product under contract manufacturing agreements.
In 2010, 2009 and 2008, our royalty revenues totaled $137.4 million, $124.4 million and $116.2 million, respectively, and our corporate partner revenues totaled $31.7 million, $5.1 million and $13.4 million, respectively.
6
Research and Development Programs
We intend to continue committing significant resources to research and development opportunities, focusing on high-potential treatments for select disorders where there is a significant unmet need and where the drug candidate has the potential to be highly differentiated. The table below highlights our research and development programs. Drug development involves a high degree of risk and investment, and the status, timing and scope of our development programs are subject to change. Important factors that could adversely affect our drug development efforts are discussed in the " Risk Factors " section of this report.
| Therapeutic Area | Product Candidate | Targeted Indications | Status | |||
Neurology | FAMPYRA | MS (walking ability) | In registration (our rights exclude the U.S.) | |||
| ||||||
| BG-12 | MS (monotherapy) | Phase 3 | ||||
| ||||||
| Daclizumab | MS | Phase 3 | ||||
| ||||||
| PEGylated Interferon Beta 1a | MS | Phase 3 | ||||
| ||||||
| BG-12 | MS (combination therapy) | Phase 2 | ||||
| ||||||
| Dexpramipexole | Amyotrophic Lateral Sclerosis | Phase 3 planned | ||||
| ||||||
| Anti-LINGO | MS | Phase 1 | ||||
| ||||||
| Baminercept | MS | Phase 1b | ||||
| ||||||
| Neublastin | Neuropathy | Phase 1 | ||||
| ||||||
| BIIB034 | Parkinson's Disease | Preclinical | ||||
| ||||||
| BART | Alzheimer's Disease | Preclinical | ||||
| ||||||
| Gamma Secretase Modulator | Alzheimer's Disease | Preclinical | ||||
| ||||||
Immunology | Anti-TWEAK | Lupus | Phase 2 planned | |||
| ||||||
| Baminercept | Ulcerative Colitis | Phase 2a | ||||
| ||||||
| Anti-TWEAK | RA | Phase 1 | ||||
| ||||||
| CD40L - Fab | Lupus | Phase 1 | ||||
| ||||||
Hemophilia | Factor VIII Fc | Hemophilia A | Phase 3 | |||
| ||||||
| Factor IX Fc | Hemophilia B | Phase 3 | ||||
| ||||||
Oncology | GA101 | Chronic Lymphocytic Leukemia | Phase 3 | |||
| ||||||
| Non-Hodgkin's Lymphoma | Phase 3 | |||||
| ||||||
Additional information about our product candidates in or near registrational stage development by therapeutic area is set forth below:
Neurology
FAMPYRA
FAMPYRA (prolonged-release fampridine) is an oral compound that is being developed as a treatment to improve walking ability in people with MS. We have filed for approval of FAMPYRA for this indication in the E.U., Canada, Australia and other jurisdictions. FAMPYRA was approved in the U.S. in January 2010 and is marketed by Acorda Therapeutics, Inc. under the trade name AMPYRA (dalfampridine) Extended Release Tablets 10 mg. AMPYRA is indicated to improve walking in patients with MS. This was demonstrated by an increase in walking speed. We collaborate with Acorda on the development and commercialization of FAMPYRA in markets outside the U.S. For a more detailed description of this collaboration, please read Note 19, Collaborations to our consolidated financial statements included in this report.
In January 2011, the EMA's Committee for Medicinal Products for Human Use (CHMP) issued a negative opinion recommending against approval of FAMPYRA to improve walking ability in adult patients with MS in the
7
E.U. We intend to appeal this opinion and request a re-examination of the decision by the CHMP. We also received a Notice of Deficiency from Health Canada for our application to sell FAMPYRA in Canada.
BG-12
BG-12 is an oral compound that is being tested in relapsing MS. During 2009, we completed patient enrollment in two Phase 3 studies of BG-12 in relapsing MS, known as DEFINE and CONFIRM, one of which includes a glatiramer acetate (COPAXONE) reference comparator arm. The two studies were designed to have a two year endpoint with each study involving approximately 1,000 to 1,200 patients. The FDA has granted BG-12 fast track status, which may result in an expedited review.
Daclizumab
Daclizumab is a monoclonal antibody that is being tested in relapsing MS. A Phase 2b trial of daclizumab in MS, known as SELECT, completed enrollment in 2010. The SELECT trial has a one year end point and is expected to involve approximately 600 patients worldwide. In May 2010, we began patient enrollment in a Phase 3 study of daclizumab in relapsing MS, known as DECIDE, evaluating the efficacy and safety of daclizumab compared to interferon beta-1a (AVONEX). The DECIDE trial is designed to have a two year endpoint and is expected to involve approximately 1,500 patients.
We collaborate with Abbott Biotherapeutics Corporation (Abbott), on the development and commercialization of daclizumab. In January 2010, we amended our collaboration agreement with Abbott whereby we assumed full development and manufacturing responsibility for daclizumab. For a more detailed description of this collaboration, please read Note 19, Collaborations to our consolidated financial statements included in this report.
PEGylated interferon beta-1a
PEGylated interferon beta-1a is designed to prolong the effects and reduce the dosing frequency of interferon beta-1a. During the first half of 2009, we began patient enrollment in a Phase 3 trial of PEGylated interferon beta-1a in relapsing MS, known as ADVANCE. The study is designed to have a one year endpoint and involve approximately 1,200 patients. The FDA has granted PEGylated interferon beta-1a fast track status, which may result in an expedited review.
Dexpramipexole
Dexpramipexole is an orally administered small molecule in clinical development for the treatment of amyotrophic lateral sclerosis (ALS). ALS, also known as Lou Gehrig's disease, is a neurodegenerative disorder characterized by progressive muscle weakness and wasting.
We have agreed with the FDA on a Special Protocol Assessment for the design of a registrational study of dexpramipexole and expect to begin patient enrollment in the first half of 2011. Dexpramipexole has been granted fast track status by the FDA, which may result in an expedited review, and has received orphan drug designation for the treatment of ALS from both the FDA and EMA.
We have entered into a license agreement with Knopp Neurosciences, Inc. for the development, manufacture and commercialization of dexpramipexole. For a more detailed description of this collaboration, please read Note 18, Investments in Variable Interest Entities to our consolidated financial statements included in this report.
Hemophilia
Long-Lasting Recombinant Factors VIII and IX.
We collaborate with Swedish Orphan Biovitrum AB (Biovitrum) on the development and commercialization of long-lasting recombinant Factor VIII and Factor IX. In February 2010, we amended our collaboration agreement with Biovitrum to provide that we will assume full development responsibilities and costs and perform all manufacturing for the Factor VIII and Factor XI programs, among other matters. For a more detailed description of this collaboration, please read Note 19, Collaborations to our consolidated financial statements included in this report.
8
Factor VIII is a proprietary fusion protein that is being tested in hemophilia A, a disorder in which blood clotting is impaired. In December 2010, we began patient enrollment in a registrational trial of Factor VIII in hemophilia A, known as A-LONG. This study will involve approximately 150 patients. Factor VIII has received orphan drug designation for the treatment of hemophilia A from both the FDA and EMA.
Factor IX is a proprietary fusion protein that is being tested in hemophilia B, a disorder in which blood clotting is impaired. During the first half of 2010, we began patient enrollment in a registrational trial of Factor IX in hemophilia B, known as B-LONG. This study will involve approximately 100 patients. Factor IX has received orphan drug designation for the treatment of hemophilia B from both the FDA and EMA.
Oncology
GA101
GA101 is a monoclonal antibody that is being tested in CLL and NHL. During the second half of 2009, we began patient enrollment in a Phase 3 trial of GA101 in combination with chlorambucil as compared to rituximab plus chlorambucil or chlorambucil alone in patients with previously untreated CLL. The study has a 6 month end point, with a minimum five year follow-up period, and is expected to involve approximately 800 patients worldwide. In April 2010, we began patient enrollment in a Phase 3 trial of GA101 combined with bendamustine compared with bendamustine alone in patients with rituximab-refractory, indolent NHL. The study has a six to twelve month end point and is expected to involve approximately 360 patients.
We collaborate with Genentech on the development and commercialization of GA101. In October 2010, we amended our collaboration agreement with Genentech to specify the terms for the development of GA101, among other matters. For a more detailed description of this collaboration and the recent amendment, please read Note 19, Collaborations to our consolidated financial statements included in this report.
Former Registrational Programs
In October 2010, we agreed to terminate our collaboration with Cardiokine Biopharma, LLC (Cardiokine ) for the development of lixivaptan in hyponatremia effective November 1, 2010. Under the terms of the agreement, we have funded our share of development costs through the effective date and made a final payment of $25.0 million to Cardiokine. The termination was consistent with our broader strategic decision to terminate our efforts in cardiovascular medicine described above under the heading " Overview - Framework for Growth. "
In May 2010, we and the Roche Group announced our decision to discontinue the ocrelizumab clinical development program for the treatment of patients with RA. Following a detailed analysis of the efficacy and safety results from the RA program, we concluded that the overall benefit to risk profile of ocrelizumab was not favorable in RA taking into account currently available treatment options. The ocrelizumab RA program included several Phase 3 studies.
Patents and Other Proprietary Rights
Patents are important to developing and protecting our competitive position. We regularly seek patent protection in the U.S. and in selected countries outside the U.S. for inventions originating from our research and development efforts. In addition, we license rights to various patents and patent applications, generally, in return for the payment of royalties to the patent owner. U.S. patents, as well as most foreign patents, are generally effective for 20 years from the date the earliest (priority) application was filed; however, U.S. patents that issue on applications filed before June 8, 1995 may be effective until 17 years from the issue date, if that is later than the 20 year date. In some cases, the patent term may be extended to recapture a portion of the term lost during FDA regulatory review or because of U.S. Patent and Trademark Office (USPTO) delays in prosecuting the application. The duration of foreign patents varies similarly, in accordance with local law.
We also rely upon unpatented confidential information to remain competitive. We protect such information principally through confidentiality agreements with our employees, consultants, outside scientific collaborators, scientists whose research we sponsor and other advisers. In the case of our employees, these agreements also
9
provide, in compliance with relevant law, that inventions and other intellectual property conceived by such employees during their employment shall be our exclusive property.
Our trademarks, including RITUXAN and AVONEX, are important to us and are generally covered by trademark applications or registrations in the USPTO and the patent offices of other countries. We also use trademarks licensed from third parties, such as the mark TYSABRI which we license from Elan. Trademark protection varies in accordance with local law, and continues in some countries as long as the mark is used and in other countries as long as the mark is registered. Trademark registrations generally are for fixed but renewable terms.
Our patent position and proprietary rights are subject to certain risks and uncertainties. For additional information about certain risks and uncertainties that may affect our patent position and proprietary rights, please read the " Risk Factors " section of this report.
Additional information about the patents and other proprietary rights covering our marketed products is set forth below.
AVONEX and Beta Interferon
Our U.S. patent No. 7,588,755, granted in September 2009, claims the use of beta interferon for immunomodulation or treating a viral condition, viral disease, cancers or tumors. This patent, which expires in September 2026, covers, among other things, the treatment of MS with our product AVONEX. This issuance of this patent extends the expected remaining life of the intangible asset related to our AVONEX core technology. For information about legal proceedings related to this patent, please read Note 20, Litigation to our consolidated financial statements included in this report.
We have non-exclusive rights under certain third-party patents and patent applications to manufacture, use and sell AVONEX, including patents owned by the Japanese Foundation for Cancer Research which expire in 2011 and 2013 in the U.S., and a European patent owned by Rentschler Biotechnologie GmbH, which expires in 2012. Additionally, third parties own pending U.S. patent applications related to recombinant interferon-beta. These applications, which fall outside of the GATT amendments to the U.S. patent statute, are not published by the USPTO and, if they mature into granted patents, may be entitled to a term of seventeen years from the grant date. There is at least one pending interference proceeding in the USPTO involving such third party applications, and additional interferences could be declared in the future. We are unable to predict which, if any, such applications will mature into patents with claims relevant to our AVONEX product.
TYSABRI
We and our collaborator, Elan, have patents and patent applications covering TYSABRI in the U.S. and other countries. These patents and patent applications cover TYSABRI and related manufacturing methods, as well as various methods of treatment using the product. In the U.S., the principal patents covering the product and use of the product to treat MS generally expire between 2015 and 2020. Additional U.S. patents and applications covering other indications, including treatment of inflammatory bowel disease, and methods of manufacturing generally expire between 2012 and 2020. In the rest of world, patents on the product and methods of manufacturing the product generally expire between 2015 and 2020, subject to any supplemental protection (i.e., patent term extension) certificates that may be obtained. In the rest of world, patents and patent applications covering methods of treatment using TYSABRI generally expire between 2012 and 2020.
RITUXAN and Anti-CD20 Antibodies
We have several U.S. patents and patent applications, and numerous corresponding foreign counterparts, directed to anti-CD20 antibody technology, including RITUXAN. The principal patents with claims to RITUXAN or its uses expire in the U.S. between 2015 and 2018 and in the rest of the world in 2013, subject to any available patent term extensions. In addition, we and our collaborator, Genentech, have filed numerous patent applications directed to anti-CD20 antibodies and their uses to treat various diseases. These pending patent applications have the potential of issuing as patents in the U.S. and in the rest of world with claims to anti-CD20 antibody molecules for
10
periods beyond that stated above for RITUXAN. In 2008, a European patent of ours claiming the treatment with anti-CD20 antibodies of certain auto-immune indications, including RA, was revoked by the European Patent Office. We are appealing that decision.
Genentech, our collaborator on RITUXAN, has secured an exclusive license to five U.S. patents and counterpart U.S. and foreign patent applications assigned to Xoma Corporation that relate to chimeric antibodies against the CD20 antigen. These patents expire between 2007 and 2014. Genentech has granted us a non-exclusive sublicense to make, have made, use and sell RITUXAN under these patents and patent applications. We, along with Genentech, share the cost of any royalties due to Xoma in our co-promotion territory on sales of RITUXAN.
Sales, Marketing and Distribution
We focus our sales and marketing efforts on specialist physicians in private practice or at major medical centers. We use customary pharmaceutical company practices to market our products and to educate physicians, such as sales representatives calling on individual physicians, advertisements, professional symposia, direct mail, public relations and other methods. We provide customer service and other related programs for our products, such as disease and product-specific websites, insurance research services and order, delivery and fulfillment services. We have also established programs in the U.S. which provide qualified uninsured or underinsured patients with marketed products at no or reduced charge, based on specific eligibility criteria. Additional information about our sales, marketing and distribution efforts for our marketed products is set forth below.
AVONEX
We continue to focus our marketing and sales activities on maximizing the potential of AVONEX in the U.S. and the rest of world in the face of increased competition. The principal markets for AVONEX are the U.S., Germany, France and Italy. In the U.S., Canada, Brazil, Argentina, Australia, Japan and most of the major countries of the E.U., we market and sell AVONEX through our own sales forces and marketing groups and distribute AVONEX principally through wholesale distributors of pharmaceutical products, mail order specialty distributors or shipping service providers. In other countries, we sell AVONEX to distribution partners who are then responsible for most marketing and distribution activities.
TYSABRI
The principal markets for TYSABRI are the U.S., Germany, France and Italy.
In the U.S., we are principally responsible for marketing TYSABRI for MS and use our own sales force and marketing group for this. Elan is responsible for TYSABRI distribution in the U.S. and uses a third party distributor to ship TYSABRI directly to customers.
In the rest of world, we are responsible for TYSABRI marketing and distribution and we use a combination of our own sales force and marketing group and third party service providers.
FUMADERM
FUMADERM is marketed only in Germany. We have been marketing and distributing FUMADERM directly in Germany since February 2009 and previously used a third party service provider.
RITUXAN
The Roche Group and its sublicensees market and sell RITUXAN worldwide. In the U.S, we had previously contributed a sales force and other resources to the marketing of RITUXAN. In connection with our framework for growth initiative, we reached an agreement with Genentech to eliminate our RITUXAN oncology and rheumatology sales force, with Genentech assuming sole responsibility for the U.S. sales and marketing efforts related to RITUXAN. Notwithstanding this operational decision, we continue to collaborate with Genentech on the development and commercialization of RITUXAN. RITUXAN is generally sold to wholesalers, specialty distributors and directly to hospital pharmacies.
11
Competition
Competition in the biotechnology and pharmaceutical industries is intense and comes from many and varied sources, including specialized biotechnology firms and large pharmaceutical companies. Many of our competitors are working to develop products similar to those we are developing or already market and have considerable experience in undertaking clinical trials and in obtaining regulatory approval to market pharmaceutical products. Certain of these companies have substantially greater financial, marketing and research and development resources than we do.
We believe that competition and leadership in the industry will be based on managerial and technological superiority and establishing patent and other proprietary positions through research and development. The achievement of a leadership position also depends largely upon our ability to identify and exploit commercially the products resulting from research and the availability of adequate financial resources to fund facilities, equipment, personnel, clinical testing, manufacturing and marketing. Another key aspect of remaining competitive within the industry is recruiting and retaining qualified scientists and technicians. We believe that we have been successful in attracting skilled and experienced scientific personnel.
Competition among products approved for sale may be based, among other things, on patent position, product efficacy, safety, convenience, reliability, availability and price. In addition, early entry of a new pharmaceutical product into the market may have important advantages in gaining product acceptance and market share. Accordingly, the relative speed with which we can develop products, complete the testing and approval process and supply commercial quantities of products will have an important impact on our competitive position.
We may face increased competitive pressures as a result of the emergence of biosimilars. In the United States, most of our marketed products, including AVONEX, RITUXAN and TYSABRI, are licensed under the Public Health Service Act (PHSA) as biological products. In March 2010, U.S. healthcare reform legislation amended the PHSA to authorize the FDA to approve biological products, known as biosimilars or follow-on biologics, that are shown to be highly similar to previously approved biological products based upon potentially abbreviated data packages. The approval pathway for biosimilars does, however, grant a biologics manufacturer a 12 year period of exclusivity from the date of approval of its biological product before biosimilar competition can be introduced. Biosimilars legislation has also been in place in the E.U. since 2003. In November 2010, draft guidelines issued by the EMA for approving biosimilars of marketed monoclonal antibody products were adopted by the CHMP. These guidelines are now out for public consultation. If a biosimilar version of one of our products were approved, it could reduce our sales of that product.
Additional information about the competition that our marketed products face is set forth below.
AVONEX AND TYSABRI
AVONEX and TYSABRI both compete with the following products:
| • | COPAXONE (glatiramer acetate), which is marketed by Teva Pharmaceutical Industries Ltd. in the U.S. and copromoted by Teva Pharmaceutical Industries and Sanofi-Aventis in Europe. COPAXONE generated worldwide revenues of approximately $2.8 billion in 2009. | |
| • | REBIF (interferon-beta-1a), which is co-promoted by EMD Serono, a subsidiary of Merck Serono, and Pfizer Inc. in the U.S. and is marketed by Merck Serono in the E.U. REBIF generated worldwide revenues of approximately $2.0 billion in 2009. | |
| • | BETASERON (interferon-beta-1b), which is marketed by Bayer HealthCare Pharmaceuticals, the U.S. pharmaceuticals affiliate of Bayer Schering Pharma AG, in the U.S. and is marketed under the name BETAFERON by Bayer Schering Pharma AG in the E.U. BETASERON and BETAFERON together generated worldwide revenues of approximately $1.6 billion in 2009. | |
| • | EXTAVIA (interferon-beta-1b), which is marketed by Novartis AG in the E.U. and other markets. EXTAVIA was launched in the U.S. in September 2009. EXTAVIA generated worldwide revenue of approximately $49.0 million in 2009. |
12
| • | GILENYA (fingolimod), which is marketed by Novartis AG in the U.S. GILENYA is the first oral MS drug approved in the U.S., and was launched in the U.S. in October 2010. In January 2011, GILENYA was recommended for approval in the E.U. by the CHMP, and is either approved or under review in other countries. |
Along with us, a number of companies are working to develop additional treatments for MS that may in the future compete with AVONEX and TYSABRI. For example, an oral formulation of cladribine (developed by Merck Serono) has recently been approved for use in Australia and Russia. LEMTRADA (alemtuzumab) (developed by Genzyme Corporation), teriflunomide (developed by Sanofi-Aventis) and laquinimod (developed by Teva Pharmaceutical Industries) are in late-stage development for the treatment of MS. In addition, the commercialization of certain of our own pipeline product candidates, such as BG-12, may also negatively impact future sales of AVONEX and TYSABRI.
FUMADERM
FUMADERM competes with several different types of therapies in the psoriasis market within Germany, including oral systemics such as methotrexate and cyclosporine.
RITUXAN IN ONCOLOGY
RITUXAN competes with several different types of therapies in the oncology market, including:
| • | CAMPATH (alemtuzumab) (marketed by Bayer HealthCare Pharmaceuticals), which is indicated for B-cell CLL. | |
| • | TREANDA (bendamustine HCL) (marketed by Cephalon) and ARZERRA (ofatumumab) (marketed by GenMab in collaboration with GlaxoSmithKline), which is indicated for refractory CLL patients to both alemtuzumab and fludarabine. |
We are also aware of other anti-CD20 molecules in development that, if successfully developed and registered, may compete with RITUXAN in the oncology market.
RITUXAN IN RA
RITUXAN competes with several different types of therapies in the RA market, including:
| • | traditional therapies for RA, including disease-modifying anti-rheumatic drugs such as steroids, methotrexate and cyclosporine, and pain relievers such as acetaminophen. | |
| • | TNF inhibitors, such as REMICADE (infliximab) and SIMPONI (golimumab) (marketed by Johnson & Johnson), HUMIRA (adalimumab) (marketed by Abbott Laboratories), ENBREL (etanercept) (marketed by Amgen, Inc. and Pfizer) and CIMZIA (certolizumab pegol) (marketed by UCB, S.A.). | |
| • | ORENCIA (abatacept) (marketed by Bristol-Myers Squibb Company). | |
| • | ACTEMRA (tocilizumab) (marketed by the Roche Group). |
We are also aware of other products in development that, if successfully developed and registered, may compete with RITUXAN in the RA market.
Regulatory
Our current and contemplated activities and the products and processes that will result from such activities are subject to substantial government regulation.
Regulation of Pharmaceuticals
Before new pharmaceutical products may be sold in the U.S. and other countries, preclinical studies and clinical trials of the products must be conducted and the results submitted to appropriate regulatory agencies for approval. Clinical trial programs must establish efficacy, determine an appropriate dose and regimen, and define the conditions for safe use. This is a high-risk process that requires stepwise clinical studies in which the candidate product must successfully meet predetermined endpoints. In the U.S., the results of the preclinical and clinical
13
testing of a product are then submitted to the FDA in the form of a Biologics License Application (BLA) or a New Drug Application (NDA). In response to a BLA or NDA, the FDA may grant marketing approval, request additional information or deny the application if it determines the application does not provide an adequate basis for approval. Similar submissions are required by authorities in other jurisdictions who independently assess the product and may reach the same or different conclusions. Our initial focus for obtaining marketing approval outside the U.S. is typically the E.U. There are currently three potential tracks for marketing approval in E.U. countries: mutual recognition, decentralized procedures, and centralized procedures. These review mechanisms may ultimately lead to approval in all countries within the E.U., but each method grants all participating countries some decision-making authority in product approval.
The receipt of regulatory approval often takes a number of years, involves the expenditure of substantial resources and depends on a number of factors, including the severity of the disease in question, the availability of alternative treatments, potential safety signals observed in preclinical or clinical tests, and the risks and benefits demonstrated in clinical trials. On occasion, regulatory authorities may require larger or additional studies, leading to unanticipated delay or expense. Even after initial FDA approval or approvals from other regulatory agencies have been obtained, further clinical trials may be required to provide additional data on safety and effectiveness. Additional trials are required to gain approval for the use of a product as a treatment for indications other than those initially approved. Furthermore, the FDA and other regulatory agencies require companies to register clinical trials and disclose clinical trial results in public databases. Failure to register a trial or disclose study results within the required time periods could result in penalties, including civil monetary penalties.
In the U.S., the FDA may grant "accelerated approval" status to products that treat serious or life-threatening illnesses and that provide meaningful therapeutic benefits to patients over existing treatments. Under this pathway, the FDA may approve a product based on surrogate endpoints, or clinical endpoints other than survival or irreversible morbidity. When approval is based on surrogate endpoints or clinical endpoints other than survival or morbidity, the sponsor will be required to conduct additional post-approval clinical studies to verify and describe clinical benefit. Under the Agency's Accelerated Approval regulations, FDA may also provide approval with restrictions to assure safe use. Within this section of the Accelerated Approval regulations, if FDA concludes that a drug that has shown to be effective can be safely used only if distribution or use is restricted, they will require such post-marketing restrictions as necessary to assure safe use. When a drug approved under these conditions requires restricted use or distribution to ensure its safe use, the sponsor may be required to establish rigorous systems to assure use of the product under safe conditions. These systems are usually referred to as Risk Evaluation and Mitigation Strategies (REMS). In addition, for all products approved under accelerated approval, sponsors must submit all copies of their promotional materials, including advertisements, to the FDA at least thirty days prior to initial dissemination. The FDA may withdraw approval under accelerated approval after a hearing if, for instance, post-marketing studies fail to verify any clinical benefit or it becomes clear that restrictions on the distribution of the product are inadequate to ensure its safe use. TYSABRI was initially approved in MS under the accelerated approval pathway and, following such approval and after efficacy was confirmed, a stringent restricted distribution program was agreed upon. We cannot be certain that the FDA will approve any products for their proposed indications whether under accelerated approval or another pathway.
In addition, the FDA may grant "fast track" status to products that treat serious diseases and fill an unmet medical need. Fast track is a process designed to expedite the review of such products by providing, among other things, more frequent meetings with the FDA to discuss the product's development plan, more frequent written correspondence from the FDA about trial design, eligibility for accelerated approval, and rolling review, which allows submission of individually completed sections of a NDA for FDA review before the entire NDA is completed. Fast track status does not ensure that a product will be developed more quickly or receive FDA approval.
If the FDA or other regulatory agency approves a product or new indication, the agency may require us to conduct additional post-marketing studies. If we fail to conduct the required studies or otherwise fail to comply with the conditions of accelerated approval, the agency may withdraw its approval. In addition, the FDA and EMA can impose financial penalties for failing to comply with certain post-marketing commitments, including REMS.
Regulatory authorities track information on side effects and adverse events reported during clinical studies and after marketing approval. Non-compliance with regulatory authorities' safety reporting requirements may result in civil or criminal penalties. Side effects or adverse events that are reported during clinical trials can delay, impede, or
14
prevent marketing approval. Regulatory authorities may conduct post-marketing safety surveillance and may require additional post-approval studies or clinical trials. These requirements may affect our ability to maintain marketing approval of our products or require us to make significant expenditures to obtain or maintain such approvals. In addition, adverse events that are reported after marketing approval can result in changes to the product's labeling, additional limitations being placed on the product's use and, potentially, withdrawal or suspension of the product from the market.
If we seek to make certain types of changes to an approved product, such as adding a new indication, making certain manufacturing changes, or changing manufacturers or suppliers of certain ingredients or components, regulatory authorities, including the FDA and EMA, will need to review and approve such changes in advance. Such regulatory reviews can result in denial or modification of the planned changes, or requirements to conduct additional tests or evaluations that can substantially delay or increase the cost of the planned changes.
In addition, the FDA regulates all advertising and promotion activities for products under its jurisdiction both before and after approval. A company can make only those claims relating to safety and efficacy that are approved by the FDA. However, physicians may prescribe legally available drugs for uses that are not described in the drug's labeling. Such off-label uses are common across medical specialties, and often reflect a physician's belief that the off-label use is the best treatment for patients. The FDA does not regulate the behavior of physicians in their choice of treatments, but the FDA regulations do impose stringent restrictions on manufacturers' communications regarding off-label uses. Failure to comply with applicable FDA requirements may subject a company to adverse publicity, enforcement action by the FDA, corrective advertising, and the full range of civil and criminal penalties available to the FDA. Similar regulations are in place in outside the U.S.
Good Manufacturing Practices
The FDA, the EMA and other regulatory agencies regulate and inspect equipment, facilities, and processes used in the manufacturing of pharmaceutical and biologic products prior to approving a product. If, after receiving clearance from regulatory agencies, a company makes a material change in manufacturing equipment, location, or process, additional regulatory review and approval may be required. We also must adhere to current Good Manufacturing Practices and product-specific regulations enforced by the FDA following product approval. The FDA, the EMA and other regulatory agencies also conduct periodic visits to re-inspect equipment, facilities, and processes following the initial approval of a product. If, as a result of these inspections, it is determined that our equipment, facilities, or processes do not comply with applicable regulations and conditions of product approval, regulatory agencies may seek civil, criminal, or administrative sanctions or remedies against us, including the suspension of our manufacturing operations.
Good Clinical Practices
The FDA, the EMA and other regulatory agencies promulgate regulations and standards, commonly referred to as current Good Clinical Practices (cGCP), for designing, conducting, monitoring, auditing and reporting the results of clinical trials to ensure that the data and results are accurate and that the rights and welfare of trial participants are adequately protected. The FDA, the EMA and other regulatory agencies enforce cGCP through periodic inspections of trial sponsors, principal investigators and trial sites, contract research organizations (CROs), and institutional review boards. If our studies fail to comply with applicable cGCP, the clinical data generated in our clinical trials may be deemed unreliable and relevant regulatory agencies may require us to perform additional clinical trials before approving our marketing applications. Noncompliance can also result in civil or criminal sanctions. We rely on third parties, including CROs, to carry out many of our clinical trial-related activities. Failure of such third party to comply with cGCP can likewise result in rejection of our clinical trial data or other sanctions.
Orphan Drug Act
Under the U.S. Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a "rare disease or condition," which generally is a disease or condition that affects fewer than 200,000 individuals in the U.S. If a product which has an orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, i.e., the FDA may not approve any other applications to market the same drug for the same indication for a period of seven years following marketing approval, except in certain very limited circumstances, such as if the later product is shown to be
15
clinically superior to the orphan product. Legislation similar to the U.S. Orphan Drug Act has been enacted in other countries, including within the E.U.
Regulation Pertaining to Sales, Marketing and Product Pricing
The U.S. and foreign governments regularly consider reforming health care coverage and costs. Such reform may include changes to the coverage and reimbursement of our products which may have a significant impact on our business.
In 2010, significant healthcare reform legislation was enacted in the U.S., which has had and will continue to have an impact our business by:
| • | expanding the coverage of and increasing the rate of rebates on sales of our products, including (1) increasing the Medicaid rebate from 15.1% to 23.1% of the average manufacturer price (AMP) on our branded prescription drugs, (2) extending the Medicaid rebate to Managed Care Organizations, and (3) expanding the 340B Public Health Service (PHS) drug pricing program, which provides outpatient drugs at reduced rates, to include additional hospitals, clinics, and healthcare centers; | |
| • | requiring drug manufactures to provide a 50% discount to Medicare beneficiaries whose prescription drug costs cause them to be subject to the Medicare Part D coverage gap (i.e. the "donut hole"); | |
| • | assessing a new fee allocated to all manufacturers and importers of branded prescription drugs paid for pursuant to coverage provided under specified government programs; | |
| • | including an abbreviated approval pathway for biosimilars; and | |
| • | changing the calculation of AMP for injectable drugs not generally dispensed through retail community pharmacies. |
Considerable uncertainty remains surrounding determinations necessary to implement the new legislation. For example, determinations as to how the Medicare coverage gap will operate remain to be clarified. In addition, uncertainty also exists as to when and how discounts will be provided to the additional hospitals eligible to participate under the 340B program. In addition, in November 2010 the Centers for Medicare and Medicaid Services (CMS) amended and then withdrew current regulations governing calculation of AMP; however, no replacement regulations have been proposed.
Medicaid is a joint federal and state program that is administered by the states for low income and disabled beneficiaries. Under the Medicaid Drug Rebate Program, we are required to pay a rebate for each unit of product reimbursed by the state Medicaid programs. The amount of the rebate for each product is set by law as the larger of 23.1% of AMP or the difference between AMP and the best price available from us to any commercial or non-governmental customer. The rebate amount must be adjusted upward where the AMP for a product's first full quarter of sales, when adjusted for increases in the CPI-U, or Consumer Price Index - Urban, exceeds the AMP for the current quarter with the upward adjustment equal to the excess amount. The rebate amount is required to be recomputed each quarter based on our report of current AMP and best price for each of our products to the Centers for Medicare and Medicaid Services. The terms of our participation in the program impose a requirement for us to report revisions to AMP or best price within a period not to exceed 12 quarters from the quarter in which the data was originally due. Any such revisions could have the impact of increasing or decreasing our rebate liability for prior quarters, depending on the direction of the revision. In addition, if we were found to have knowingly submitted false information to the government, the statute provides for civil monetary penalties in an amount not to exceed $100,000 per item of false information, in addition to other penalties available to the government.
Medicare is a federal program that is administered by the federal government that covers individuals age 65 and over as well as those with certain disabilities. Medicare Part B pays physicians that administer our products under a payment methodology using average sales price, or ASP, information. Manufacturers, including us, are required to provide ASP information to the Centers for Medicare and Medicaid Services on a quarterly basis. The manufacturer-submitted information is used to compute Medicare payment rates, which are set at ASP plus 6 percent and updated quarterly. There is a mechanism for comparison of such payment rates to widely available market prices, which could cause further decreases in Medicare payment rates, although this mechanism has yet to be utilized. Effective January 1, 2006, Medicare began to use the same ASP plus 6 percent payment methodology to determine Medicare rates paid for products furnished by hospital outpatient departments. As of January 1, 2009, the
16
reimbursement rate in the hospital outpatient setting was ASP plus 4 percent. The reimbursement rate in the hospital outpatient setting was increased to ASP plus 5 percent effective January 1, 2011. If a manufacturer is found to have made a misrepresentation in the reporting of ASP, the statute provides for civil monetary penalties of up to $10,000 for each misrepresentation and for each day in which the misrepresentation was applied.
The Medicare Prescription Drug Improvement and Modernization Act of 2003 established the Medicare Part D program to provide voluntary prescription drug benefit to enrolled Medicare patients. This is a voluntary benefit that is being implemented through private plans under contractual arrangements with the federal government. Like pharmaceutical coverage through private health insurance, Part D plans establish formularies that govern the drugs and biologicals that will be offered and the out-of-pocket obligations for such products. In addition, plans negotiate discounts from drug manufacturers and pass on some of those savings to Medicare beneficiaries.
The availability of federal funds to pay for our products under the Medicaid and Medicare Part B programs requires that we extend discounts under the 340B/PHS drug pricing program. The 340B/PHS drug pricing program extends discounts to a variety of community health clinics and other entities that receive health services grants from the PHS, as well as hospitals that serve a disproportionate share of poor Medicare beneficiaries.
We also make our products available for purchase by authorized users of the Federal Supply Schedule (FSS) of the General Services Administration pursuant to our FSS contract with the Department of Veterans Affairs. Under the Veterans Health Care Act of 1992 (VHC Act) we are required to offer deeply discounted FSS contract pricing to four Federal agencies - the Department of Veterans Affairs, the Department of Defense, the Coast Guard and the PHS (including the Indian Health Service) - for federal funding to be made available for reimbursement of any of our products under the Medicaid program and for our products to be eligible to be purchased by those four Federal agencies and certain Federal grantees. FSS pricing to those four Federal agencies must be equal to or less than the "Federal Ceiling Price," which is, at a minimum, 24% below the Non-Federal Average Manufacturer Price (Non-FAMP) for the prior fiscal year. In addition, if we are found to have knowingly submitted false information to the government, the VHC Act provides for civil monetary penalties up to $100,000 per false item of information in addition to other penalties available to the government.
Under the 2008 National Defense Authorization Act, we are required to treat the TRICARE retail pharmacy program, which reimburses military personnel for drug purchases from retail pharmacies, as an element of the Department of Defense to ensure the application of the VHC Act's pricing standards.
We are also subject to various federal and state laws pertaining to health care "fraud and abuse," including anti-kickback laws and false claims laws. Anti-kickback laws make it illegal for a prescription drug manufacturer to solicit, offer, receive, or pay any remuneration in exchange for, or to induce, the referral of business, including the purchase or prescription of a particular drug. Due to the breadth of the statutory provisions and the absence of guidance in the form of regulations and very few court decisions addressing industry practices, it is possible that our practices might be challenged under anti-kickback or similar laws. False claims laws prohibit anyone from knowingly and willingly presenting, or causing to be presented for payment to third party payors (including Medicare and Medicaid) claims for reimbursed drugs or services that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services. In addition, several states require that companies implement compliance programs or comply with industry ethics codes, adopt spending limits, and report to state governments any gifts, compensation, and other remuneration provided to physicians. Federal legislation, the Physician Payments Sunshine Act of 2009, also has been proposed that would require disclosure to the federal government of payments to physicians. Our activities relating to the sale and marketing of our products may be subject to scrutiny under these laws. Violations of fraud and abuse laws may be punishable by criminal or civil sanctions, including fines and civil monetary penalties, as well as the possibility of exclusion from federal health care programs (including Medicare and Medicaid). If the government were to allege or convict us of violating these laws, our business could be harmed. In addition, private individuals may bring similar actions.
Our activities could be subject to challenge for the reasons discussed above and due to the broad scope of these laws and the increasing attention being given to them by law enforcement authorities. Further, there are an increasing number of state laws that require manufacturers to make reports to states on pricing and marketing information. Many of these laws contain ambiguous requirements. Given the lack of clarity in laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent state authorities.
17
Future legislation or regulatory actions implementing recent or future legislation may have a significant effect on our business. Our ability to successfully commercialize products may depend in part on the extent to which reimbursement for the costs of our products and related treatments will be available in the U.S. and worldwide from government health administration authorities, private health insurers and other organizations. Substantial uncertainty exists as to the reimbursement status by third party payors of newly approved health care products.
Other Regulations
Foreign Anti-Corruption
We are subject to the U.S. Foreign Corrupt Practices Act (FCPA), which prohibits U.S. corporations and their representatives from paying, offering to pay, promising, authorizing, or making payments of anything of value to any foreign government official, government staff member, political party, or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in an official capacity. In many countries, the health care professionals we regularly interact with may meet the FCPA's definition of a foreign government official. The FCPA also requires public companies to make and keep books and records that accurately and fairly reflect their transactions and to devise and maintain an adequate system of internal accounting controls.
In 2010, the Bribery Act was passed in the United Kingdom, which proscribes giving and receiving bribes in the public and private sectors, bribing a foreign public official, and failing to have adequate procedures to prevent employees and other agents from giving bribes. U.S. corporations that conduct business in the United Kingdom generally will be subject to the Bribery Act. Penalties under the Bribery Act include potentially unlimited fines for corporations and criminal sanctions for corporate officers under certain circumstances.
NIH Guidelines
We conduct research at our U.S. facilities in compliance with the current U.S. National Institutes of Health Guidelines for Research Involving Recombinant DNA Molecules (NIH Guidelines) and all other applicable federal and state regulations. By local ordinance, we are required to, among other things, comply with the NIH Guidelines in relation to our facilities in Cambridge, Massachusetts and Research Triangle Park, North Carolina and are required to operate pursuant to certain permits.
Other Laws
Our present and future business has been and will continue to be subject to various other laws and regulations. Various laws, regulations and recommendations relating to safe working conditions, laboratory practices, the experimental use of animals, and the purchase, storage, movement, import and export and use and disposal of hazardous or potentially hazardous substances, including radioactive compounds and infectious disease agents, used in connection with our research work are or may be applicable to our activities. Certain agreements entered into by us involving exclusive license rights may be subject to national or international antitrust regulatory control, the effect of which cannot be predicted. The extent of government regulation, which might result from future legislation or administrative action, cannot accurately be predicted.
Manufacturing and Raw Materials
We are focused on the manufacture of biologics. The chart below outlines the location of our primary manufacturing locations and products manufactured therein.
| Research | ||||||||||||
| Triangle | ||||||||||||
| Product | Park, NC | Cambridge, MA | Third Party | |||||||||
AVONEX | ü | ü | ||||||||||
| ||||||||||||
TYSABRI | ü | |||||||||||
| ||||||||||||
FUMADERM | ü | |||||||||||
| ||||||||||||
CLINICAL PRODUCTS | ü | ü | ü | |||||||||
| ||||||||||||
We currently produce all of our bulk AVONEX at our manufacturing facilities located in Research Triangle Park, North Carolina (RTP) and Cambridge, Massachusetts. We currently produce TYSABRI at our RTP facility. In April 2009, the FDA approved our high titer process for the production of TYSABRI. Similar approval was obtained from
18
the EMA in December 2008. Genentech is responsible for all worldwide manufacturing activities for bulk RITUXAN and has sourced the manufacturing of certain bulk RITUXAN requirements to an independent third party.
We plan to stop further validation of our large-scale manufacturing facility in Hillerød, Denmark following completion of the facility's operational qualification activities in the first half of 2011 as we continue to evaluate our current manufacturing utilization strategy. This facility is intended to manufacture large molecule products. Recent manufacturing improvements have resulted in favorable production yields on TYSABRI, that along with slower than expected TYSABRI growth, have reduced our expected capacity requirements. As a result, we have decided to delay the start of manufacturing activities at this site until additional capacity is required by the business.
We source all of our fill-finish and the majority of final product storage operations for our products, along with a substantial part of our packaging operations, to a concentrated group of third party contractors. Many of the raw materials and supplies required for the production of AVONEX, TYSABRI and FUMADERM are available from various suppliers in quantities adequate to meet our needs. However, due to the unique nature of the production of our products, we do have single source providers of several raw materials. We make efforts to qualify new vendors and to develop contingency plans so that production is not impacted by short-term issues associated with single source providers. Each of our third party service providers, suppliers and manufacturers is subject to continuing inspection by the FDA or comparable agencies in other jurisdictions. Any delay, interruption or other issues that arise in the manufacture, fill-finish, packaging or storage of our products, including as a result of a failure of our facilities or the facilities or operations of third parties to pass any regulatory agency inspection, could significantly impair our ability to sell our products.
Important factors that could adversely affect our manufacturing operations are discussed in the " Risk Factors " section of this report.
Our Employees
As of December 31, 2010, we had approximately 4,850 employees worldwide. We are in the process of completing a 13% reduction in our workforce as part of our framework for growth initiatives. This workforce reduction impacts our sales, research and development and administrative functions.
Our Executive Officers (as of February 4, 2011)
George A. Scangos , Ph.D. , 62, is our Chief Executive Officer and has served in this position since July 2010. Prior to that, Dr. Scangos served as President and Chief Executive Officer of Exelixis, Inc., a life sciences company, since October 1996, where he continues to serve on the board. From 1993 to 1996, Dr. Scangos served as President of Bayer Biotechnology, where he was responsible for research, business development, process development, manufacturing, engineering and quality assurance of Bayer's biological products. Before joining Bayer in 1987, Dr. Scangos was a Professor of Biology at Johns Hopkins University for six years. Dr. Scangos served as non-executive Chairman of Anadys Pharmaceuticals, Inc., a biopharmaceutical company, from 2005 to July 2010 and was a director of the company since 2003. Dr. Scangos served as the Chair of the California Healthcare Institute in 2010, was a member of the Board of the Global Alliance for TB Drug Developments until 2010, and is a director of Foundation Sante. He is also a member of the Board of Visitors of the University of California, San Francisco School of Pharmacy, and the National Board of Visitors of the University of California, Davis School of Medicine. He is currently an Adjunct Professor of Biology at Johns Hopkins. Dr. Scangos was a Jane Coffin Childs Post-Doctoral Fellow at Yale University. Dr. Scangos holds a B.A. in Biology from Cornell University and a Ph.D. in Microbiology from the University of Massachusetts.
Susan H. Alexander , 54, is our Executive Vice President, General Counsel and Corporate Secretary and has served in these positions since January 2006. Prior to that, Ms. Alexander served as the Senior Vice President, General Counsel and Corporate Secretary of PAREXEL International Corporation, a biopharmaceutical services company, since September 2003. From June 2001 to September 2003, Ms. Alexander served as General Counsel of IONA Technologies, a software company. Prior to that, Ms. Alexander served as Counsel at Cabot Corporation, a specialty chemicals and performance materials company, from January 1995 to May 2001. Prior to that, Ms. Alexander was a partner at the law firms of Hinckley, Allen & Snyder and Fine & Ambrogne.
Paul J. Clancy , 49, is our Executive Vice President, Finance and Chief Financial Officer and has served in these positions since August 2007. Mr. Clancy joined Biogen, Inc. in 2001 and has held several senior executive positions with us, including Vice President of Business Planning, Portfolio Management and U.S. Marketing, and Senior Vice President
19
of Finance with responsibilities for leading the Treasury, Tax, Investor Relations and Business Planning groups. Prior to that, he spent 13 years at PepsiCo, a food and beverage company, serving in a range of financial and general management positions. He holds a B.S. in finance from Babson College and a M.B.A. from Columbia University.
John G. Cox , 48, is our Executive Vice President, Pharmaceutical Operations and Technology and has served in this position since June 2010. Mr. Cox joined Biogen, Inc. in 2003 and has held several senior executive positions with us, including Senior Vice President of Technical Operations, Senior Vice President of Global Manufacturing, and Vice President of Manufacturing and General Manager of Biogen Idec's operations in RTP. Prior to that, Mr. Cox held a number of senior operational roles at Diosynth, a life sciences manufacturing and services company, where he worked in technology transfer, validation and purification. Prior to that, Mr. Cox focused on the same areas at Wyeth Corporation, a life sciences company, from 1993 to 2000.
Robert E. Gagnon , 36, is our Vice President, Finance, Chief Accounting Officer and Controller and has served in these positions since November 2010. Prior to that, Mr. Gagnon served as Vice President, Finance and Controller from July 2007 to November 2010, and Director of Corporate Accounting from October 2005 to July 2007. Prior to that, Mr. Gagnon worked in the business advisory and assurance practices of PricewaterhouseCoopers LLP and Deloitte & Touche LLP. Mr. Gagnon is a certified public accountant and holds an M.B.A. from the MIT Sloan School of Management.
Francesco Granata, M.D. , 60, is our Executive Vice President, Global Commercial Operations and has served in this position since January 2010. Prior to that, Dr. Granata served as Group Vice President and President of EUCAN Region in the Global Pharmaceutical Business at Schering-Plough Corporation, a pharmaceutical company, from September 2005 to November 2010. Prior to that, Dr. Granata worked in commercial leadership positions at Pfizer, Inc., a pharmaceutical company, from 2003 to 2005 and at Pharmacia Corporation, a life sciences company, from 1999 to 2003.
Stephen H. Holtzman , 56, is our Executive Vice President, Corporate Development and has served in this position since January 2011. Prior to that, Mr. Holtzman was a founder of Infinity Pharmaceuticals, Inc., a drug discovery and development company, where he has served as Chair of the Board of Directors since 2001, and served as Executive Chair of the Board of Directors in 2010 and as Chief Executive Officer from 2001 to December 2009. From 1994 to 2001, Mr. Holtzman was Chief Business Officer at Millenium Pharmaceuticals Inc., a biopharmaceutical company. From 1986 to 1994, he was the co-founder, member of the Board of Directors and Executive Vice President of DNX Corporation, a biotechnology company. From 1996 to 2001, Mr. Holtzman served as presidential appointee to the national Bioethics Advisory Commission.
Craig Eric Schneier, Ph.D. , 63, is our Executive Vice President, Human Resources, Public Affairs and Communications and has served in this position since October 2007. Dr. Schneier joined Biogen, Inc. in 2001 and has held several senior executive positions with us, including Executive Vice President, Human Resources and Senior Vice President, Strategic Organization Design and Effectiveness. Prior to that, Dr. Schneier was president of his own management consulting firm in Princeton, NJ, where he provided consulting services to over 70 of the Fortune 100 companies, as well as several of the largest European and Asian firms. Dr. Schneier held a tenured professorship at the University of Maryland's Smith School of Business and has held teaching positions at the business schools of the University of Michigan, Columbia University, and at the Tuck School of Business, Dartmouth College.
Douglas E. Williams, Ph.D. , 52, is our Executive Vice President, Research and Development and has served in this position since January 2011. Prior to that, Dr. Williams held several senior executive positions at ZymoGenetics Inc., a biopharmaceutical company, including Chief Executive Officer and a director from January 2009 to October 2010, President and Chief Scientific Officer from July 2007 to January 2009, and Executive Vice President, Research and Development and Chief Scientific Officer from 2004 to July 2007. Prior to that, he held leadership positions within the biotechnology industry, including Chief Scientific Officer and Executive Vice President of Research and Development at Seattle Genetics Inc., a biotechnology company, from 2003 to 2004, and Senior Vice President and Washington Site Leader at Amgen Inc., a biotechnology company, in 2002. Dr. Williams also served in a series of scientific and senior leadership positions over a decade at Immunex Corp., a biopharmaceutical company, including Executive Vice President and Chief Technology Officer, Senior Vice President of Discovery Research, Vice President of Research and Development and as a director. Prior to that, Dr. Williams served on the faculty of the Indiana University School of Medicine and the Department of Laboratory Medicine at the Roswell Park Memorial Institute in Buffalo, New York.
20
| Item 1A. | Risk Factors |
We are substantially dependent on revenues from our three principal products.
Our current and future revenues depend upon continued sales of our three principal products, AVONEX, RITUXAN and TYSABRI, which represented substantially all of our total revenues during 2010. Although we have developed and continue to develop additional products for commercial introduction, we may be substantially dependent on sales from these three products for many years. Any negative developments relating to any of these products, such as safety or efficacy issues, the introduction or greater acceptance of competing products, including biosimilars, or adverse regulatory or legislative developments may reduce our revenues and adversely affect our results of operations. New competing products for use in multiple sclerosis are beginning to enter the market and if they have a similar or more attractive profile in terms of efficacy, convenience or safety, future sales of AVONEX and TYSABRI could be limited, which would reduce our revenues.
TYSABRI's sales growth is important to our success.
We expect that our revenue growth over the next several years will be dependent in part upon sales of TYSABRI. If we are not successful in growing sales of TYSABRI, our future business plans, revenue growth and results of operations may be adversely affected.
TYSABRI's sales growth cannot be certain given the significant restrictions on use and the significant safety warnings in the label, including the risk of developing progressive multifocal leukoencephalopathy (PML), a serious brain infection. The risk of developing PML increases with prior immunosuppressant use, which may cause patients who have previously received immunosuppressants or their physicians to refrain from using or prescribing TYSABRI. The risk of developing PML also increases with longer treatment duration, with limited experience beyond three years. This may cause prescribing physicians or patients to suspend treatment with TYSABRI. Increased incidences of PML could limit sales growth, prompt regulatory review, require significant changes to the label or result in market withdrawal. Additional regulatory restrictions on the use of TYSABRI or safety-related label changes, including enhanced risk management programs, whether as a result of additional cases of PML or otherwise, may significantly reduce expected revenues and require significant expense and management time to address the associated legal and regulatory issues. In addition, ongoing or future clinical trials involving TYSABRI and efforts at stratifying patients into groups with lower or higher risk for developing PML, including evaluating the potential clinical utility of a JC virus antibody assay, may have an adverse impact on prescribing behavior and reduce sales of TYSABRI.
If we fail to compete effectively, our business and market position would suffer.
The biotechnology and pharmaceutical industry is intensely competitive. We compete in the marketing and sale of our products, the development of new products and processes, the acquisition of rights to new products with commercial potential and the hiring and retention of personnel. We compete with biotechnology and pharmaceutical companies that have a greater number of products on the market and in the product pipeline, greater financial and other resources and other technological or competitive advantages. One or more of our competitors may benefit from significantly greater sales and marketing capabilities, may develop products that are accepted more widely than ours and may receive patent protection that dominates, blocks or adversely affects our product development or business. In addition, recently enacted healthcare reform legislation in the U.S. has created a pathway for the FDA to approve biosimilars, which could compete on price and differentiation with products that we now or could in the future market. The introduction of more efficacious, safer, cheaper, or more convenient alternatives to our products could reduce our revenues and the value of our product development efforts.
Our long-term success depends upon the successful development and commercialization of other product candidates.
Our long-term viability and growth will depend upon the successful development and commercialization of other products from our research and development activities, including products licensed from third parties. In addition, we have several late-stage clinical programs expected to have near-term data readouts that could impact our prospects for additional revenue growth. Product development and commercialization are very expensive and
21
involve a high degree of risk. Only a small number of research and development programs result in the commercialization of a product. Success in preclinical work or early stage clinical trials does not ensure that later stage or larger scale clinical trials will be successful. Even if later stage clinical trials are successful, product candidates may not receive marketing approval if regulatory authorities disagree with our view of the data or require additional studies.
Conducting clinical trials is a complex, time-consuming and expensive process. Our ability to complete our clinical trials in a timely fashion depends in large part on a number of key factors including protocol design, regulatory and institutional review board approval, the rate of patient enrollment in clinical trials, and compliance with extensive current good clinical practice requirements. We have opened clinical sites and are enrolling patients in a number of new countries where our experience is more limited, and we are in many cases using the services of third-party clinical trial providers. If we fail to adequately manage the design, execution and regulatory aspects of our large, complex and diverse clinical trials, our studies and ultimately our regulatory approvals may be delayed or we may fail to gain approval for our product candidates altogether.
Our product pipeline includes several small molecule drug candidates. Our small molecule drug discovery platform is not as well developed as our biologics platform and we expect to rely on third party manufacturers to supply substantially all of our clinical requirements for small molecules. If these manufacturers fail to deliver sufficient quantities of such drug candidates in a timely and cost-effective manner, it could adversely affect our small molecule drug discovery efforts.
Adverse safety events can negatively affect our business and stock price.
Adverse safety events involving our marketed products may have a negative impact on our commercialization efforts. Later discovery of safety issues with our products that were not known at the time of their approval by the FDA or other regulatory agencies worldwide could cause product liability events, additional regulatory scrutiny and requirements for additional labeling, withdrawal of products from the market and the imposition of fines or criminal penalties. Any of these actions could result in, among other things, material write-offs of inventory and impairments of intangible assets, goodwill and fixed assets and material restructuring charges. In addition, the reporting of adverse safety events involving our products and public rumors about such events could cause our stock price to decline or experience periods of volatility.
We depend, to a significant extent, on reimbursement from third party payors and a reduction in the extent of reimbursement could reduce our product sales and revenue.
Sales of our products are dependent, in large part, on the availability and extent of reimbursement from government health administration authorities, private health insurers and other organizations. Changes in government regulations or private third-party payors' reimbursement policies may reduce reimbursement for our products and adversely affect our future results. In addition, when a new medical product is approved, the availability of government and private reimbursement for that product is uncertain, as is the amount for which that product will be reimbursed. We cannot predict the availability or amount of reimbursement for our product candidates.
The U.S. Congress recently enacted legislation to reform the health care system. This legislation imposes cost containment measures that have adversely affected the amount of reimbursement for our products. These measures include increasing the minimum rebates we pay to state Medicaid programs for our drugs covered by Medicaid programs; extending such rebates to drugs dispensed to Medicaid beneficiaries enrolled in Medicaid managed care organizations; and expanding the 340B Public Health Service drug discount program under which we are obligated to provide certain discounts on our drugs to certain purchasers. Additional provisions of the health care reform legislation, which become effective in 2011, may negatively affect our revenues and prospects for profitability in the future. Beginning in 2011, a new fee will be payable by all branded prescription drug manufacturers and importers. This fee will be calculated based upon each organization's percentage share of total branded prescription drugs sales to qualifying U.S. government programs, including Medicare and Medicaid. As part of the health care reform legislation's provisions closing a coverage gap that currently exists in the Medicare Part D prescription drug
22
program (commonly known as the "donut hole"), we will also be required to provide a 50% discount on brand name prescription drugs sold to beneficiaries who fall within the donut hole.
Economic pressure on state budgets may result in states increasingly seeking to achieve budget savings through mechanisms that limit coverage or payment for our drugs. In recent years, some states have considered legislation that would control the prices of drugs. State Medicaid programs are increasingly requesting manufacturers to pay supplemental rebates and requiring prior authorization by the state program for use of any drug for which supplemental rebates are not being paid. Managed care organizations continue to seek price discounts and, in some cases, to impose restrictions on the coverage of particular drugs. Government efforts to reduce Medicaid expenses may lead to increased use of managed care organizations by Medicaid programs. This may result in managed care organizations influencing prescription decisions for a larger segment of the population and a corresponding constraint on prices and reimbursement for our products. It is likely that federal and state legislatures and health agencies will continue to focus on additional health care reform in the future.
We encounter similar regulatory and legislative issues in most other countries. In the European Union and some other international markets, the government provides health care at low cost to consumers and regulates pharmaceutical prices, patient eligibility or reimbursement levels to control costs for the government-sponsored health care system. Many countries are reducing their public expenditures and we expect to see strong efforts to reduce healthcare costs in our international markets, including patient access restrictions, suspensions on price increases, prospective and possibly retroactive price reductions and increased mandatory discounts or rebates, recoveries of past price increases, and greater importation of drugs from lower-cost countries to higher-cost countries. We expect that our revenues would be negatively impacted if similar measures are or are continued to be implemented in other countries in which we operate. In addition, certain countries set prices by reference to the prices in other countries where our products are marketed. Thus, our inability to secure adequate prices in a particular country may also impair our ability to obtain acceptable prices in existing and potential new markets. This may create the opportunity for third party cross border trade or influence our decision to sell or not to sell a product, thus affecting our geographic expansion plans.
Adverse market and economic conditions may exacerbate certain risks affecting our business.
Sales of our products are dependent on reimbursement from government health administration authorities, private health insurers, distribution partners and other organizations. As a result of adverse conditions affecting the U.S. and global economies and credit and financial markets, including the current sovereign debt crisis in certain countries in Europe, these organizations may be unable to satisfy their reimbursement obligations or may delay payment. In addition, governmental health authorities may reduce the extent of reimbursements, and private insurers may increase their scrutiny of claims. A reduction in the availability or extent of reimbursement could reduce our product sales and revenue, or result in additional allowances or significant bad debts, which may adversely affect our results of operations.
We depend on collaborators and other third-parties for both product and royalty revenue and the clinical development of future products, which are outside of our full control.
Collaborations between companies on products or programs are a common business practice in the biotechnology industry. Out-licensing typically allows a partner to collect up front payments and future milestone payments, share the costs of clinical development and risk of failure at various points, and access sales and marketing infrastructure and expertise in exchange for certain financial rights to the product or program going to the in-licensing partner. In addition, the obligation of in-licensees to pay royalties or share profits generally terminates upon expiration of the related patents. We have a number of collaborators and partners, and have both in-licensed and out-licensed several products and programs. These collaborations are subject to several risks:
| • | Our RITUXAN revenues are substantially dependent on the efforts of Genentech and the Roche Group. Their interests may not always be aligned with our interests and they may not market RITUXAN in the same manner or to the same extent that we would, which could adversely affect our RITUXAN revenues. |
23
| • | Under our collaboration agreement with Genentech, the successful development and commercialization of GA101 and certain other anti-CD20 products will decrease our percentage of the collaboration's co-promotion profits. | |
| • | We are not fully in control of the royalty or profit sharing revenues we receive from collaborators, which may be adversely affected by patent expirations, pricing or health care reforms, other legal and regulatory developments, and the introduction of competitive products, and new indication approvals which may affect the sales of collaboration products. | |
| • | Any failure on the part of our collaboration partners to comply with applicable laws and regulatory requirements in the sale and marketing of our products could have an adverse effect on our revenues as well as involve us in possible legal proceedings. | |
| • | Collaborations often require the parties to cooperate, and failure to do so effectively could have an adverse impact on product sales by our collaborators and partners, and could adversely affect the clinical development or regulatory approvals of products under joint control. |
In addition, we rely on third parties for several other aspects of our business. As a sponsor of clinical trials of our products, we rely on third party contract research organizations to carry out many of our clinical trial related activities. These activities include initiating the conduct of studies at clinical trial sites, regularly monitoring the conduct of the study at study sites, and identifying instances of noncompliance with the study protocol or current Good Clinical Practices. The failure of a contract research organization to conduct these activities with proper vigilance and competence and in accordance with Good Clinical Practices can result in regulatory authorities rejecting our clinical trial data or, in some circumstances, the imposition of civil or criminal sanctions against us.
If we do not successfully execute our growth initiatives through the acquisition, partnering and in-licensing of products, technologies or companies, our future performance could be adversely affected.
We anticipate growing through internal development projects as well as external opportunities, which include the acquisition, partnering and in-licensing of products, technologies and companies or the entry into strategic alliances and collaborations. The availability of high quality opportunities is limited and we are not certain that we will be able to identify candidates that we and our shareholders consider suitable or complete transactions on terms that are acceptable to us and our shareholders. In order to pursue such opportunities, we may require significant additional financing, which may not be available to us on favorable terms, if at all. Even if we are able to successfully identify and complete acquisitions, we may not be able to integrate them or take full advantage of them and therefore may not realize the benefits that we expect. If we are unsuccessful in our external growth program, we may not be able to grow our business significantly and we may incur asset impairment or restructuring charges as a result of unsuccessful transactions.
If we fail to comply with the extensive legal and regulatory requirements affecting the health care industry, we could face increased costs, penalties and a loss of business.
Our activities, and the activities of our collaborators and third party providers, are subject to extensive government regulation and oversight both in the U.S. and in foreign jurisdictions. The FDA and comparable agencies in other jurisdictions directly regulate many of our most critical business activities, including the conduct of preclinical and clinical studies, product manufacturing, advertising and promotion, product distribution, adverse event reporting and product risk management. Our interactions in the U.S. or abroad with physicians and other health care providers that prescribe or purchase our products are also subject to government regulation designed to prevent fraud and abuse in the sale and use of the products. In the U.S., states increasingly have been placing greater restrictions on the marketing practices of health care companies. In addition, pharmaceutical and biotechnology companies have been the target of lawsuits and investigations alleging violations of government regulation, including claims asserting submission of incorrect pricing information, impermissible off-label promotion of pharmaceutical products, payments intended to influence the referral of federal or state health care business, submission of false claims for government reimbursement, antitrust violations, or violations related to environmental matters. Violations of governmental regulation may be punishable by criminal and civil sanctions against us, including fines and civil monetary penalties and exclusion from participation in government programs, including
24
Medicare and Medicaid, as well as against executives overseeing our business. In addition to penalties for violation of laws and regulations, we could be required to repay amounts we received from government payors, or pay additional rebates and interest if we are found to have miscalculated the pricing information we have submitted to the government. Whether or not we have complied with the law, an investigation into alleged unlawful conduct could increase our expenses, damage our reputation, divert management time and attention and adversely affect our business. Recent changes in U.S. fraud and abuse laws have strengthened government regulation, increased the investigative powers of government enforcement agencies, and enhanced penalties for non-compliance.
If we fail to meet the stringent requirements of governmental regulation in the manufacture of our products, we could incur substantial costs and a reduction in sales.
We and our third party providers are generally required to maintain compliance with current Good Manufacturing Practice and are subject to inspections by the FDA and comparable agencies in other jurisdictions to confirm such compliance. In addition, the FDA must approve any significant changes to our suppliers or manufacturing methods. If we or our third party service providers cannot demonstrate ongoing current Good Manufacturing Practice compliance, we may be required to withdraw or recall product, interrupt commercial supply of our products, undertake costly remediation efforts or seek more costly manufacturing alternatives. Any delay, interruption or other issues that arise in the manufacture, fill-finish, packaging, or storage of our products as a result of a failure of our facilities or the facilities or operations of third parties to pass any regulatory agency inspection could significantly impair our ability to develop and commercialize our products. Significant noncompliance could also result in the imposition of monetary penalties or other civil or criminal sanctions. This non-compliance could increase our costs, cause us to lose revenue or market share and damage our reputation.
Our investments in properties, including our manufacturing facilities, may not be fully realizable.
We own or lease real estate primarily consisting of buildings that contain research laboratories, office space, and biologic manufacturing operations, some of which are located in markets that are experiencing high vacancy rates and decreasing property values. If we decide to consolidate or co-locate certain aspects of our business operations, for strategic or other operational reasons, we may dispose of one or more of our properties.
Due to reduced expectations of product demand, improved yields on production and other factors, we may not fully utilize our manufacturing facilities at normal levels resulting in idle time at facilities or substantial excess manufacturing capacity. We regularly evaluate our current manufacturing strategy, and may pursue alternatives that include disposing of manufacturing facilities.
If we determine that the fair value of any of our owned properties, including any properties we may classify as held for sale, is lower than their book value we may not realize the full investment in these properties and incur significant impairment charges. In addition, if we decide to fully or partially vacate a leased property, we may incur significant cost, including lease termination fees, rent expense in excess of sublease income and impairment of leasehold improvements.
Problems with manufacturing or with inventory planning could result in inventory shortages, product recalls and increased costs.
Biologics manufacturing is extremely susceptible to product loss due to contamination, equipment failure, or vendor or operator error. In addition, we may need to close a manufacturing facility for an extended period of time due to microbial, viral or other contamination. Any of these events could result in shipment delays or product recalls, impairing our ability to supply products in existing markets or expand into new markets. In the past, we have taken inventory write-offs and incurred other charges and expenses for products that failed to meet specifications, and we may incur similar charges in the future.
We rely solely on our manufacturing facility in Research Triangle Park, North Carolina for the production of TYSABRI. Our global bulk supply of TYSABRI depends on the uninterrupted and efficient operation of this facility, which could be adversely affected by equipment failures, labor shortages, natural disasters, power failures and numerous other factors. If we are unable to meet demand for TYSABRI for any reason, we would need to rely on a limited number of qualified third party contract manufacturers. We cannot be certain that we could reach agreement on reasonable terms, if at all, with those manufacturers or that the FDA or other regulatory authorities
25
would approve our use of such manufacturers on a timely basis, if at all. Moreover, the transition of our manufacturing process to a third party could take a significant amount of time, involve significant expense and increase our manufacturing costs.
We rely on third parties to provide services in connection with the manufacture of our products and, in some instances, manufacture the product itself.
We rely on Genentech for all RITUXAN manufacturing. Genentech relies on a third party to manufacture certain bulk RITUXAN requirements. If Genentech or any third party upon which it relies does not manufacture or fill-finish RITUXAN in sufficient quantities and on a timely and cost-effective basis, or if Genentech or any third party does not obtain and maintain all required manufacturing approvals, our business could be harmed.
We also source all of our fill-finish and the majority of our final product storage operations, along with a substantial portion of our packaging operations, to a concentrated group of third party contractors. Any third party we use to fill-finish, package or store our products to be sold in the U.S. must be licensed by the FDA. As a result, alternative third party providers may not be readily available on a timely basis or, if available, may be more costly than current providers. The manufacture of products and product components, fill-finish, packaging and storage of our products require successful coordination among us and multiple third party providers. Our inability to coordinate these efforts, the lack of capacity available at a third party contractor or any other problems with the operations of these third party contractors could require us to delay shipment of saleable products or recall products previously shipped or impair our ability to supply products at all. This could increase our costs, cause us to lose revenue or market share, diminish our profitability or damage our reputation.
Due to the unique manner in which our products are manufactured, we rely on single source providers of several raw materials. We make efforts to qualify new vendors and to develop contingency plans so that production is not impacted by short-term issues associated with single source providers. Nonetheless, our business could be materially impacted by long-term or chronic issues associated with single source providers.
Changes in laws affecting the health care industry could adversely affect our revenues and profitability.
We and our collaborators and third party providers operate in a highly regulated industry. As a result, governmental actions may adversely affect our business, operations or financial condition, including:
| • | new laws, regulations or judicial decisions, or new interpretations of existing laws, regulations or decisions, related to health care availability, method of delivery and payment for health care products and services; | |
| • | changes in the FDA and foreign regulatory approval processes that may delay or prevent the approval of new products and result in lost market opportunity; | |
| • | changes in FDA and foreign regulations that may require additional safety monitoring, labeling changes, restrictions on product distribution or use, or other measures after the introduction of our products to market, which could increase our costs of doing business, adversely affect the future permitted uses of approved products, or otherwise adversely affect the market for our products; | |
| • | new laws, regulations and judicial decisions affecting pricing or marketing practices; and | |
| • | changes in the tax laws relating to our operations. |
The enactment in the U.S. of health care reform, potential regulations easing the entry of competing follow-on biologics in the marketplace, new legislation or implementation of existing statutory provisions on importation of lower-cost competing drugs from other jurisdictions, and legislation on comparative effectiveness research are examples of previously enacted and possible future changes in laws that could adversely affect our business.
Our effective tax rate may fluctuate and we may incur obligations in tax jurisdictions in excess of accrued amounts.
As a global biotechnology company, we are subject to taxation in numerous countries, states and other jurisdictions. As a result, our effective tax rate is derived from a combination of applicable tax rates in the various
26
places that we operate. In preparing our financial statements, we estimate the amount of tax that will become payable in each of such places. Our effective tax rate, however, may be different than experienced in the past due to numerous factors, including changes in the mix of our profitability from country to country, the results of audits of our tax filings, changes in accounting for income taxes and changes in tax laws. Any of these factors could cause us to experience an effective tax rate significantly different from previous periods or our current expectations.
In addition, our inability to secure or sustain acceptable arrangements with tax authorities and previously enacted or future changes in the tax laws, among other things, may result in tax obligations in excess of amounts accrued in our financial statements.
In the U.S., there are several proposals under consideration to reform tax law, including proposals that may reduce or eliminate the deferral of U.S. income tax on our unrepatriated earnings, scrutinize certain transfer pricing structures, and reduce or eliminate certain foreign tax credits. Our future reported financial results may be adversely affected by tax law changes which restrict or eliminate certain foreign tax credits or deduct expenses attributable to foreign earnings, or otherwise affect the treatment of our unrepatriated earnings.
The growth of our business depends on our ability to attract and retain qualified personnel and key relationships.
The achievement of our commercial, research and development and external growth objectives depends upon our ability to attract and retain qualified scientific, manufacturing, sales and marketing and executive personnel and to develop and maintain relationships with qualified clinical researchers and key distributors. Competition for these people and relationships is intense and comes from a variety of sources, including pharmaceutical and biotechnology companies, universities and non-profit research organizations.
Our sales and operations are subject to the risks of doing business internationally.
We are increasing our presence in international markets, which subjects us to many risks, such as:
| • | the inability to obtain necessary foreign regulatory or pricing approvals of products in a timely manner; | |
| • | fluctuations in currency exchange rates; | |
| • | difficulties in staffing and managing international operations; | |
| • | the imposition of governmental controls; | |
| • | less favorable intellectual property or other applicable laws; | |
| • | restrictions on direct investments by foreign entities and trade restrictions; and | |
| • | changes in tax laws and tariffs. |
In addition, our international operations are subject to regulation under U.S. law. For example, the Foreign Corrupt Practices Act prohibits U.S. companies and their representatives from offering, promising, authorizing or making payments to foreign officials for the purpose of obtaining or retaining business abroad. In many countries, the health care professionals we regularly interact with may meet the definition of a foreign government official for purposes of the Foreign Corrupt Practices Act. Failure to comply with domestic or foreign laws could result in various adverse consequences, including possible delay in approval or refusal to approve a product, recalls, seizures, withdrawal of an approved product from the market, the imposition of civil or criminal sanctions and the prosecution of executives overseeing our international operations.
Uncertainty over intellectual property in the biotechnology industry has been the source of litigation, which is inherently costly and unpredictable.
We are aware that others, including various universities and companies working in the biotechnology field, have filed patent applications and have been granted patents in the U.S. and in other countries claiming subject matter potentially useful to our business. Some of those patents and patent applications claim only specific products or methods of making such products, while others claim more general processes or techniques useful or now used in
27
the biotechnology industry. There is considerable uncertainty within the biotechnology industry about the validity, scope and enforceability of many issued patents in the U.S. and elsewhere in the world, and, to date, there is no consistent policy regarding the breadth of claims allowed in biotechnology patents. We cannot currently determine the ultimate scope and validity of patents which may be granted to third parties in the future or which patents might be asserted to be infringed by the manufacture, use and sale of our products.
There has been, and we expect that there may continue to be, significant litigation in the industry regarding patents and other intellectual property rights. Litigation and administrative proceedings concerning patents and other intellectual property rights may be protracted, expensive and distracting to management. Competitors may sue us as a way of delaying the introduction of our products. Any litigation, including any interference proceedings to determine priority of inventions, oppositions to patents in foreign countries or litigation against our partners may be costly and time consuming and could harm our business. We expect that litigation may be necessary in some instances to determine the validity and scope of certain of our proprietary rights. Litigation may be necessary in other instances to determine the validity, scope or non-infringement of certain patent rights claimed by third parties to be pertinent to the manufacture, use or sale of our products. Ultimately, the outcome of such litigation could adversely affect the validity and scope of our patent or other proprietary rights or hinder our ability to manufacture and market our products.
If we are unable to adequately protect and enforce our intellectual property rights, our competitors may take advantage of our development efforts or our acquired technology.
We have filed numerous patent applications in the U.S. and various other countries seeking protection of the processes, products and other inventions originating from our research and development. Patents have been issued on many of these applications. We have also obtained rights to various patents and patent applications under licenses with third parties, which provide for the payment of royalties by us. The ultimate degree of patent protection that will be afforded to biotechnology products and processes, including ours, in the U.S. and in other important markets remains uncertain and is dependent upon the scope of protection decided upon by the patent offices, courts and lawmakers in these countries. Our patents may not afford us substantial protection or commercial benefit. Similarly, our pending patent applications or patent applications licensed from third parties may not ultimately be granted as patents and we may not prevail if patents that have been issued to us are challenged in court. In addition, pending legislation to reform the patent system and court decisions or patent office regulations that place additional restrictions on patent claims or that facilitate patent challenges could also reduce our ability to protect our intellectual property rights. If we cannot prevent others from exploiting our inventions, we will not derive the benefit from them that we currently expect.
We also rely upon unpatented trade secrets and other proprietary information, and we cannot ensure that others will not independently develop substantially equivalent proprietary information and techniques or otherwise gain access to our trade secrets or disclose such technology, or that we can meaningfully protect such rights. We require our employees, consultants, outside scientific collaborators, scientists whose research we sponsor and other advisers to execute confidentiality agreements upon the commencement of employment or consulting relationships with us. These agreements may not provide meaningful protection or adequate remedies for our unpatented proprietary information in the event of use or disclosure of such information.
If our products infringe the intellectual property rights of others, we may incur damages and be required to incur the expense of obtaining a license.
A substantial number of patents have already been issued to other biotechnology and pharmaceutical companies. To the extent that valid third party patent rights cover our products or services, we or our strategic collaborators would be required to seek licenses from the holders of these patents in order to manufacture, use or sell these products and services, and payments under them would reduce our profits from these products and services. We are currently unable to predict the extent to which we may wish or be required to acquire rights under such patents and the availability and cost of acquiring such rights, or whether a license to such patents will be available on acceptable terms or at all. There may be patents in the U.S. or in foreign countries or patents issued in the future that are unavailable to license on acceptable terms. Our inability to obtain such licenses may hinder our ability to manufacture and market our products.
28
Pending and future product liability claims may adversely affect our business and our reputation.
The administration of drugs in humans, whether in clinical studies or commercially, carries the inherent risk of product liability claims whether or not the drugs are actually the cause of an injury. Our products or product candidates may cause, or may appear to have caused, injury or dangerous drug interactions, and we may not learn about or understand those effects until the product or product candidate has been administered to patients for a prolonged period of time.
We are subject from time to time to lawsuits based on product liability and related claims. We cannot predict with certainty the eventual outcome of any pending or future litigation. We may not be successful in defending ourselves in the litigation and, as a result, our business could be materially harmed. These lawsuits may result in large judgments or settlements against us, any of which could have a negative effect on our financial condition and business if in excess of our insurance coverage. Additionally, lawsuits can be expensive to defend, whether or not they have merit, and the defense of these actions may divert the attention of our management and other resources that would otherwise be engaged in managing our business.
Our operating results are subject to significant fluctuations.
Our quarterly revenues, expenses and net income (loss) have fluctuated in the past and are likely to fluctuate significantly in the future due to the timing of charges and expenses that we may take. In recent periods, for instance, we have recorded charges that include:
| • | the cost of restructurings; | |
| • | impairments that we are required to take with respect to investments; | |
| • | impairments that we are required to take with respect to fixed assets, including those that are recorded in connection with the sale of fixed assets; | |
| • | inventory write-downs for failed quality specifications, charges for excess or obsolete inventory and charges for inventory write downs relating to product suspensions; | |
| • | milestone payments under license and collaboration agreements; and | |
| • | payments in connection with acquisitions and other business development activity. |
Our revenues are also subject to foreign exchange rate fluctuations due to the global nature of our operations. We recognize foreign currency gains or losses arising from our operations in the period in which we incur those gains or losses. Although we have foreign currency forward contracts to hedge specific forecasted transactions denominated in foreign currencies, our efforts to reduce currency exchange losses may not be successful. As a result, currency fluctuations among our reporting currency, the U.S. dollar, and the currencies in which we do business will affect our operating results, often in unpredictable ways. Our net income may also fluctuate due to the impact of charges we may be required to take with respect to foreign currency hedge transactions. In particular, we may incur higher charges from hedge ineffectiveness than we expect or from the termination of a hedge relationship.
These examples are only illustrative and other risks, including those discussed in these " Risk Factors ," could also cause fluctuations in our reported earnings. In addition, our operating results during any one period do not necessarily suggest the anticipated results of future periods.
Our portfolio of marketable securities is significant and subject to market, interest and credit risk that may reduce its value.
We maintain a significant portfolio of marketable securities. Changes in the value of this portfolio could adversely affect our earnings. In particular, the value of our investments may decline due to increases in interest rates, downgrades in the corporate bonds and other securities included in our portfolio, instability in the global financial markets that reduces the liquidity of securities included in our portfolio, declines in the value of collateral underlying the mortgage and asset-backed securities included in our portfolio, and other factors. Each of these events may cause us to record charges to reduce the carrying value of our investment portfolio or sell investments for
29
less than our acquisition cost. Although we attempt to mitigate these risks by investing in high quality securities and continuously monitoring our portfolio's overall risk profile, the value of our investments may nevertheless decline.
Our level of indebtedness could adversely affect our business and limit our ability to plan for or respond to changes in our business.
As of December 31, 2010, we had $1.2 billion of outstanding indebtedness, and we may incur additional debt in the future. Our level of indebtedness could adversely affect our business by, among other things:
| • | requiring us to dedicate a substantial portion of our cash flow from operations to payments on our indebtedness, thereby reducing the availability of our cash flow for other purposes, including business development efforts and research and development; | |
| • | limiting our flexibility in planning for, or reacting to, changes in our business and the industry in which we operate, thereby placing us at a competitive disadvantage compared to our competitors that may have less debt; and | |
| • | increasing our vulnerability to adverse economic and industry conditions. |
Our business involves environmental risks, which include the cost of compliance and the risk of contamination or injury.
Our business and the business of several of our strategic partners, including Genentech and Elan, involve the controlled use of hazardous materials, chemicals, biologics and radioactive compounds. Although we believe that our safety procedures for handling and disposing of such materials comply with state and federal standards, there will always be the risk of accidental contamination or injury. If we were to become liable for an accident, or if we were to suffer an extended facility shutdown, we could incur significant costs, damages and penalties that could harm our business. Biologics manufacturing also requires permits from government agencies for water supply and wastewater discharge. If we do not obtain appropriate permits, or permits for sufficient quantities of water and wastewater, we could incur significant costs and limits on our manufacturing volumes that could harm our business.
Several aspects of our corporate governance and our collaboration agreements may discourage a third party from attempting to acquire us.
Several factors might discourage a takeover attempt that could be viewed as beneficial to shareholders who wish to receive a premium for their shares from a potential bidder. For example:
| • | Our Board of Directors has the authority to issue, without a vote or action of shareholders, shares of preferred stock and to fix the price, rights, preferences and privileges of those shares, which shares could be used to dilute the interest of a potential bidder. | |
| • | Our collaboration agreements with Elan and Genentech respectively allow Elan to purchase our rights to TYSABRI and Genentech to purchase our rights to RITUXAN and certain anti-CD20 products developed under the agreement if we undergo a change of control and certain other conditions are met, which may limit our attractiveness to potential acquirers. | |
| • | Our directors are elected to staggered terms, which prevents the entire board from being replaced in any single year. |
The possibility that activist shareholders may gain additional representation on or control of our Board of Directors could result in costs and disruption to our operations and cause uncertainty about the direction of our business.
Entities affiliated with Carl Icahn commenced proxy contests in 2008, 2009 and 2010, resulting in three of their director nominees being elected to our Board of Directors. In addition, recent SEC rulemaking gives certain shareholders or groups of shareholders the ability to include director nominees and proposals relating to a shareholder nomination process in company proxy materials. As a result, we may face an increase in the number of shareholder nominees for election to our Board of Directors. Future proxy contests could be costly and time-
30
consuming, disrupt our operations and divert the attention of management and our employees from executing our strategic plan. Disagreement among our directors may create uncertainty regarding the direction of our business and could impair our ability to effectively execute our strategic plan.
| Item 1B. | Unresolved Staff Comments |
None.
| Item 2. | Properties |
Below is a summary of our owned and leased properties as of December 31, 2010. In connection with our recent restructuring initiative, described above under the heading "Overview - Framework for Growth," we are in the process of closing the San Diego, California facility and consolidating our Massachusetts facilities.
Massachusetts
In Cambridge, we own approximately 508,000 square feet of real estate space, consisting of a building that houses a research laboratory, office space and a cogeneration plant which total approximately 263,000 square feet and a building that contains research, development and quality laboratories totaling approximately 245,000 square feet.
In addition, we lease a total of approximately 885,000 square feet in Massachusetts, which is summarized as follows:
| • | 356,000 square feet of office space housing our principal executive offices in Weston; | |
| • | 347,000 square feet in Cambridge, which is comprised of a 67,000 square foot biologics manufacturing facility, laboratory space of 127,000 square feet and office space of 153,000 square feet; | |
| • | 105,000 square feet of office space in Wellesley, which we expect to vacate in the first quarter of 2011; | |
| • | 41,000 square feet of office and laboratory space in Waltham; and | |
| • | 36,000 square feet of warehouse space in Somerville. |
Our Massachusetts lease agreements expire at various dates through the year 2025.
California
On October 1, 2010, we sold the San Diego facility, which was comprised of 43 acres of land and buildings totaling approximately 355,000 square feet of laboratory and office space for cash proceeds, net of transaction costs, of approximately $127.0 million. As part of this transaction, we agreed to lease back the San Diego facility for a period of 15 months, however in January 2011, we entered into an agreement to terminate this lease effective August 31, 2011.
North Carolina
We manufacture bulk AVONEX, TYSABRI and other products in our pipeline at our facilities located in Research Triangle Park, North Carolina, where we own approximately 550,000 square feet of real estate space, which is summarized as follows:
| • | 175,000 square feet related to a large-scale biologics manufacturing facility; | |
| • | 167,000 square feet of laboratory and office space; | |
| • | 105,000 square feet related to a biologics manufacturing facility; | |
| • | 60,000 square feet of warehouse space; and | |
| • | 43,000 square feet related to a large-scale purification facility. |
In addition, we lease approximately 50,000 square feet of office space in Durham, North Carolina.
31
We are planning to increase the laboratory space in our Research Triangle Park campus and consolidate all of our North Carolina activities by moving local general and administrative offices and patient services to a 180,000 square foot office building to be built on the campus, with a planned occupancy around mid-year 2012.
Denmark
We own approximately 60 acres of land in Hillerød, Denmark, upon which we have been constructing a large-scale biologics manufacturing facility totaling approximately 225,000 square feet. We plan to stop further validation on this facility following completion of the facility's operational qualification activities in the first half of 2011 as we continue to evaluate our current manufacturing utilization strategy.
We own approximately 310,000 square feet of additional space, which is currently in use at this location and is summarized as follows:
| • | 140,000 square feet of warehouse, utilities and support space; | |
| • | 70,000 square feet related to a label and packaging facility; | |
| • | 50,000 square feet of administrative space; and | |
| • | 50,000 square feet related to a laboratory facility. |
Other International
We lease office and laboratory space in Zug, Switzerland, our international headquarters, the United Kingdom, Germany, France, Denmark, and numerous other countries. Our international lease agreements expire at various dates through the year 2023.
| Item 3. | Legal Proceedings |
Please refer to Note 20, Litigation to our consolidated financial statements included in this report, which is incorporated into this item by reference.
| Item 4. | [Reserved] |
32
PART II
| Item 5. | Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Market and Stockholder Information
Our common stock trades on The NASDAQ Global Select Market under the symbol "BIIB." The following table shows the high and low sales price for our common stock as reported by The NASDAQ Global Select Market for each quarter in the years ended December 31, 2010 and 2009:
| Common Stock Price | ||||||||||||||||
| 2010 | 2009 | |||||||||||||||
| High | Low | High | Low | |||||||||||||
First Quarter | $ | 60.28 | $ | 52.16 | $ | 53.66 | $ | 42.92 | ||||||||
Second Quarter | $ | 57.99 | $ | 45.96 | $ | 55.34 | $ | 44.56 | ||||||||
Third Quarter | $ | 58.64 | $ | 46.15 | $ | 52.12 | $ | 44.41 | ||||||||
Fourth Quarter | $ | 68.60 | $ | 55.63 | $ | 54.00 | $ | 41.75 | ||||||||
As of January 31, 2011, there were approximately 1,052 stockholders of record of our common stock.
In addition, as of January 31, 2011, 128 stockholders of record of Biogen, Inc. common stock have yet to exchange their shares of Biogen, Inc. common stock for our common stock as contemplated by the merger of Biogen, Inc. and IDEC Pharmaceuticals Corporation in November 2003.
Dividends
We have not paid cash dividends since our inception. We do not anticipate paying any cash dividends in the near term.
Issuer Purchases of Equity Securities
During the fourth quarter of 2010, we did not repurchase any common stock.
33
Stock Performance Graph
The graph below compares the five-year cumulative total stockholder return on our common stock, the S&P 500 Index and the Nasdaq Pharmaceutical Index, assuming the investment of $100.00 on December 31, 2005 with dividends being reinvested. The stock price performance in the graph below is not necessarily indicative of future price performance.
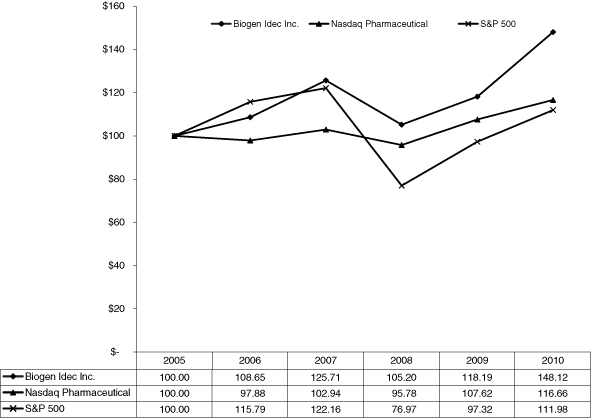
34
| Item 6. | Selected Consolidated Financial Data |
BIOGEN
IDEC INC. AND SUBSIDIARIES
SELECTED FINANCIAL DATA
| For the Years Ended December 31, | ||||||||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||
| (In millions, except per share amounts) | (8) (9) (10) (11) | (5) (6) (7) | (4) | (2) (3) | (1) | |||||||||||||||
Results of Operations | ||||||||||||||||||||
Product revenues | $ | 3,470.1 | $ | 3,152.9 | $ | 2,839.7 | $ | 2,136.8 | $ | 1,781.3 | ||||||||||
Revenue from unconsolidated joint business | 1,077.2 | 1,094.9 | 1,128.2 | 926.1 | 810.9 | |||||||||||||||
Other revenues | 169.1 | 129.5 | 129.6 | 108.7 | 90.8 | |||||||||||||||
Total revenues | 4,716.4 | 4,377.3 | 4,097.5 | 3,171.6 | 2,683.0 | |||||||||||||||
Cost and expenses: | ||||||||||||||||||||
Cost of sales, excluding amortization of acquired intangible assets | 400.3 | 382.1 | 402.0 | 335.2 | 274.4 | |||||||||||||||
Research and development | 1,248.6 | 1,283.1 | 1,072.1 | 925.2 | 718.4 | |||||||||||||||
Selling, general and administrative | 1,031.5 | 911.0 | 925.3 | 776.1 | 685.1 | |||||||||||||||
Collaboration profit sharing | 258.1 | 215.9 | 136.0 | 14.1 | (9.7 | ) | ||||||||||||||
Amortization of acquired intangible assets | 208.9 | 289.8 | 332.7 | 257.5 | 267.0 | |||||||||||||||
Restructuring charge | 75.2 | - | - | - | - | |||||||||||||||
Acquired in process research and development | 245.0 | - | 25.0 | 84.2 | 330.5 | |||||||||||||||
Facility impairments and gain on dispositions, net | - | - | (9.2 | ) | (0.4 | ) | (16.5 | ) | ||||||||||||
Gain on settlement of license agreements, net | - | - | - | - | (6.1 | ) | ||||||||||||||
Total costs and expenses | 3,467.5 | 3,081.9 | 2,883.9 | 2,391.8 | 2,243.0 | |||||||||||||||
Income from operations | 1,248.9 | 1,295.4 | 1,213.6 | 779.8 | 440.0 | |||||||||||||||
Other income (expense), net | (19.0 | ) | 37.3 | (57.7 | ) | 72.4 | 58.9 | |||||||||||||
Income before income tax expense and cumulative effect of accounting change | 1,229.9 | 1,332.7 | 1,155.9 | 852.2 | 498.9 | |||||||||||||||
Income tax expense | 331.3 | 355.6 | 365.8 | 272.4 | 278.4 | |||||||||||||||
Cumulative effect of accounting change, net of income tax | - | - | - | - | 3.8 | |||||||||||||||
Net income | 898.6 | 977.1 | 790.1 | 579.8 | 224.3 | |||||||||||||||
Net income (loss) attributable to noncontrolling interest, net of tax | (106.7 | ) | 6.9 | 6.9 | (58.4 | ) | 6.8 | |||||||||||||
Net income attributable to Biogen Idec Inc. | $ | 1,005.3 | $ | 970.1 | $ | 783.2 | $ | 638.2 | $ | 217.5 | ||||||||||
Diluted earnings per share | ||||||||||||||||||||
Income before cumulative effect of accounting change | $ | 3.94 | $ | 3.35 | $ | 2.65 | $ | 1.99 | $ | 0.62 | ||||||||||
Cumulative effect of accounting change, net of income tax | - | - | - | - | 0.01 | |||||||||||||||
Diluted earnings per share | $ | 3.94 | $ | 3.35 | $ | 2.65 | $ | 1.99 | $ | 0.63 | ||||||||||
Weighted-average shares used in calculating diluted earnings per share | 254.9 | 289.5 | 295.0 | 320.2 | 345.3 | |||||||||||||||
35
Financial Condition | ||||||||||||||||||||
Cash, cash equivalents and marketable securities | $ | 1,950.8 | $ | 2,457.8 | $ | 2,262.8 | $ | 2,115.8 | $ | 2,314.9 | ||||||||||
Total assets | $ | 8,092.5 | $ | 8,551.9 | $ | 8,479.0 | $ | 8,628.8 | $ | 8,552.8 | ||||||||||
Notes payable and line of credit, less current portion | $ | 1,066.4 | $ | 1,080.2 | $ | 1,085.4 | $ | 51.8 | $ | 96.7 | ||||||||||
Total Biogen Idec Inc. shareholders' equity | $ | 5,449.4 | $ | 6,221.5 | $ | 5,806.1 | $ | 5,534.3 | $ | 7,149.8 |
In addition to the following notes, the financial data included within the tables above should be read in conjunction with our consolidated financial statements and related notes and the "Management's Discussion and Analysis of Financial Condition and Results of Operations" sections of this report and our previously filed Forms 10-K. Certain totals may not sum due to rounding.
| (1) | Included in total cost and expenses in 2006 is a charge of $207.4 million for in process research and development from the acquisition of Fumapharm AG, a net gain of $6.1 million on the settlement of license agreements associated with Fumapharm AG and Fumedica GmbH and a charge of $123.1 million for in process research and development related to the acquisition of Conforma Therapeutics, Inc. | |
| (2) | Included in total cost and expenses in 2007 is a charge of $18.4 million for in process research and development related to the acquisition of Syntonix Pharmaceuticals Inc. and $64.3 million related to our collaborations with Cardiokine Biopharma LLC and Neurimmune SubOne AG, which we consolidated as we determined that we were the primary beneficiary of these relationships. The $64.3 million was offset by an equal amount of noncontrolling interest, resulting in no net impact to the results of our operations. | |
| (3) | In July 2007, we purchased approximately 56.4 million shares of our common stock pursuant to a tender offer. We funded the transaction through existing cash and cash equivalents of $1,490.5 million and a short term loan of $1,500.0 million. | |
| (4) | Included in total cost and expenses in 2008 is $25.0 million for in process research and development related to a milestone payment made to the former shareholders of Conforma Therapeutics pursuant to the terms of our acquisition of Conforma Therapeutics in 2006. | |
| (5) | Total cost and expenses in 2009 includes the $110.0 million upfront payment made to Acorda Therapeutics, Inc. pursuant to our June 30, 2009 collaboration and license agreement to develop and commercialize products containing fampridine in markets outside the U.S. | |
| (6) | Changes in tax law in certain state jurisdictions in which we operate and the resolution of multiple federal, state and foreign tax audits, including the effective settlement of several uncertain tax positions resulted in a $58.3 million reduction to our 2009 income tax expense. | |
| (7) | In 2009, we repurchased 16.0 million shares of our common stock at a cost of $751.2 million under our 2006 and 2009 share repurchase programs. | |
| (8) | Included in total cost and expenses in 2010 is a charge to acquired in process research and development of $40.0 million related to the achievement of a milestone by Biogen Idec Hemophilia, Inc. (formerly Syntonix Pharmaceuticals, Inc.). | |
| (9) | Included in total cost and expenses in 2010 is a charge to acquired in process research and development of $205.0 million incurred in connection with the license agreement entered into with Knopp Neurosciences Inc. (Knopp), which we consolidated as we determined that we are the primary beneficiary of the entity. The $205.0 million charge was partially offset by an attribution of $145.0 million to the noncontrolling interest. | |
| (10 ) | Net income attributable to noncontrolling interest also includes a charge of $25.0 million related to the payment made in 2010 to Cardiokine Biopharma LLC pursuant to the termination of our lixivaptan collaboration. | |
| (11) | During 2010, we repurchased approximately 40.3 million shares at a cost of approximately $2.1 billion under our 2010 and 2009 share repurchase authorizations. |
36
| Item 7. | Management's Discussion and Analysis of Financial Condition and Results of Operations |
The following discussion should be read in conjunction with our consolidated financial statements and related notes beginning on page F-1 of this report.
Executive Summary
Introduction
Biogen Idec is a global biotechnology company focused on discovering, developing, manufacturing and marketing products for the treatment of neurological disorders and other serious diseases. We have four marketed products: AVONEX, TYSABRI, RITUXAN and FUMADERM. Patients worldwide benefit from our significant products used for the treatment of multiple sclerosis (MS), non-Hodgkin's lymphoma (NHL), rheumatoid arthritis (RA), Crohn's disease, chronic lymphocytic leukemia (CLL) and psoriasis.
In the near term, our current and future revenues are dependent upon continued sales of our three principal products, AVONEX, RITUXAN and TYSABRI. In the longer term, our revenue growth will be dependent upon the successful pursuit of external business development opportunities and clinical development, regulatory approval and launch of new commercial products as well as upon our ability to protect our patents related to our marketed products and assets originating from our research and development efforts. As part of our ongoing research and development efforts, we have devoted significant resources to conducting clinical studies to advance the development of new pharmaceutical products and to explore the utility of our existing products in treating disorders beyond those currently approved in their labels.
On November 3, 2010, we announced a number of strategic, operational and organizational initiatives, which are described below under the heading "Restructuring Charges." We expect to incur charges totaling approximately $110.0 million associated with the implementation of these initiatives, which are anticipated to be substantially completed by the end of 2011.
Financial Highlights
The following table is a summary of financial results achieved:
| For the Years Ended | % Change | |||||||||||
| December 31, | 2010 | |||||||||||
| 2010 | 2009 | Compared | ||||||||||
| (In millions, except per share amounts and percentages) | (1) (2) | (3) | to 2009 | |||||||||
Total revenues | $ | 4,716.4 | $ | 4,377.3 | 7.7 | % | ||||||
Income from operations | $ | 1,248.9 | $ | 1,295.4 | (3.6 | )% | ||||||
Net income attributable to Biogen Idec Inc. | $ | 1,005.3 | $ | 970.1 | 3.6 | % | ||||||
Diluted earnings per share attributable to Biogen Idec Inc | $ | 3.94 | $ | 3.35 | 17.6 | % | ||||||
| (1) | Income from operations for 2010 was reduced by approximately $40.0 million related to the achievement of a milestone by Biogen Idec Hemophilia, Inc. (formerly Syntonix Pharmaceuticals, Inc.) and a $205.0 million charge incurred in connection with the collaboration and license agreement entered into with Knopp Neurosciences Inc. (Knopp), which we consolidated as we determined that we were the primary beneficiary of this relationship. The $205.0 million was partially offset by an attribution of $145.0 million to the noncontrolling interest. Net income attributable to noncontrolling interest also includes a charge of $25.0 million related to the payment made in 2010 to Cardiokine Biopharma LLC (Cardiokine) pursuant to the termination of our lixivaptan collaboration. | |
| (2) | Income from operations, as well as net income attributable to Biogen Idec Inc. for 2010, were reduced by the $75.2 million restructuring charge recognized during the fourth quarter of 2010. | |
| (3) | Income from operations for 2009 was reduced by the $110.0 million upfront payment made to Acorda Therapeutics, Inc. related to our collaboration and license agreement dated June 30, 2009. |
As described below under "Results of Operations ," our operating results for the year ended December 31, 2010, reflect the following:
| • | Worldwide AVONEX revenues totaled $2,518.4 million for 2010, representing an increase of 8.4% over 2009. |
37
| • | Our share of TYSABRI revenues totaled $900.2 million for 2010, representing an increase of 16.0% over 2009. | |
| • | Our share of RITUXAN revenues totaled $1,077.2 million for 2010, representing a decrease of 1.6% from 2009. This decrease was primarily driven by royalty expirations in our rest of world markets. Our share of revenue on sales of RITUXAN in the rest of world decreased 33.2% or $84.8 million from 2009. Our share of co-promotion profits in the U.S. increased 9.6% or $74.4 million over 2009. Selling and development expenses incurred by us and reimbursed by Genentech, which are also included within our total unconsolidated joint business revenues, decreased 11.1% to $58.3 million from the prior year comparative period. | |
| • | Total cost and expenses increased 12.5% for 2010, compared to 2009. This increase was primarily driven by the $245.0 million IPR&D charge and the $75.2 million restructuring charges recognized in 2010 as well as a 13.2% increase in selling, general and administrative costs and a 19.5% increase in collaboration profit sharing expense due to TYSABRI revenue growth, offset by a 27.9% decrease in amortization of acquired intangible assets. |
In addition, we generated $1,624.7 million of net cash flows from operations for 2010, which were primarily driven by earnings. Cash and cash equivalents and marketable securities totaled approximately $1,950.8 million as of December 31, 2010.
In 2010, we repurchased approximately 40.3 million shares at a cost of approximately $2.1 billion under our 2010 and 2009 share repurchase authorizations. We retired all of these shares as they were acquired. Our 2010 and 2009 share repurchase programs were completed during the third and first quarters of 2010, respectively.
Business Development Highlights
| • | In December 2010, we completed our acquisition of Panima Pharmaceuticals AG (Panima), an affiliate of Neurimmune AG. The purchase price is comprised of a $32.5 million cash payment, plus contingent consideration in the form of development milestones of up to $395.0 million in cash. Panima is involved in the discovery of antibodies designed to treat neurological disorders. For a more detailed description of this transaction, please read Note 2, Acquisitions to our consolidated financial statements included in this report. | |
| • | In October 2010, we amended our collaboration agreement with Genentech with regard to the development of ocrelizumab and agreed to terms for the development of GA101. Under the terms of the amended agreement, Genentech is responsible for the further development and commercialization of ocrelizumab and funding future costs. We will receive tiered royalties between 13.5% and 24% on U.S. sales of ocrelizumab. Commercialization of ocrelizumab will not impact our percentage of the co-promotion profits for RITUXAN. In addition, we will pay 35% of the development and commercialization expenses of GA101 and will receive between 35% and 39% of the profits of GA101 based upon the achievement of certain sales milestones. Commercialization of GA101 will impact our percentage of the co-promotion profits for RITUXAN. This amendment did not have an impact on our share of the co-promotion operating profits of RITUXAN in 2010. For a more detailed description of this collaboration, please read Note 19, Collaborations to our consolidated financial statements included in this report. | |
| • | In August 2010, we entered into a license agreement with Knopp Neurosciences, Inc. (Knopp), for the development, manufacture and commercialization of dexpramipexole, an orally administered small molecule in clinical development for the treatment of amyotrophic lateral sclerosis (ALS). Under the terms of the license agreement we made a $26.4 million upfront payment and agreed to pay Knopp up to an additional $265.0 million in development and sales-based milestone payments, as well as royalties on future commercial sales. For a more detailed description of this transaction, please read Note 18, Investments in Variable Interest Entities to our consolidated financial statements included in this report. |
Business Environment
We conduct our business primarily within the biotechnology and pharmaceutical industries, which are highly competitive. Many of our competitors are working to develop products similar to those we are developing or already market. We may also face increased competitive pressures as a result of the emergence of biosimilars. In the U.S., AVONEX, RITUXAN and TYSABRI are licensed under the Public Health Service Act (PHSA) as biological
38
products. In March 2010, U.S. healthcare reform legislation amended the PHSA to authorize the FDA to approve biological products, known as biosimilars or follow-on biologics, that are shown to be highly similar to previously approved biological products based upon potentially abbreviated data packages.
In addition, the recently enacted U.S. healthcare reform legislation contained additional provisions, including cost containment measures. We have encountered similar efforts to reform health care coverage and costs in other countries in which we operate. Moreover, the economic environment in Europe has become increasingly challenging. Many of the countries in which we operate are also seeking to reduce their public expenditures in light of the recent global economic downturn. The deterioration of the credit and economic conditions in certain countries in Europe has delayed reimbursement for our products and led to additional austerity measures aimed at reducing healthcare costs. Global efforts to reduce healthcare costs continue to exert pressure on product pricing and have negatively impacted our revenues and results of operations. For additional information about certain risks that could negatively impact our financial position or future results of operations, please read the "Risk Factors" section of this report.
Results of Operations
Revenues
Revenues are summarized as follows:
| % Change | ||||||||||||||||||||
| For the Years Ended | 2010 | 2009 | ||||||||||||||||||
| December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
Product revenue | ||||||||||||||||||||
United States | $ | 1,744.4 | $ | 1,638.0 | $ | 1,472.9 | 6.5 | % | 11.2 | % | ||||||||||
Rest of world | 1,725.7 | 1,514.9 | 1,366.8 | 13.9 | % | 10.8 | % | |||||||||||||
Total product revenues | 3,470.1 | 3,152.9 | 2,839.7 | 10.1 | % | 11.0 | % | |||||||||||||
Unconsolidated joint business | 1,077.2 | 1,094.9 | 1,128.2 | (1.6 | )% | (3.0 | )% | |||||||||||||
Other revenues | 169.1 | 129.5 | 129.6 | 30.6 | % | (0.1 | )% | |||||||||||||
Total revenues | $ | 4,716.4 | $ | 4,377.3 | $ | 4,097.5 | 7.7 | % | 6.8 | % | ||||||||||
Product Revenues
Product revenues are summarized as follows:
| % Change | ||||||||||||||||||||
| For the Years Ended | 2010 | 2009 | ||||||||||||||||||
| December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
AVONEX | $ | 2,518.4 | $ | 2,322.9 | $ | 2,202.6 | 8.4 | % | 5.5 | % | ||||||||||
TYSABRI | 900.2 | 776.0 | 588.6 | 16.0 | % | 31.8 | % | |||||||||||||
Other | 51.5 | 54.0 | 48.5 | (4.6 | )% | 11.3 | % | |||||||||||||
Total product revenues | $ | 3,470.1 | $ | 3,152.9 | $ | 2,839.7 | 10.1 | % | 11.0 | % | ||||||||||
39
AVONEX
Revenues from AVONEX are summarized as follows:
| % Change | ||||||||||||||||||||
| 2010 | 2009 | |||||||||||||||||||
| For the Years Ended December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
United States | $ | 1,491.6 | $ | 1,406.2 | $ | 1,276.5 | 6.1 | % | 10.2 | % | ||||||||||
Rest of world | 1,026.8 | 916.7 | 926.1 | 12.0 | % | (1.0 | )% | |||||||||||||
Total AVONEX revenues | $ | 2,518.4 | $ | 2,322.9 | $ | 2,202.6 | 8.4 | % | 5.5 | % | ||||||||||
For 2010 compared to 2009, as well as for 2009 compared to 2008, the increase in U.S. AVONEX revenue was due to price increases offset by decreased commercial demand. Decreased commercial demand resulted in declines of approximately 6% and 8% in U.S. AVONEX unit sales volume for 2010 and 2009, respectively, from the prior year comparative periods. Our 2010 U.S. AVONEX revenue was also negatively impacted by reserves established for rebates and allowances related to the newly enacted healthcare reform legislation in the U.S. In addition, we continued to experience higher participation in our Access Program, which provides free product to eligible patients for both the 2010 and 2009 comparative periods.
For 2010 compared to 2009, the increase in rest of world AVONEX revenue was due to increased commercial demand offset by price decreases in some countries and the negative impact of foreign currency exchange rates resulting from the relative strengthening of the U.S. dollar against relevant foreign currencies, primarily the Euro. For 2009 compared to 2008, the decrease in rest of world AVONEX revenue was primarily due to the negative impact of foreign exchange rate changes, offset by increased commercial demand and price increases in some countries. Increased commercial demand resulted in increases of approximately 6% in rest of world AVONEX sales volume for 2010 and 2009 in both periods.
AVONEX rest of world revenues for 2010 also include gains recognized in relation to the settlement of certain cash flow hedge instruments under our foreign currency hedging program totaling $35.0 million, compared to losses recognized of $39.5 million and $8.5 million for 2009 and 2008, respectively.
We expect AVONEX to face increasing competition in the MS marketplace in both the U.S. and rest of world. A number of companies, including us, are working to develop products to treat MS that may compete with AVONEX now and in the future, including oral and other alternative formulations. In addition, the continued growth of TYSABRI and the commercialization of our other pipeline product candidates may negatively impact future sales of AVONEX. Increased competition may lead to reduced unit sales of AVONEX, as well as increasing price pressure.
TYSABRI
We collaborate with Elan Pharma International, Ltd (Elan) an affiliate of Elan Corporation, plc, on the development and commercialization of TYSABRI. For a more detailed description of this collaboration, please read Note 19, Collaborations to our consolidated financial statements included in this report.
Revenues from TYSABRI are summarized as follows:
| % Change | ||||||||||||||||||||
| 2010 | 2009 | |||||||||||||||||||
| For the Years Ended December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
United States | $ | 252.8 | $ | 231.8 | $ | 196.4 | 9.1 | % | 18.0 | % | ||||||||||
Rest of world | 647.4 | 544.2 | 392.2 | 19.0 | % | 38.8 | % | |||||||||||||
Total TYSABRI revenues | $ | 900.2 | $ | 776.0 | $ | 588.6 | 16.0 | % | 31.8 | % | ||||||||||
For 2010 compared to 2009, as well as for 2009 compared to 2008, the increase in U.S. TYSABRI revenue was due to increased commercial demand. Increased commercial demand resulted in increases of approximately 10%
40
and 16% in U.S. TYSABRI unit sales volume for 2010 and 2009, respectively, over the prior year comparative periods. For 2010 compared to 2009, the increase was also due to price increases. This increase was offset by the impact of the sale of previously written-down TYSABRI inventory, which became saleable following the approval of our higher-yielding manufacturing process. As our sales price to Elan in the U.S. is set to effect an approximate equal sharing of the gross margin with Elan plus reimbursement for our cost of goods sold, the distribution of this specific inventory reduced our cost of sales, which reduced the price per unit we charged to Elan and reduced our revenues by $7.5 million compared to 2009. This inventory was fully utilized during 2010.
Net sales of TYSABRI from our collaboration partner, Elan, to third-party customers in the U.S. for 2010, 2009 and 2008 totaled $593.1 million, $508.5 million and $421.6 million, respectively.
For 2010 compared to 2009, as well as for 2009 compared to 2008, the increase in rest of world TYSABRI revenue was due to increased commercial demand of TYSABRI in our rest of world markets offset by the negative impact of foreign currency exchange rates resulting from the relative strengthening of the U.S. dollar against relevant foreign currencies, primarily the Euro. For 2010 compared to 2009, the increase in rest of world TYSABRI revenue was partially offset by price decreases in some countries. Increased commercial demand resulted in increases of 23% and 49% in rest of world TYSABRI sales volume for 2010 and 2009, respectively, over the prior year comparative periods.
TYSABRI rest of world revenues for 2010 also include gains recognized in relation to the settlement of certain cash flow hedge instruments under our foreign currency hedging program totaling $10.7 million, compared to losses recognized of $10.1 million for 2009. No such losses were recognized in 2008 as we did not designate hedges against TYSABRI rest of world revenues in that period.
The prescribing information for TYSABRI contains significant safety warnings, including:
| • | TYSABRI increases the risk of developing progressive multifocal leukoencephalopathy (PML), a serious brain infection. | |
| • | The risk of PML is increased in patients who have been treated with an immunosuppressant prior to receiving TYSABRI. | |
| • | The risk of developing PML increases with longer treatment duration, with limited experience beyond three years. | |
| • | Immune Reconstitution Inflammatory Syndrome (IRIS) may occur in patients who developed PML and subsequently discontinued TYSABRI. |
These safety warnings, and any future safety-related label changes, may limit the growth of TYSABRI unit sales. We continue to research and develop protocols and therapies that may reduce risk and improve outcomes of PML in patients. For example, our efforts have included working to identify patient or viral characteristics which contribute to the risk of developing PML, including the presence of asymptomatic JC virus infection with an assay to detect an immune response against the JC virus. We have initiated two clinical studies in the U.S., known as STRATIFY-1 and STRATIFY-2, that collectively, are intended to define the prevalence of serum JC virus antibody in patients with relapsing MS receiving or considering treatment with TYSABRI and to evaluate the potential to stratify patients into lower or higher risk for developing PML based on antibody status. Our efforts to stratify patients into lower or higher risk for developing PML, and other ongoing or future clinical trials involving TYSABRI may have a negative impact on prescribing behavior in at least the short term which may result in decreased product revenues from sales of TYSABRI.
We also expect TYSABRI to face increasing competition in the MS marketplace in both the U.S. and rest of world. A number of companies, including us, are working to develop products to treat MS that may compete with TYSABRI now and in the future, including oral and other alternative formulations. In addition, the commercialization of our other pipeline product candidates may negatively impact future sales of TYSABRI. Increased competition may also lead to reduced unit sales of TYSABRI, as well as increasing price pressure.
We have initiated the five year renewal process for TYSABRI's marketing authorization in the E.U. This marketing authorization review by E.U. regulators, in addition to ongoing label discussions with U.S. regulators,
41
includes assessment of the criteria for confirming PML diagnosis, the number of PML cases, the incidence of PML in TYSABRI patients, the risk factors for PML, as well as an overall assessment of TYSABRI's benefit-risk profile. Our interactions with E.U. and U.S. regulators could result in modifications to the respective labels or other restrictions for TYSABRI. Upon completion of the assessment of the TYSABRI renewal in the E.U. the marketing authorization is expected to be valid for either an unlimited period or for an additional five year term.
Other Product Revenues
Other product revenues primarily consist of revenues derived from sales of FUMADERM and are summarized as follows:
| % Change | ||||||||||||||||||||
| 2010 | 2009 | |||||||||||||||||||
| For the Years Ended December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
FUMADERM | $ | 51.2 | $ | 49.6 | $ | 43.4 | 3.2 | % | 14.3 | % | ||||||||||
Other | $ | 0.3 | $ | 4.4 | $ | 5.1 | (93.2 | )% | (13.7 | )% | ||||||||||
Total other product revenues | $ | 51.5 | $ | 54.0 | $ | 48.5 | (4.6 | )% | 11.3 | % | ||||||||||
Unconsolidated Joint Business Revenues
We collaborate with Genentech on the development and commercialization of RITUXAN. On October 19, 2010, we and Genentech amended and restated our Amended and Restated Collaboration Agreement dated June 19, 2003 with regard to the development of ocrelizumab, a humanized anti-CD20 antibody, and agreed to terms for the development of GA101, a next-generation anti-CD20 antibody. This amendment did not have an impact on our share of the co-promotion operating profits recognized for RITUXAN in 2010. For a more detailed description of this collaboration and additional information regarding the pretax co-promotion profit sharing formula for RITUXAN and its impact on future unconsolidated joint business revenues, please read Note 19, Collaborations to our consolidated financial statements included in this report.
In the fourth quarter of 2010, as part of our recent restructuring initiative, which is described below under the heading "Restructuring Charge," we reached an agreement with Genentech to eliminate our RITUXAN oncology and rheumatology sales force, with Genentech assuming the sole responsibility for the U.S. sales and marketing efforts related to RITUXAN. We believe that centralizing the sales force will enhance the sales effectiveness and profitability of our collaboration for the sale of RITUXAN in the U.S. As a result of this change, we expect that the amount of reimbursement for selling and development expense in the U.S. to decrease in future periods to a negligible amount. For 2010, 2009, and 2008, we were reimbursed $58.3 million, $65.6 million and $59.7 million, respectively, primarily for sales and marketing activities performed in support of RITUXAN.
Revenues from unconsolidated joint business are summarized as follows:
| % Change | ||||||||||||||||||||
| For the Years Ended | 2010 | 2009 | ||||||||||||||||||
| December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
Biogen Idec's share of co-promotion profits in the U.S. | $ | 848.0 | $ | 773.6 | $ | 733.5 | 9.6 | % | 5.5 | % | ||||||||||
Reimbursement of selling and development expenses in the U.S. | 58.3 | 65.6 | 59.7 | (11.1 | )% | 9.9 | % | |||||||||||||
Revenue on sales of RITUXAN in the rest of world | 170.9 | 255.7 | 335.0 | (33.2 | )% | (23.7 | )% | |||||||||||||
Total unconsolidated joint business revenues | $ | 1,077.2 | $ | 1,094.9 | $ | 1,128.2 | (1.6 | )% | (3.0 | )% | ||||||||||
42
Biogen Idec's Share of Co-Promotion Profits in the U.S.
The following table provides a summary of amounts comprising our share of co-promotion profits in the U.S.:
| % Change | ||||||||||||||||||||
| 2010 | 2009 | |||||||||||||||||||
| For the Years Ended December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
Product revenues, net | $ | 2,759.2 | $ | 2,665.5 | $ | 2,587.4 | 3.5 | % | 3.0 | % | ||||||||||
Costs and expenses | 626.8 | 724.1 | 741.0 | (13.4 | )% | (2.3 | )% | |||||||||||||
Co-promotion profits in the U.S. | $ | 2,132.4 | $ | 1,941.4 | $ | 1,846.4 | 9.8 | % | 5.1 | % | ||||||||||
Biogen Idec's share of co-promotion profits in the U.S. | $ | 848.0 | $ | 773.6 | $ | 733.5 | 9.6 | % | 5.5 | % | ||||||||||
For 2010 compared to 2009, as well as for 2009 compared to 2008, the increase in U.S. RITUXAN product revenues was primarily due to price increases and an increase in commercial demand, which resulted in an increase in unit sales volume of approximately 2%. For 2009 compared to 2008, the increase in U.S. RITUXAN product revenue was primarily due to price increases offset by a decrease in commercial demand of approximately 1%.
For 2010 compared to 2009, as well as for 2009 compared to 2008, the decrease in collaboration costs and expenses primarily resulted from a decline in expenditures for the development of RITUXAN for use in other indications. As described below under the heading "Provision for Discounts and Allowances - Healthcare Reform" , beginning in 2011, a new fee will be payable by all prescription drug manufacturers and importers. We estimate that the fee assessed Genentech on qualifying sales of RITUXAN will result in a reduction of our share of pre-tax co-promotion profits in the U.S. of approximately $15.0 million in 2011.
Under our collaboration agreement, our current pretax co-promotion profit-sharing formula, which resets annually, provides for a 40% share of co-promotion profits if co-promotion operating profits exceed $50.0 million. For 2010, 2009 and 2008, the 40% threshold was met during the first quarter.
Reimbursement of Selling and Development Expense in the U.S.
As discussed in Note 19, Collaborations to our consolidated financial statements included in this report, Genentech incurs the majority of continuing development costs for RITUXAN. Expenses incurred by Genentech in the development of RITUXAN are not recorded as research and development expense, but rather reduce our share of co-promotion profits recorded as a component of unconsolidated joint business revenue. For 2010 compared to 2009, the decrease in selling and development expenses incurred by us in the U.S. and reimbursed by Genentech was primarily the result of the elimination of our RITUXAN oncology and rheumatology sales force in the fourth quarter 2010. For 2009 compared to 2008, the increase in selling and development expenses incurred by us in the U.S. and reimbursed by Genentech was primarily the result of our increased sales and marketing activities in support of RITUXAN.
Revenue on Sales of RITUXAN in the Rest of World
Revenue on sales of RITUXAN in the rest of world consists of our share of pretax co-promotion profits in Canada and royalty revenue on sales of RITUXAN outside the U.S. and Canada. For 2010 compared to 2009, as well as for 2009 compared to 2008, revenues on sales of RITUXAN in the rest of world continue to decline due to royalty expirations in certain of our rest of world markets. The royalty period for sales in the rest of world with respect to all products is 11 years from the first commercial sale of such product on a country-by-country basis. Specifically, the royalty periods with respect to sales in France, Spain, Germany and the United Kingdom expired in 2009. The royalty period with respect to sales in Italy expired in 2010. The royalty periods for substantially all of the remaining royalty-bearing sales of RITUXAN in the rest of the world will expire through 2012. As a result of these expirations, we expect royalty revenues derived from sales of RITUXAN in the rest of world to continue to decline in future periods. The decreases experienced during 2010 were offset by a payment from Genentech totaling $21.3 million representing a cumulative underpayment of royalties owed to us on sales of RITUXAN in the rest of world.
43
Other Revenues
Other revenues are summarized as follows:
| % Change | ||||||||||||||||||||
| For the Years Ended | 2010 | 2009 | ||||||||||||||||||
| December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
Royalty revenues | $ | 137.4 | $ | 124.4 | $ | 116.2 | 10.5 | % | 7.1 | % | ||||||||||
Corporate partner revenues | 31.7 | 5.1 | 13.4 | 521.6 | % | (61.9 | )% | |||||||||||||
Total other revenues | $ | 169.1 | $ | 129.5 | $ | 129.6 | 30.6 | % | (0.1 | )% | ||||||||||
Royalty Revenues
We receive royalties on sales by our licensees of products covered under patents that we own. Sales of licensed products could vary significantly due to competition, manufacturing difficulties and other factors that are not within our control. In addition, the expiration or invalidation of any underlying patents could reduce or eliminate the royalty revenues derived from such patents.
For 2010 compared to 2009, as well as for 2009 compared to 2008, the increase in royalty revenues was primarily driven by increased sales of ANGIOMAX (bivalirudin) licensed to The Medicines Company (TMC). The increase for 2009 compared to 2008 was offset by a decline in royalties from sales of other licensed products and the expiration of certain contracts and license agreements.
Our most significant source of royalty revenue is derived from sales of ANGIOMAX by TMC. TMC sells ANGIOMAX in the U.S., Europe, Canada, Central America, South America, Israel and Australia. Royalty revenues related to the sales of ANGIOMAX are recognized in an amount equal to the level of net sales achieved during a calendar year multiplied by the royalty rate in effect for that tier under our agreement with TMC. The royalty rate increases based upon which tier of total net sales are earned in any calendar year. The increased royalty rate is applied retroactively to the first dollar of net sales achieved during the year. This formula has the effect of increasing the amount of royalty revenue to be recognized in later quarters and, as a result, an adjustment is recorded in the period in which an increase in royalty rate has been achieved.
Under the terms of our agreement, TMC is obligated to pay us royalties earned, on a country-by-country basis, until the later of (1) twelve years from the date of the first commercial sale of ANGIOMAX in such country or (2) the date upon which the product is no longer covered by a patent in such country. The annual royalty rate is reduced by a specified percentage in any country where the product is no longer covered by a patent and where sales have been reduced to a certain volume-based market share. TMC began selling ANGIOMAX in the U.S. in January 2001. The principal U.S. patent that covers ANGIOMAX was due to expire in March 2010 and TMC applied for an extension of the term of this patent. Initially, the U.S. Patent and Trademark Office (PTO) rejected TMC's application because in its view the application was not timely filed. TMC sued the PTO in federal district court seeking to extend the term of the principal U.S. patent to December 2014. On August 3, 2010, the federal district court ordered the PTO to deem the application as timely filed. The PTO did not appeal the order, but a generic manufacturer is seeking the right to intervene and file an appeal. The PTO has granted an interim extension of the patent term until August 13, 2011. In the event that TMC is unsuccessful in obtaining a patent term extension thereafter and third parties sell products comparable to ANGIOMAX, we would expect a significant decrease in royalty revenues due to increased competition, which may impact sales and result in lower royalty tiered rates.
Corporate Partner Revenues
We have also sold or exclusively licensed to third parties rights to certain products previously included within our product line. Royalty or supply agreement revenues received based upon those products are recorded as corporate partner revenue. Amounts recorded as corporate partner revenue also include amounts earned upon delivery of product under contract manufacturing agreements.
For 2010 compared to 2009, the increase in corporate partner revenues was primarily due to amounts earned under the terms of our 2006 contract manufacturing agreement with Astellas Pharma US, Inc. for the supply of
44
AMEVIVE. For 2009 compared to 2008, the decrease in corporate partner revenues was primarily due to milestone and royalty payments received in 2008 totaling $7.0 million related to ZEVALIN.
Provisions for Discounts and Allowances
Revenues from product sales are recorded net of applicable allowances for trade term discounts, wholesaler incentives, Medicaid rebates, Veterans Administration (VA) and Public Health Service (PHS) discounts, managed care rebates, product returns, and other applicable allowances. Reserves established for these discounts and allowances are classified as reductions of accounts receivable (if the amount is payable to our customer) or a liability (if the amount is payable to a party other than our customer). Reserves for discounts, contractual adjustments and returns that reduced gross product revenues are summarized as follows:
| % Change | ||||||||||||||||||||
| For the Years Ended | 2010 | 2009 | ||||||||||||||||||
| December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
Discounts | $ | 77.9 | $ | 74.0 | $ | 67.1 | 5.3 | % | 10.3 | % | ||||||||||
Contractual adjustments | 282.6 | 192.5 | 149.0 | 46.8 | % | 29.2 | % | |||||||||||||
Returns | 14.3 | 16.6 | 12.2 | (13.9 | )% | 36.1 | % | |||||||||||||
Total allowances | $ | 374.8 | $ | 283.1 | $ | 228.3 | 32.4 | % | 24.0 | % | ||||||||||
Gross product revenues | $ | 3,844.9 | $ | 3,436.0 | $ | 3,068.0 | 11.9 | % | 12.0 | % | ||||||||||
Percent of gross product revenues | 9.7 | % | 8.2 | % | 7.4 | % | 18.3 | % | 10.8 | % | ||||||||||
Discount reserves include trade term discounts and wholesaler incentives. For 2010 compared to 2009, as well as for 2009 compared to 2008, the increase in discounts was primarily driven by increases in trade term discounts and wholesaler incentives as a result of price increases and increased sales.
Contractual adjustment reserves relate to Medicaid and managed care rebates, VA and PHS discounts and other applicable allowances. For 2010 compared to 2009, as well as for 2009 compared to 2008, the increase in contractual adjustments was due to the impact of higher reserves for managed care and Medicaid and VA programs primarily associated with price increases in the U.S. For 2010 compared to 2009, the increase in contractual adjustments was also due to the impact of higher contractual rebates and discounts resulting from U.S. healthcare reform legislation passed in March 2010, as further discussed below.
Product return reserves are established for returns made by wholesalers. In accordance with contractual terms, wholesalers are permitted to return product for reasons such as damaged or expired product. The majority of wholesaler returns are due to product expiration. We also accept returns from our patients for various reasons. Reserves for product returns are recorded in the period the related revenue is recognized, resulting in a reduction to product sales. For 2010 compared to 2009, as well as for 2009 compared to 2008, return reserves remained relatively unchanged.
Healthcare Reform
In 2010, healthcare reform legislation was enacted in the U.S. This legislation contains several provisions that affect our business. Although many provisions of the new legislation do not take effect immediately, several provisions became effective in 2010. These include (1) an increase in the minimum Medicaid rebate to states participating in the Medicaid program from 15.1% to 23.1% on our branded prescription drugs; (2) the extension of the Medicaid rebate to Managed Care Organizations that dispense drugs to Medicaid beneficiaries; and (3) the expansion of the 340B PHS drug pricing program, which provides outpatient drugs at reduced rates, to include additional hospitals, clinics, and healthcare centers.
Beginning in 2011, the new law also requires drug manufacturers to provide a 50% discount to Medicare beneficiaries whose prescription drug costs cause them to be subject to the Medicare Part D coverage gap (i.e., the "donut hole"). Also, in 2011, a new fee will be payable by all branded prescription drug manufacturers and importers. This fee will be calculated based upon each organization's percentage share of total branded prescription
45
drug sales to qualifying U.S. government programs (such as Medicare, Medicaid and VA and PHS discount programs). As defined by the Act, "branded prescription drug sales" exclude the sales of any drug or biologic for which an orphan drug tax credit was allowed and was not subsequently approved for a non-orphan indication. As AVONEX has no other labeled indications, other than that for which it received its orphan designation, we believe that AVONEX sales are considered exempt from the fee. We estimate that the fee assessed to Genentech on qualifying sales of RITUXAN will result in a reduction of our share of pre-tax co-promotion profits in the U.S. of approximately $15.0 million in 2011. We will reflect our share of the fee assessed to Elan on qualifying sales of TYSABRI as selling, general and administrative expense, which we do not expect to be significant based on expected sales for qualifying U.S. government programs.
This new legislation contains a number of provisions that affect existing government programs and has required the creation of new programs, policies and processes, many of which remain under development and have not been fully implemented. For example, we do not yet fully know the extent of additional entities eligible to participate under the 340B program or when and how discounts will be provided to these entities. In addition, in November 2010, the Centers for Medicare and Medicaid Services (CMS) amended and then withdrew current regulations governing calculation of Average Manufacture Price; however, no replacement regulations have been proposed. Accordingly, our discounts and allowances are based on several assumptions about the implementation of this legislation. Actual results may differ from our estimates.
In addition, we anticipate that many countries outside the U.S. will continue to implement austerity measures including efforts aimed at reducing healthcare costs as these countries attempt to manage increasing healthcare expenditures, especially in light of the global economic downturn and the deterioration of the credit and economic conditions in certain countries in Europe. For example, certain governments of countries in which we operate have already implemented or may implement measures to reduce or control healthcare costs that, among other things, include imposed price reductions, suspensions on pricing increases on pharmaceuticals, increased mandatory discounts and rebates or seek recoveries of past price increases. Certain measures already implemented have negatively impacted our revenues. Our revenues and results of operations will be further negatively impacted if these, similar or more extensive measures continue to be implemented.
Cost and Expenses
A summary of total cost and expenses is as follows:
| % Change | ||||||||||||||||||||
| For the Years Ended | 2010 | 2009 | ||||||||||||||||||
| December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
Cost of sales, excluding amortization of acquired intangible assets | $ | 400.3 | $ | 382.1 | $ | 402.0 | 4.8 | % | (5.0 | )% | ||||||||||
Research and development | 1,248.6 | 1,283.1 | 1,072.1 | (2.7 | )% | 19.7 | % | |||||||||||||
Selling, general and administrative | 1,031.5 | 911.0 | 925.3 | 13.2 | % | (1.5 | )% | |||||||||||||
Collaboration profit sharing | 258.1 | 215.9 | 136.0 | 19.5 | % | 58.8 | % | |||||||||||||
Amortization of acquired intangible assets | 208.9 | 289.8 | 332.7 | (27.9 | )% | (12.9 | )% | |||||||||||||
Restructuring charge | 75.2 | - | - | ** | ** | |||||||||||||||
Acquired in process research and development | 245.0 | - | 25.0 | ** | (100.0 | )% | ||||||||||||||
Gain on dispositions, net | - | - | (9.2 | ) | ** | (100.0 | )% | |||||||||||||
Total cost and expenses | $ | 3,467.5 | $ | 3,081.9 | $ | 2,883.9 | 12.5 | % | 6.9 | % | ||||||||||
46
Cost of Sales, Excluding Amortization of Acquired Intangible Assets (Cost of Sales)
| % Change | ||||||||||||||||||||
| For the Years Ended | 2010 | 2009 | ||||||||||||||||||
| December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
Cost of sales, excluding amortization of acquired intangible assets | $ | 400.3 | $ | 382.1 | $ | 402.0 | 4.8 | % | (5.0 | )% | ||||||||||
For 2010 compared to 2009, the increase in cost of sales was primarily due to higher unit sales volume. The increase for the comparative period was also driven by a $5.7 million increase in costs associated with contract manufacturing activity for the supply of AMEVIVE as well as $6.7 million of period expense incurred related to the shutdown for capital upgrades of our manufacturing facility in Research Triangle Park, North Carolina. This comparative increase was offset by the sale of previously written-down TYSABRI inventory, which became saleable following approval of our new higher-yielding manufacturing process. The distribution of this inventory, which was fully utilized during 2010, reduced our cost of sales by $11.4 million compared to 2009. In addition, the sale of inventory produced under our new high-titer production process reduced our cost of sales by $8.4 million compared to 2009.
For 2009 compared to 2008, the decrease in cost of sales was primarily due to a $12.9 million decrease in write-downs from unmarketable inventory, a $10.9 million decrease in production costs due to the implementation of a new high-titer production process which produces higher yields of TYSABRI and an $8.8 million decrease in royalty payments on sales of licensed product due mainly to the expiration of certain contracts and license agreements. These decreases were offset by a $17.0 million increase in costs associated with higher TYSABRI unit sales volume. In addition, during 2008 we also incurred a $4.3 million period expense related to the shutdown of our manufacturing facility in Research Triangle Park, North Carolina for the implementation of the high-titer production process upgrades.
We expect an increase in total cost of sales for 2011, as a result of an increase in expected contract manufacturing activity and increased production costs.
Write-downs from Unmarketable Inventory
Our products are subject to strict quality control and monitoring which we perform throughout the manufacturing process. Periodically, certain batches or units of product may no longer meet quality specifications or may expire. The expiry associated with our inventory is generally between 6 months and 5 years, depending on the product. Obsolescence due to expiration has historically been insignificant.
Amounts written down related to unmarketable inventory are charged to cost of sales, and totaled $11.8 million, $16.9 million and $29.8 million for the years ended December 31, 2010, 2009 and 2008, respectively.
Research and Development
| % Change | ||||||||||||||||||||
| For the Years Ended | 2010 | 2009 | ||||||||||||||||||
| December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
Research and development | $ | 1,248.6 | $ | 1,283.1 | $ | 1,072.1 | (2.7 | )% | 19.7 | % | ||||||||||
For 2010 compared to 2009, research and development expense decreased by $34.5 million. Our research and development spend in 2010 included a $26.4 million upfront payment made to Knopp under a license agreement, increased clinical activity for our daclizumab, PEGylated interferon beta-1a, Neublastin, Factor VIII and Factor IX programs, and efforts to research and develop protocols that may reduce risk and improve outcomes of PML in patients treated with TYSABRI. In addition, our costs for the Factor VIII and Factor IX programs increased in 2010 following the restructuring of our collaboration agreement with Swedish Orphan Biovitrum, whereby we assumed full development and manufacturing responsibilities for these programs. These increases were offset by a reduction in spending in certain deprioritized programs.
47
For 2009 compared to 2008, research and development expenses increased by $211.0 million, driven primarily by the $110.0 million upfront payment made to Acorda, as well as a net increase of $100.2 million related to the ramp up of clinical trial activity for certain development stage product candidates including lixivaptan, BG-12, humanized anti-CD20 and ADENTRI. In addition, in 2009, we also initiated registrational trials in our PEGylated interferon program. The aforementioned increases were offset by a reduction of spending across several programs including baminercept in RA, lumiliximab and volociximab.
As part of our recent restructuring initiative, which is described below under the heading " Restructuring Charge ," we are in the process of reducing our overall headcount by approximately 13% and have terminated or are in the process of discontinuing certain research and development programs, including substantially all of our cardiovascular and oncology programs and select programs in neurology and immunology. Our workforce reduction efforts impact all sales, research and development and administrative functions.
We expect total research and development expense in 2011 to be between 22% and 24% of total revenue.
Milestone and Upfront Payments
Milestone and upfront payments to our collaboration partners, included within research and development expense, totaled $68.9 million, $151.5 million and $47.6 million for 2010, 2009 and 2008, respectively. The change for each of the comparative periods was primarily the result of the $110.0 million upfront payment made to Acorda in 2009. The timing of future upfront fees and milestone payments may cause variability in future research and development expense.
Selling, General and Administrative
| % Change | ||||||||||||||||||||
| For the Years Ended | 2010 | 2009 | ||||||||||||||||||
| December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
Selling, general and administrative | $ | 1,031.5 | $ | 911.0 | $ | 925.3 | 13.2 | % | (1.5 | )% | ||||||||||
Selling, general and administrative expenses are primarily comprised of compensation and benefits associated with sales and marketing, finance, legal and other administrative personnel, outside marketing and legal expenses and other general and administrative costs.
For 2010 compared to 2009, selling, general and administrative expenses increased primarily due to increased sales and marketing activities in support of AVONEX and TYSABRI and increased grant and sponsorship activity. The increase for the comparative periods also includes an incremental charge of approximately $18.6 million recognized in 2010 related to the modification of equity based compensation in accordance with the transition agreement entered into with James C. Mullen, who retired as our President and Chief Executive Officer on June 8, 2010.
For 2009 compared to 2008, the decrease in selling, general and administrative expenses was primarily driven by the positive impact of foreign currency exchange rates and a reduction of expenses reimbursed to Elan for their marketing of TYSABRI for Crohn's disease in the U.S. These decreases were offset by costs incurred associated with our geographic expansion into new markets.
As part of our recent restructuring initiative, which is described below under the heading "Restructuring Charge," we are in the process of reducing our overall headcount by approximately 13%. This workforce reduction impacts all sales, research and development and administrative functions.
We expect total selling, general and administrative expense in 2011 to be between 20% and 21% of total revenue.
48
Collaboration Profit Sharing
| % Change | ||||||||||||||||||||
| For the Years Ended | 2010 | 2009 | ||||||||||||||||||
| December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
Collaboration profit sharing | $ | 258.1 | $ | 215.9 | $ | 136.0 | 19.5 | % | 58.7 | % | ||||||||||
For 2010 compared to 2009, as well as for 2009 compared to 2008, the increases in collaboration profit sharing expense were due to the continued increase in TYSABRI rest of world sales resulting in higher rest of world net operating profits to be shared with Elan and resulting in growth in the third-party royalties Elan paid on behalf of the collaboration. For 2010, 2009 and 2008, our collaboration profit sharing expense included $45.5 million, $40.0 million and $28.4 million related to the reimbursement of third-party royalty payments made by Elan. For a more detailed description of this collaboration, please read Note 19, Collaborations to our consolidated financial statements included in this report.
Amortization of Acquired Intangible Assets
| % Change | ||||||||||||||||||||
| For the Years Ended | 2010 | 2009 | ||||||||||||||||||
| December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
Amortization of acquired intangible assets | $ | 208.9 | $ | 289.8 | $ | 332.7 | (27.9 | )% | (12.9 | )% | ||||||||||
Our most significant intangible asset is the core technology related to our AVONEX product. Our amortization policy reflects our belief that the economic benefit of our core technology is consumed as revenue is generated from our AVONEX product. We refer to this amortization methodology as the economic consumption model, which involves calculating a ratio of actual current period sales to total anticipated sales for the life of the product and applying this ratio to the carrying amount of the intangible asset. An analysis of the anticipated lifetime revenue of AVONEX is performed at least annually during our long range planning cycle, and this analysis serves as the basis for the calculation of our economic consumption amortization model. Although we believe this process has allowed us to reliably determine the best estimate of the pattern in which we will consume the economic benefits of our core technology intangible asset, the model could result in deferring amortization charges to future periods in certain instances, due to continued sales of the product at a nominal level after patent expiration or otherwise. In order to ensure that amortization charges are not unreasonably deferred to future periods, we compare the amount of amortization determined under the economic consumption model against the minimum amount of amortization recalculated each year under the straight-line method and record the higher amount.
We completed our most recent long range planning cycle in the third quarter of 2010. This analysis is based upon certain assumptions that we evaluate on a periodic basis, such as the anticipated product sales of AVONEX and expected impact of competitor products and our own pipeline product candidates, as well as the issuance of new patents or the extension of existing patents. Based upon this analysis, we have continued to amortize this asset on the economic consumption model for the third and fourth quarters of 2010, and expect to apply the same model for the next two quarters. In addition, since we do not currently expect a significant change in the expected lifetime revenue of AVONEX, amortization expected to be recorded in relation to our core intangible asset for the first two quarters of 2011 is anticipated to be comparable to the amounts recorded during the third and fourth quarters of 2010. Amortization of our core intangible asset related to AVONEX totaled $162.4 million, $229.3 million and $271.7 million in 2010, 2009 and 2008, respectively.
For 2009 compared to 2008, amortization recorded for the third and fourth quarters of 2009 decreased significantly from their respective prior year comparative periods. This decrease was driven by the issuance of the AVONEX '755 Patent in September 2009. The issuance of this patent, expiring in September 2026, resulted in an increase in the total expected lifetime revenue of AVONEX and an extension of the assumed remaining life of our core intangible asset.
Based upon our most recent analysis, amortization for acquired intangible assets is expected to be in the range of approximately $170.0 million to $210.0 million annually through 2015.
49
We monitor events and expectations on product performance. If there are any indications that the assumptions underlying our most recent analysis would be different than those utilized within our current estimates, our analysis would be updated and may result in a significant change in the anticipated lifetime revenue of AVONEX determined during our most recent annual review. For example, the occurrence of an adverse event, such as the invalidation of our AVONEX '755 Patent issued in September 2009, could substantially increase the amount of amortization expense associated with our acquired intangible assets as compared to previous periods or our current expectations, which may result in a significant negative impact on our future results of operations.
Restructuring Charge
| % Change | ||||||||||||||||||||
| For the Years Ended | 2010 | 2009 | ||||||||||||||||||
| December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
Restructuring charge | $ | 75.2 | $ | - | $ | - | ** | ** | ||||||||||||
On November 3, 2010, we announced a number of strategic, operational and organizational initiatives designed to provide a framework for the future growth of our business, which are summarized as follows:
| • | We intend to focus our business on neurology and leverage our strengths in biologics research, development and manufacturing to pursue select biological therapies where there is a significant unmet need and where the drug candidate has the potential to be highly differentiated. Accordingly, during the fourth quarter of 2010, we began to reallocate resources within our research and development organization to maximize our investment in our highest-potential programs. As a result, we have terminated or are in the process of discontinuing certain research and development programs, including substantially all of our oncology programs (which we are looking to spin out or out-license), our cardiovascular programs and select neurology and immunology programs. In addition, we have substantially reduced our small molecule discovery activities in favor of outsourcing these efforts. | |
| • | We are in the process of vacating the San Diego, California facility and consolidating our Massachusetts facilities. | |
| • | We eliminated our RITUXAN oncology and rheumatology sales force and Genentech, Inc., a wholly-owned member of the Roche Group, has assumed sole responsibility for the U.S. sales and marketing efforts related to RITUXAN. | |
| • | We are in the process of completing a 13% reduction in our workforce and realigning our overall structure to become a more efficient and cost-effective organization. The workforce reduction spans our sales, research and development and administrative functions. |
As a result of these initiatives, we expect to realize annual savings of approximately $300.0 million. The substantial majority of the savings will be realized within research and development and selling, general and administrative expense and are expected to be fully realized beginning in the latter half of 2011. These expected savings may be offset to some degree by costs associated with initiatives to grow our business.
We expect to incur total restructuring charges of approximately $110.0 million, comprised of $90.0 million for workforce reduction and $20.0 million for facility consolidation.
We recognized $75.2 million of these charges within our consolidated statement of income during 2010, which are summarized as follows:
| For the Year Ended | ||||
| (In millions) | December 31, 2010 | |||
Workforce reduction | $ | 67.2 | ||
Facility consolidation | 8.0 | |||
Total restructuring charges | $ | 75.2 | ||
50
We expect that our restructuring efforts will be substantially completed, and that substantially all of the remaining restructuring charges will be incurred by the end of 2011.
Costs associated with our workforce reduction primarily relate to employee severance and benefits. Facility consolidation costs are primarily comprised of charges associated with the closing of facilities, related lease obligations and additional depreciation recognized when the expected useful lives of certain assets have been shortened due to the consolidation and closing of related facilities and the discontinuation of certain research and development programs.
The following table summarizes the charges and spending related to our restructuring efforts during 2010:
| Workforce | Facility | |||||||||||
| (In millions) | Reduction | Consolidation | Total | |||||||||
Reserves established | $ | 67.2 | $ | 8.0 | $ | 75.2 | ||||||
Amounts paid | (6.6 | ) | - | (6.6 | ) | |||||||
Additional depreciation and other non-cash charges | - | (2.2 | ) | (2.2 | ) | |||||||
Restructuring reserves at December 31, 2010 | $ | 60.6 | $ | 5.8 | $ | 66.4 | ||||||
We expect that substantially all remaining payments will be made, by the end of 2011.
Acquired In Process Research and Development (IPR&D)
| % Change | ||||||||||||||||||||
| For the Years Ended | 2010 | 2009 | ||||||||||||||||||
| December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
Acquired in process research and development | $ | 245.0 | $ | - | $ | 25.0 | ** | (100.0 | )% | |||||||||||
In August 2010, we entered into a license agreement with Knopp for the development, manufacture and commercialization of dexpramipexole, an orally administered small molecule in clinical development for the treatment of ALS. As we determined that we are the primary beneficiary of Knopp, we consolidate the results of Knopp and recorded an IPR&D charge of approximately $205.0 million upon initial consolidation. We have attributed approximately $145.0 million of the total IPR&D charge to the noncontrolling interest, representing the noncontrolling interest's ownership interest in the equity of Knopp. For a more detailed description of this transaction, please read Note 18, Investments in Variable Interest Entities to our consolidated financial statements included in this report.
In connection with our acquisition of Biogen Idec Hemophilia Inc., formerly Syntonix Pharmaceuticals, Inc. (Syntonix), in January 2007, we agreed to make additional future consideration payments based upon the achievement of certain milestone events. One of these milestones was achieved when, in January 2010, we initiated patient enrollment in a registrational trial of Factor IX in hemophilia B. As a result of the achievement of this we paid approximately $40.0 million to the former shareholders of Syntonix.
In 2008, we recorded an IPR&D charge of $25.0 million related to a HSP90-related milestone payment made to the former shareholders of Conforma Therapeutics, Inc. (Conforma) pursuant to the terms of our acquisition of Conforma in 2006.
51
Other Income (Expense), Net
Components of other income (expense), net, are summarized as follows:
| % Change | ||||||||||||||||||||
| For the Years Ended | 2010 | 2009 | ||||||||||||||||||
| December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
Interest income | $ | 22.3 | $ | 48.5 | $ | 72.1 | (54.0 | )% | (32.7 | )% | ||||||||||
Interest expense | (36.1 | ) | (35.8 | ) | (52.0 | ) | (0.8 | )% | 31.2 | % | ||||||||||
Impairments of investments | (21.3 | ) | (10.6 | ) | (60.3 | ) | (100.9 | )% | 82.4 | % | ||||||||||
Gain (loss) on sales of investments, net | 16.3 | 22.8 | (1.1 | ) | (28.5 | )% | 2172.7 | % | ||||||||||||
Foreign exchange gains (losses), net | (3.5 | ) | 11.4 | (9.8 | ) | (130.7 | )% | 216.3 | % | |||||||||||
Other, net | 3.3 | 1.0 | (6.6 | ) | 230.0 | % | 114.9 | % | ||||||||||||
Total other income (expense), net | $ | (19.0 | ) | $ | 37.3 | $ | (57.7 | ) | (151.0 | )% | 164.5 | % | ||||||||
Interest Income
For 2010 compared to 2009, as well as for 2009 compared to 2008, interest income decreased primarily due to lower yields on cash, cash equivalents, and marketable securities. The decrease for 2010 compared to 2009, was also due to lower average cash balances. For 2009 compared to 2008, these decreases were offset by higher average cash balances.
Interest Expense
For 2010 compared to 2009, interest expense remained relatively unchanged. For 2009 compared to 2008, interest expense decreased primarily due to decreased average debt balances. In addition, approximately $5.7 million and $5.4 million was recorded in 2010 and 2009, respectively, as a reduction of interest expense due to the amortization of the deferred gain associated with the termination of an interest rate swap in December 2008.
Capitalized Interest Costs
For 2010, 2009, and 2008, we capitalized interest costs related to construction in progress totaling approximately $28.6 million, $28.5 million and $23.2 million, respectively, which reduced our interest expense by the same amount. Capitalized interest costs are primarily related to the development of our large-scale biologic manufacturing facility in Hillerød, Denmark.
We plan to stop further validation on this facility following completion of facility's operational qualification activities in the first half of 2011 as we continue to evaluate our current manufacturing utilization strategy. Recent manufacturing improvements have resulted in favorable production yields on TYSABRI, that along with slower than expected TYSABRI growth, have reduced our expected capacity requirements. As a result, we have decided to delay the start of manufacturing activities at this site until additional capacity is required by the business. Accordingly, we expect to cease capitalizing interest in relation to this project at that time.
Impairment on Investments
In 2010, we recognized $21.3 million in charges for the other-than-temporary impairment of our publicly held strategic investments, investments in venture capital funds and investments in privately held companies. The increase over amounts recognized in 2009 was primarily the result of AVEO Pharmaceuticals, Inc., one of our strategic investments, executing an equity offering at a price below our cost basis during the first quarter of 2010.
In 2009, we recognized impairment losses of $7.0 million on our publicly-held strategic investments and non-marketable securities and an additional $3.6 million in charges for the other-than-temporary impairment on our marketable debt securities primarily related to mortgage and asset-backed securities.
52
In 2008, we recognized impairment losses of $18.6 million on our publicly-held strategic investments and non-marketable securities and an additional $41.7 million in impairment on our marketable debt securities primarily related to mortgage and asset-backed and corporate securities.
We may incur additional impairment charges on these investments in the future.
Income Tax Provision
| % Change | ||||||||||||||||||||
| 2010 | 2009 | |||||||||||||||||||
| For the Years Ended December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
Effective tax rate on pre-tax income | 26.9 | % | 26.7 | % | 31.6 | % | 0.7 | % | (15.5 | )% | ||||||||||
Income tax expense | $ | 331.3 | $ | 355.6 | $ | 365.8 | (6.8 | )% | (2.8 | )% | ||||||||||
Our effective tax rate fluctuates from year to year due to the nature of our global operations. The factors that most significantly impact our effective tax rate include variability in the allocation of our taxable earnings between multiple jurisdictions, changes in tax laws, acquisitions and licensing transactions.
For 2010 compared to 2009, our effective tax rate was negatively impacted due to the attribution to noncontrolling interest of $145.0 million of the IPR&D charge related to our license agreement with Knopp Neurosciences, Inc. As such, the attributed amount will not generate a tax deduction, causing our tax rate to be unfavorably impacted by 2.8%. The impact of the Knopp transaction was partially offset by a higher percentage of our profits being earned in lower rate international jurisdictions in 2010. This change in the location of our relative profits was caused by the growth of our international operations and lower 2010 domestic earnings as a proportion of total consolidated earnings. For a more detailed description of our transaction with Knopp, please read Note 18, Investments in Variable Interest Entities to our consolidated financial statements included in this report.
During 2010, we also experienced a favorable impact on our effective tax rates due to a statutory increase in the U.S. manufacturers' tax deduction and an increase in expenditures eligible for our orphan drug credit. In December 2010, an extension of the research and development tax credit was enacted for years 2010 and 2011. Upon enactment, we recognized an income tax benefit of $14.9 million for qualifying expenditures from the full year 2010. In addition, our 2009 effective tax rate was increased by 2.1% as a result of the $110.0 million upfront payment incurred in connection with the collaboration and license agreement entered into with Acorda Therapeutics, Inc. (Acorda) in the second quarter of 2009. Our effective tax rate for 2009 was also favorably impacted by 2.3% for changes in tax law which became effective during the first quarter of 2009 in certain state jurisdictions in which we operate and the favorable resolution of certain federal, state and foreign tax audits. The resolution of these tax audits resulted in a reduction of our reserves for several uncertain tax positions, which had a favorable impact of 2.1% on our 2009 effective tax rate.
Our effective tax rate in 2009 was lower than in 2008 due to the net effect of changes in tax laws and the resolution of certain tax audits discussed above, as well as a higher percentage of our foreign earnings being subject to U.S. income taxation in 2008 partially offset by the effect of the Acorda licensing transaction. The effect of the allocation of earnings was partially offset by certain tax credits and deferred tax assets realized as a result of our 2008 domestic reorganization.
Our 2008 domestic and foreign reorganizations to our corporate structure involved the movement of certain personnel, operations and processes amongst our affiliates. Our effective tax rate will continue to be dependent upon the allocation of our profits amongst jurisdictions and the percentage of our foreign earnings which are subject to taxation in the U.S. We expect our 2011 effective tax rate to be between 26% and 28%.
53
Noncontrolling Interest
| % Change | ||||||||||||||||||||
| 2010 | 2009 | |||||||||||||||||||
| For the Years Ended December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
Net income (loss) attributable to noncontrolling interests, net of tax | $ | (106.7 | ) | $ | 6.9 | $ | 6.9 | (1,639.7 | )% | 0.0 | % | |||||||||
For 2010 compared to 2009, net income attributable to noncontrolling interests decreased by $113.6 million. This decrease was primarily the result of the attribution of $145.0 million of the $205.0 million IPR&D charge recognized upon consolidation of the Knopp variable interest entity to the noncontrolling interest. This decrease was partially offset by the $25.0 million payment made to Cardiokine upon the termination of our license agreement and an attribution of earnings from our foreign joint ventures.
For 2009 compared to 2008, net income (loss) attributable to noncontrolling interests primarily consisted of the attribution of earnings from our foreign joint ventures, which were relatively consistent in each year.
Market Risk
We conduct business globally. As a result, our international operations are subject to certain opportunities and risks which may affect our results of operations, including volatility in foreign currency exchange rates or weak economic conditions in the foreign market in which we operate.
Foreign Currency Exchange Risk
Our results of operations are subject to foreign currency exchange rate fluctuations due to the global nature of our operations. While the financial results of our global activities are reported in U.S. dollars, the functional currency for most of our foreign subsidiaries is their local currency. Fluctuations in the foreign currency exchange rates of the countries in which we do business will affect our operating results, often in ways that are difficult to predict. For example, when the U.S. dollar strengthens against foreign currencies, the relative value of sales made in the respective foreign currencies decreases, conversely, when the U.S. dollar weakens against foreign currencies, the relative amount of such sales in U.S. dollars increases.
Our net income may also fluctuate due to the impact of our foreign currency hedging program. Our foreign currency management program is designed to mitigate, over time, a portion of the impact on volatility in exchange rate changes on net income and earnings per share. We use foreign currency forward contracts to manage foreign currency risk with the majority of our forward contracts used to hedge certain forecasted revenue transactions denominated in foreign currencies. Foreign currency gains or losses arising from our operations are recognized in the period in which we incur those gains or losses.
Pricing Pressure
We operate in certain countries where the economic conditions continue to present significant challenges. Many countries are reducing their public expenditures in light of the global economic downturn and the deterioration of the credit and economic conditions in certain countries in Europe. As a result, we expect to see continued efforts to reduce healthcare costs, particularly in certain of the international markets in which we operate. The implementation of pricing actions varies by country and certain measures already implemented, which include among other things, mandatory price reductions and suspensions on pricing increases on pharmaceuticals, have negatively impacted our revenues. In addition, certain countries set prices by reference to the prices in other countries where our products are marketed. Thus, our inability to secure adequate prices in a particular country may also impair our ability to obtain acceptable prices in existing and potential new markets. We expect that our revenues and results of operations will be further negatively impacted if these, similar or more extensive measures are, or continue to be, implemented in other countries in which we operate.
54
Credit Risk
We are subject to credit risk from our accounts receivable related to our product sales. The majority of our accounts receivable arise from product sales in the U.S. and Europe with concentrations of credit risk generally limited due to the wide variety of customers and markets using our products, as well as their dispersion across many different geographic areas. Our accounts receivable are primarily due from wholesale distributors, large pharmaceutical companies and public hospitals. We monitor the financial performance and credit worthiness of our large customers so that we can properly assess and respond to changes in their credit profile. We operate in certain countries where the economic conditions continue to present significant challenges. We continue to monitor these conditions, including the volatility associated with international economies and associated impacts on the relevant financial markets and our business. Our historical write-offs of accounts receivable have not been significant.
Within the European Union, our product sales in Italy, Spain and Portugal continue to be subject to significant payment delays due to government funding and reimbursement practices. The credit and economic conditions within these countries have continued to deteriorate throughout 2010. These conditions have resulted in, and may continue to result in, an increase in the average length of time that it takes to collect on our accounts receivable outstanding in these countries. As of December 31, 2010, our accounts receivable balances in Italy, Spain and Portugal totaled $118.0 million, $100.6 million and $23.3 million, respectively, totaling approximately $241.9 million. Approximately $45.0 million of this amount was outstanding for greater than one year. As of December 31, 2010, we had $50.1 million of receivables that are expected to be collected beyond one year, which are included as a component of investments and other assets within our consolidated balance sheet.
Our concentrations of credit risk related to our accounts receivable from product sales in Greece to date have been limited as our receivables within this market are due from our wholesale distributor, for which related accounts receivable balances as of December 31, 2010, remain current and substantially in compliance with their contractual due dates. As of December 31, 2010 our accounts receivable balances due from our distributor in Greece totaled $3.9 million. However, the majority of our sales by our distributor are to government funded hospitals and as a result our distributor maintains significant outstanding receivables with the government of Greece. Furthermore, the government of Greece has recently required financial support from both the European Union and the International Monetary Fund to avoid defaulting on its debt. In the event that Greece defaults on its debt, and could not pay our distributor, we may be unable to collect some or all of our remaining amounts due from the distributor. The government of Greece may also require pharmaceutical creditors to accept mandatory, retroactive, price deductions in settlement of outstanding receivables and we could be required to repay our distributor a portion of the amounts they have previously remitted to us. The potential impact resulting from such mandatory actions remains uncertain, although delays or changes in the availability of government funding may adversely impact the operations of our distributor. To date, we have not been required to repay such amounts to our distributor or take a discount in settlement of any outstanding receivables and do not intend to do so.
We believe that our allowance for doubtful accounts was adequate as of December 31, 2010; however, if significant changes occur in the availability of government funding or the reimbursement practices of these or other governments, we may not be able to collect on amounts due to us from customers in such countries and our results of operations could be adversely affected.
55
Financial Condition and Liquidity
Our financial condition is summarized as follows:
| % Change | ||||||||||||
| 2010 | ||||||||||||
| As of December 31, | Compared | |||||||||||
| (In millions, except percentages) | 2010 | 2009 | to 2009 | |||||||||
Financial assets: | ||||||||||||
Cash and cash equivalents | $ | 759.6 | $ | 581.9 | 30.5 | % | ||||||
Marketable securities - current | 448.1 | 681.8 | (34.3 | )% | ||||||||
Marketable securities - non-current | 743.1 | 1,194.1 | (37.8 | )% | ||||||||
Total financial assets | $ | 1,950.8 | $ | 2,457.8 | (20.6 | )% | ||||||
Borrowings: | ||||||||||||
Current portion of notes payable, line of credit and other financing arrangements | $ | 137.2 | $ | 19.8 | 594.0 | % | ||||||
Notes payable and line of credit | 1,066.4 | 1,080.2 | (1.3 | )% | ||||||||
Total borrowings | $ | 1,203.5 | $ | 1,100.0 | 9.4 | % | ||||||
Total working capital | $ | 1,490.3 | $ | 1,765.7 | (15.7 | )% | ||||||
For the year ended December 31, 2010, certain significant cash flows were as follows:
| • | $2,077.6 million used for share repurchases; | |
| • | $680.3 million in net proceeds received on sales and maturities of marketable securities; | |
| • | $352.0 million in total payments for domestic income taxes; | |
| • | $183.5 million in proceeds from the issuance of stock for share-based compensation arrangements; | |
| • | $173.1 million used for purchases of property, plant and equipment; | |
| • | $127.0 million in proceeds, net of transaction costs, received from sale of the San Diego facility, which has been accounted for as a financing arrangement; | |
| • | $26.4 million in upfront payments to Knopp under our license agreement dated August 17, 2010 and a $60.0 million investment in the equity of Knopp; | |
| • | $40.0 million payment made to the former shareholders of Syntonix recognized as IPR&D expense; | |
| • | $32.5 million payment made for the acquisition of Panima; | |
| • | $30.0 million milestone payment made to Abbott Biotherapeutics Corp (formerly Facet Biotech Corporation) recognized as research and development expense; and | |
| • | $25.0 million termination payment made to Cardiokine recognized as a distribution to a noncontrolling interest. |
For the year ended December 31, 2009, certain significant cash flows were as follows:
| • | $696.3 million in total payments for domestic income taxes; | |
| • | $229.1 million used for net purchases of marketable securities; | |
| • | $165.6 million used for purchases of property, plant and equipment. | |
| • | $110.0 million upfront payment made to Acorda on July 1, 2009; | |
| • | $751.2 million used for share repurchases; and | |
| • | $47.8 million in proceeds from the issuance of stock for share-based compensation arrangements. |
56
We have historically financed our operating and capital expenditures primarily through positive cash flows earned through our operations. We expect to continue funding our current and planned operating requirements principally through our cash flows from operations, as well as our existing cash resources. We believe that existing funds, when combined with cash generated from operations and our access to additional financing resources, if needed, are sufficient to satisfy our operating, working capital, strategic alliance, milestone payment, capital expenditure and debt service requirements for the foreseeable future. In addition, we may choose to opportunistically return cash to shareholders and pursue other business initiatives, including acquisition and licensing activities. We may, from time to time, also seek additional funding through a combination of new collaborative agreements, strategic alliances and additional equity and debt financings or from other sources should we identify a significant new opportunity.
We consider the unrepatriated cumulative earnings of certain of our foreign subsidiaries to be invested indefinitely outside the U.S. Of the total cash, cash equivalents and marketable securities at December 31, 2010, approximately $0.9 billion was generated from operations in foreign tax jurisdictions and is intended for use in our foreign operations. In managing our day-to-day liquidity in the U.S., we do not rely on the unrepatriated earnings as a source of funds and we have not provided for U.S. federal or state income taxes on these undistributed foreign earnings.
For additional information related to certain risks that could negatively impact our financial position or future results of operations, please read the " Risk Factors " and " Quantitative and Qualitative Disclosures About Market Risk " sections of this report.
Share Repurchase Programs
In April 2010, our Board of Directors authorized the repurchase of up to $1.5 billion of our common stock, with the objective of reducing shares outstanding and returning excess cash to shareholders. This repurchase authorization was completed during the third quarter of 2010. During 2010, we repurchased approximately 29.8 million shares of our common stock under this authorization. All shares repurchased under this program were retired.
In October 2009, our Board of Directors authorized the repurchase of up to $1.0 billion of our common stock with the objective of reducing shares outstanding and returning excess cash to shareholders. This repurchase program was completed during the first quarter of 2010. During the first quarter of 2010, approximately 10.5 million shares of our common stock were repurchased for approximately $577.6 million under this authorization. During 2009, approximately 8.8 million shares of our common stock were repurchased for approximately $422.4 million under this authorization. All shares repurchased under this program were retired.
In October 2006, our Board of Directors authorized the repurchase of up to 20.0 million shares of our common stock. This repurchase program was completed during the fourth quarter of 2009. During 2009, approximately 7.2 million shares of our common stock were repurchased for approximately $328.8 million under this authorization. During 2008, approximately 12.8 million shares of our common stock were repurchased for approximately $738.9 million under this authorization. We used the 2006 share repurchase program principally for share stabilization.
As a result of the approximately 40.3 million shares repurchased during 2010, common shares outstanding have decreased by approximately 15% since December 31, 2009.
Cash, Cash Equivalents and Marketable Securities
Until required for another use in our business, we invest our cash reserves in bank deposits, certificates of deposit, commercial paper, corporate notes, U.S. and foreign government instruments and other interest bearing marketable debt instruments in accordance with our investment policy. We mitigate credit risk in our cash reserves and marketable securities by maintaining a well diversified portfolio that limits the amount of investment exposure as to institution, maturity, and investment type. The value of our investments, however, may be adversely affected by increases in interest rates, downgrades in the credit rating of the corporate bonds included in our portfolio, instability in the global financial markets that reduces the liquidity of securities included in our portfolio, and by other factors which may result in declines in the value of the investments. Each of these events may cause us to
57
record charges to reduce the carrying value of our investment portfolio if the declines are other-than-temporary or sell investments for less than our acquisition cost which could adversely impact our financial position and our overall liquidity. For a summary of the fair value and valuation methods of our marketable securities please read Note 7, Fair Value Measurements to our consolidated financial statements included in this report.
The decrease in cash and marketable securities from December 31, 2009, was primarily due to the execution of our share repurchases programs, tax payments, purchases of property, plant and equipment, the $32.5 million paid upon the acquisition of Panima, and the $86.4 million in payments made to Knopp under our recent license and stock purchase agreements, along with other milestone payments. These uses of cash were offset by cash generated from operations, net proceeds received from sales and maturities of marketable securities, net proceeds recorded from the sale of the San Diego facility and proceeds from the issuance of stock under our share-based compensation arrangements.
Borrowings
We have a $360.0 million senior unsecured revolving credit facility, which we may choose to use for future working capital and general corporate purposes. The terms of this revolving credit facility include various covenants, including financial covenants that require us to not exceed a maximum leverage ratio and, under certain circumstances, an interest coverage ratio. This facility terminates in June 2012. No borrowings have been made under this credit facility and as of December 31, 2010 and 2009 we were in compliance with all applicable covenants.
In connection with our 2006 distribution agreement with Fumedica, we issued notes totaling 61.4 million Swiss Francs which were payable to Fumedica in varying amounts from June 2008 through June 2018. In June 2010, we repaid 12.0 million Swiss Francs ($10.3 million) of the outstanding amount. As of December 30, 2010, our remaining note payable to Fumedica has a present value of 20.7 million Swiss Francs ($22.0 million) and remains payable in a series of payments through June 2018. The notes are non-interest bearing, have been discounted for financial statement presentation purposes, and are being accreted at an annual rate of 5.75%.
As described in Note 10 Property, Plant & Equipment, on October 1, 2010, we sold the San Diego facility and agreed to lease back the facility for a period of 15 months. Because we do not qualify for immediate sales treatment due to our continuing involvement with the facility, we have accounted for these transactions as a financing arrangement and recorded an obligation of $127.0 million on that date reflecting cash proceeds received, net of transaction costs. As of December 31, 2010, our remaining obligation was $125.9 million, which is reflected as a component of current portion of notes payable, line of credit and other financing arrangements within our consolidated balance sheet.
There have been no other significant changes in our borrowings since December 31, 2009. For a summary of the fair and carrying value of our outstanding borrowings as of December 31, 2010 and 2009, please read Note 7, Fair Value Measurements to our consolidated financial statements included in this report.
Working Capital
We define working capital as current assets less current liabilities. The decrease in working capital from December 31, 2009, primarily reflects the overall increase in total current liabilities by $335.2 million.
The increase in total current liabilities reflects increases in accounts payable and accrued expenses offset by the June 2010 repayment of certain Fumedica notes payable as described above under Borrowings. The increase in accrued expenses is inclusive of an increase in the current portion of our Medicaid and VA accruals and accruals related to the restructuring activities we under took in the fourth quarter of 2010 and higher employee compensation accruals.
58
Cash Flows
Our net cash flows are summarized as follows:
| % Change | ||||||||||||||||||||
| 2010 | 2009 | |||||||||||||||||||
| For the Years Ended December 31, | Compared | Compared | ||||||||||||||||||
| (In millions, except percentages) | 2010 | 2009 | 2008 | to 2009 | to 2008 | |||||||||||||||
Net cash flows provided by operating activities | $ | 1,624.7 | $ | 1,074.9 | $ | 1,562.4 | 51.1 | % | (31.2 | )% | ||||||||||
Net cash flows used in investing activities | $ | 345.3 | $ | (395.0 | ) | $ | (365.9 | ) | (187.4 | %) | 8.0 | % | ||||||||
Net cash flows used in financing activities | $ | (1,784.9 | ) | $ | (724.2 | ) | $ | (1,234.6 | ) | 146.5 | % | (41.3 | )% | |||||||
Operating Activities
Cash flows from operating activities represent the cash receipts and disbursements related to all of our activities other than investing and financing activities. Cash provided by operating activities was primarily driven by our earnings and changes in working capital. We expect cash provided from operating activities will continue to be our primary source of funds to finance operating needs and capital expenditures for the foreseeable future.
Operating cash flow is derived by adjusting our net income for:
| • | Non-cash operating items such as depreciation and amortization, impairment charges and share-based compensation charges; | |
| • | Changes in operating assets and liabilities which reflect timing differences between the receipt and payment of cash associated with transactions and when they are recognized in results of operations; and | |
| • | Changes associated with the payment of contingent milestones associated with our prior acquisitions of businesses. |
For 2010 compared to 2009, the increase in net cash provided by operating activities was primarily driven by increased revenues and lower payments for U.S. federal income taxes offset by an increase in accounts receivable and receivables due from unconsolidated joint business.
For 2009 compared to 2008, the decrease in net cash provided by operating activities was primarily driven by changes in other liabilities and taxes payable, primarily due to an increase in income tax payments of $373.4 million which primarily resulted from increased earnings and the resolution of a number of audits in 2009, the $110.0 million upfront payment made to Acorda on July 1, 2009 and the payment of certain accrued expenses and other current liabilities.
On November 3, 2010, we announced a restructuring plan that involves a workforce reduction and the consolidation of facilities. During the fourth quarter of 2010, we began to record restructuring charges and currently expect to incur total pre-tax costs through the fourth quarter of 2011 totaling approximately $110.0 million. The majority of the cash expenditures associated with these charges will be paid in the first half of 2011 and we expect that substantially all payments will be made by the end of 2011.
Investing Activities
For 2010 compared to 2009, the increase in net cash provided by investing activities was primarily due to net proceeds received from sales and maturities of marketable securities, offset by the $86.4 million in payments made to Knopp under our recent license and stock purchase agreements, the $32.5 million payment made upon our acquisition of Panima, our purchases of property, plant and equipment and the milestone payment made to the former shareholders of Syntonix. Net proceeds received from sales and maturities of marketable securities in 2010 totaled $680.3 million compared to net purchases of $229.1 million made in 2009.
For 2009 compared to 2008, the increase in net cash used in investing activities was primarily due to a decrease in collateral received under our securities lending program and an increase in net purchases of marketable securities and strategic and other investments offset by a reduction in purchases of property, plant and equipment and the 2008 milestone payment made to the former shareholders of Conforma Therapeutics, Inc. The decline in purchases
59
of property, plant and equipment was primarily attributable to our Hillerød, Denmark manufacturing facility and certain other manufacturing upgrades.
Financing Activities
For 2010 compared to 2009, the increase in net cash used in financing activities was primarily due to increases in the amounts of our common stock repurchased compared to the same period in 2009. In 2010, we repurchased approximately 40.3 million shares of our common stock for approximately $2.1 billion compared to 16.0 million shares for approximately $751.2 million in 2009. Cash used in financing activities also includes the $127.0 in net proceeds from the sale of the San Diego facility, which is being accounted for as a financing arrangement and activity under our employee stock plans. We received $183.5 million in 2010 compared to $47.8 million in 2009 related to stock option exercises and stock issuances under our employee stock purchase plan.
For 2009 compared to 2008, the decrease in cash used in financing activities was primarily due to the repayment of our term loan facility of $1.5 billion in 2008 and a decrease in obligations under our securities lending program offset in part by the net proceeds of $987.0 million from the issuance of long-term debt and a decrease in proceeds received from the issuance of stock under our share-based compensation programs.
Contractual Obligations and Off-Balance Sheet Arrangements
Contractual Obligations
The following table summarizes our contractual obligations as of December 31, 2010, excluding amounts related to uncertain tax positions, amounts payable to tax authorities, funding commitments, contingent milestone payments, our financing arrangement related to the San Diego facility, and restructuring accruals, as described below.
| Payments Due by Period | ||||||||||||||||||||
| Less than | 1 to 3 | 4 to 5 | After | |||||||||||||||||
| (In millions) | Total | 1 Year | Years | Years | 5 Years | |||||||||||||||
Non-cancellable operating leases(1) | $ | 367.8 | $ | 40.9 | $ | 63.7 | $ | 57.6 | $ | 205.6 | ||||||||||
Notes payable and line of credit(2) | 1,359.7 | 76.2 | 563.2 | 81.1 | 639.2 | |||||||||||||||
Purchase and other obligations(3) | 69.6 | 52.1 | 14.8 | 2.4 | 0.3 | |||||||||||||||
Defined benefit obligation | 8.2 | - | - | - | 8.2 | |||||||||||||||
Total contractual obligations | $ | 1,805.3 | $ | 169.2 | $ | 641.7 | $ | 141.1 | $ | 853.3 | ||||||||||
| (1) | We lease properties and equipment for use in our operations. In addition to rent, the leases may require us to pay additional amounts for taxes, insurance, maintenance and other operating expenses. Amounts reflected within the table, detail future minimum rental commitments under non-cancelable operating leases as of December 31 for each of the years presented. | |
| (2) | Notes payable and line of credit includes principal and interest payments. | |
| (3) | Purchase and other obligations include our obligations of approximately $12.2 million related to the fair value of net liabilities on derivative contracts due in less than one year, approximately $4.5 million related to fixed obligations for the purchase of natural gas and approximately $16.8 million related to obligations for communication services |
Restructuring
In connection with our recent restructuring initiative, we are in the process of vacating the San Diego, California facility and consolidating our Massachusetts facilities. Costs associated with closing these facilities, including costs related to the termination of certain leases, are reflected within our consolidated statement of income as a component of total restructuring charges incurred. For a more detailed description of our restructuring efforts, including our plan to consolidate facilities, please read Note 3, Restructuring to our consolidated financial statements included in this report.
60
Financing Arrangement
As described in Note 10 Property, Plant & Equipment to our consolidated financial statements included in this report, on October 1, 2010, we sold the San Diego facility and agreed to lease back the facility for a period of 15 months. We have accounted for these transactions as a financing arrangement and recorded an obligation of $127.0 million on that date. As of December 31, 2010, our remaining obligation was $125.9 million, which is reflected as a component of current portion of notes payable, line of credit and other financing arrangements within our consolidated balance sheet.
In January 2011, we entered into an agreement to terminate our 15 month lease of the San Diego facility. Under the terms of this agreement, we will continue to make monthly rental payments through August 31, 2011 and will have no continuing involvement or remaining obligation after that date. Once the lease arrangement has concluded we will account for the San Diego facility as a sale of property and we do not expect to recognize a significant gain or loss on the sale at that time. We are scheduled to incur debt service payments and interest totaling approximately $6.9 million over the term of the revised leaseback period.
Tax Related Obligations
We exclude liabilities pertaining to uncertain tax positions from our summary of contractual obligations as we cannot make a reliable estimate of the period of cash settlement with the respective taxing authorities. As of December 31, 2010, we have approximately $137.7 million of liabilities associated with uncertain tax positions. Included in these liabilities are amounts related to the settlement of certain federal and state tax audits in the fourth quarter of 2009. As of December 31, 2010, we expect to pay approximately $76.1 million within the next twelve months in connection with such settlements.
Other Funding Commitments
As of December 31, 2010, we have funding commitments of up to approximately $19.0 million as part of our investment in biotechnology oriented venture capital funds.
As of December 31, 2010, we have several ongoing clinical studies in various clinical trial stages. Our most significant clinical trial expenditures are to clinical research organizations (CROs). The contracts with CROs are generally cancellable, with notice, at our option. We have recorded accrued expenses of $16.1 million on our consolidated balance sheet for expenditures incurred by CROs as of December 31, 2010. We have approximately $326.9 million in cancellable future commitments based on existing CRO contracts as of December 31, 2010 which are not included in the contractual obligations table above because of our termination rights.
Contingent Milestone Payments
Based on our development plans as of December 31, 2010, we have committed to make potential future milestone payments to third parties of up to approximately $1,334.3 million as part of our various collaborations, including licensing and development programs. Payments under these agreements generally become due and payable only upon achievement of certain development, regulatory or commercial milestones. Because the achievement of these milestones had not occurred as of December 31, 2010, such contingencies have not been recorded in our financial statements. We anticipate that we may pay approximately $55.6 million of milestone payments in 2011, provided various development, regulatory or commercial milestones are achieved. Amounts related to contingent milestone payments are not included in the contractual obligations table above as they are contingent on the successful achievement of certain development, regulatory approval and commercial milestones. These milestones may not be achieved.
Other Off-Balance Sheet Arrangements
We do not have any relationships with entities often referred to as structured finance or special purpose entities which would have been established for the purpose of facilitating off-balance sheet arrangements. As such, we are not exposed to any financing, liquidity, market or credit risk that could arise if we had engaged in such relationships. We consolidate variable interest entities if we are the primary beneficiary.
61
Legal Matters
For a discussion of legal matters as of December 31, 2010, please read Note 20, Litigation to our consolidated financial statements included in this report.
Critical Accounting Estimates
The preparation of our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the U.S. (U.S. GAAP), requires us to make estimates, judgments and assumptions that may affect the reported amounts of assets, liabilities, revenues and expenses, and related disclosure of contingent assets and liabilities. We believe the most complex judgments result primarily from the need to make estimates about the effects of matters that are inherently uncertain and are significant to our consolidated financial statements. We base our estimates on historical experience and on various other assumptions that we believe are reasonable, the results of which form the basis for making judgments about the carrying values of assets and liabilities. We evaluate our estimates, judgments and assumptions on an ongoing basis. Actual results may differ from these estimates under different assumptions or conditions.
The most significant areas involving estimates, judgments and assumptions used in the preparation of our consolidated financial statements are as follows:
| • | Revenue recognition and related allowances; | |
| • | Collaborative relationships; | |
| • | Clinical trial expenses; | |
| • | Consolidation of variable interest entities; | |
| • | Valuation of contingent consideration resulting from a business combination; | |
| • | Valuation of acquired intangible assets, including in process research and development; | |
| • | Inventory; | |
| • | Impairment and amortization of long-lived assets and accounting for goodwill; | |
| • | Investments, including fair value measures and impairments; | |
| • | Share-based compensation; | |
| • | Income taxes; | |
| • | Contingencies; and | |
| • | Restructuring charges. |
Revenue Recognition and Related Allowances
We recognize revenue when all of the following criteria are met: persuasive evidence of an arrangement exists; delivery of product has occurred or services have been rendered; the seller's price to the buyer is fixed or determinable; and collectability is reasonably assured.
Product Revenues
Revenues from product sales are recognized when title and risk of loss have passed to the customer, which is typically upon delivery. However, sales of TYSABRI in the U.S. are recognized on the "sell-through" model, that is, upon shipment of the product by Elan to its third party distributor rather than upon shipment to Elan. The timing of distributor orders and shipments can cause variability in earnings.
Reserves for Discounts and Allowances
We establish reserves for trade term discounts, wholesaler incentives, Medicaid rebates, Veterans Administration and PHS discounts, managed care rebates, product returns and other applicable allowances. These reserves
62
are based on estimates of the amounts earned or to be claimed on the related sales. Our estimates take into consideration our historical experience, current contractual and statutory requirements, specific known market events and trends and forecasted customer buying patterns. If actual results vary, we may need to adjust these estimates, which could have an effect on earnings in the period of the adjustment. The estimates we make with respect to these allowances represent the most significant judgments with regard to revenue recognition.
In addition to the discounts and rebates described above and classified as a reduction of revenue, we also maintain certain customer service contracts with distributors and other customers in the distribution channel that provide us with inventory management and distribution services. We have established the fair value of these services and classified these customer service contracts as sales and marketing expense. If we had concluded that we did not receive a separate identifiable benefit or have sufficient evidence that the fair value did not exist for these services, we would have been required to classify these costs as a reduction of revenue.
Healthcare Reform
In 2010, healthcare reform legislation was enacted in the U.S. This legislation contains several provisions that affect our accounting estimates. Although many provisions of the new legislation did not take effect immediately, several provisions became effective in 2010. These include (1) an increase in the minimum Medicaid rebate to states participating in the Medicaid program from 15.1% to 23.1% on our branded prescription drugs; (2) the extension of the Medicaid rebate to Managed Care Organizations that dispense drugs to Medicaid beneficiaries; and (3) the expansion of the 340B PHS drug pricing program, which provides outpatient drugs at reduced rates, to include additional hospitals, clinics, and healthcare centers. These incremental discounts have been factored into determining the amount and timing of our revenues on sales to certain customers and are based upon several assumptions about the implementation of this new legislation. Our estimates are based upon our knowledge of current events and actual results may ultimately differ from these estimates.
Revenues from Unconsolidated Joint Business
We collaborate with Genentech on the development and commercialization of RITUXAN. Revenues from unconsolidated joint business consist of (1) our share of pre-tax co-promotion profits in the U.S.; (2) reimbursement of our selling and development expense in the U.S.; and (3) revenue on sales of RITUXAN in the rest of world, which consists of our share of pretax co-promotion profits in Canada and royalty revenue on sales of RITUXAN outside the U.S. and Canada by F. Hoffmann-La Roche Ltd. (Roche) and its sublicensees. Pre-tax co-promotion profits are calculated and paid to us by Genentech in the U.S. and by Roche in Canada. Pre-tax co-promotion profits consist of U.S. and Canadian sales of RITUXAN to third-party customers net of discounts and allowances less the cost to manufacture RITUXAN, third-party royalty expenses, distribution, selling and marketing, and joint development expenses incurred by Genentech, Roche and us. We record our share of the pretax co-promotion profits in Canada and royalty revenues on sales of RITUXAN outside the U.S. on a cash basis. Additionally, our share of the pretax co-promotion profits in the U.S. includes estimates supplied by Genentech. Actual results may ultimately differ from our estimates.
Bad Debt Reserves
Bad debt reserves are based on our estimated uncollectible accounts receivable. Given our historical experiences with bad debts, combined with our credit management policies and practices, we do not presently maintain significant bad debt reserves.
Concentrations of Credit Risk
The majority of our accounts receivable arise from product sales in the United States and Europe and are primarily due from wholesale distributors, large pharmaceutical companies and public hospitals. We monitor the financial performance and credit worthiness of our large customers so that we can properly assess and respond to changes in their credit profile. We continue to monitor economic conditions, including the volatility associated with international economies, and associated impacts on the relevant financial markets and our business, especially in light of the global economic downturn. The credit and economic conditions within Italy, Spain, Portugal and Greece among other members of the European Union have deteriorated throughout 2010. These conditions have resulted in,
63
and may continue to result in, an increase in the average length of time that it takes to collect on our accounts receivable outstanding in these countries.
As of December 31, 2010, our accounts receivable balances in Italy, Spain, Portugal and Greece, were $118.0 million, $100.6 million, $23.3 million and $3.9 million, respectively, totaling approximately $245.8 million. Approximately $45.0 million of these amounts were outstanding for greater than one year, none of which related to our Greek distributor. As of December 31, 2010, we had $50.1 million of receivables that are expected to be collected beyond one year, which are included as a component of investments and other assets within our consolidated balance sheet. To date, we have not experienced any significant losses with respect to the collection of our accounts receivable. If economic conditions worsen and/or the financial condition of our customers were to further deteriorate, our risk of collectability may increase, which may result in additional allowances and/or significant bad debts.
Royalty Revenues
We receive royalty revenues under license agreements with a number of third parties that sell products based on technology we have developed or to which we own rights. The license agreements provide for the payment of royalties to us based on sales of these licensed products. There are no future performance obligations on our part under these license agreements. We record these revenues based on estimates of the sales that occurred during the relevant period. The relevant period estimates of sales are based on interim data provided by licensees and analysis of historical royalties that have been paid to us, adjusted for any changes in facts and circumstances, as appropriate. We maintain regular communication with our licensees in order to assess the reasonableness of our estimates. Differences between actual royalty revenues and estimated royalty revenues are adjusted for in the period in which they become known, typically the following quarter. Historically, adjustments have not been material when compared to actual amounts paid by licensees. To the extent we do not have sufficient ability to accurately estimate revenues; we record such revenues on a cash basis.
Collaborative Relationships
We evaluate our collaborative agreements for proper income statement classification based on the nature of the underlying activity. Amounts due from our collaborative partners related to development activities are generally reflected as a reduction of research and development expense because the performance of contract development services is not central to our operations. For collaborations with commercialized products, if we are the principal we record revenue and the corresponding operating costs in their respective line items within our consolidated statements of income. If we are not the principal, we record operating costs as a reduction of revenue.
As discussed within Note 19, Collaborations to our consolidated financial statements included in this report, Genentech incurs the majority of continuing development cost for RITUXAN. Expenses incurred by Genentech in the development of RITUXAN are not recorded as research and development expense, but rather reduce our share of co-promotion profits recorded as a component of unconsolidated joint business revenue.
Clinical Trial Expenses
Clinical trial expenses include expenses associated with CROs. The invoicing from CROs for services rendered can lag several months. We accrue the cost of services rendered in connection with CRO activities based on our estimate of site management, monitoring costs, and project management costs. We maintain regular communication with our CROs to gauge the reasonableness of our estimates. Differences between actual clinical trial expenses and estimated clinical trial expenses recorded have not been material and are adjusted for in the period in which they become known.
Consolidation of Variable Interest Entities
We consolidate variable interest entities in which we are the primary beneficiary. For such consolidated entities where we own less than a 100% interest, we record noncontrolling interest in our statement of income for the current results allocated to the third party equity interests.
64
Effective January 1, 2010, we adopted a new accounting standard related to the consolidation of variable interest entities which affected how we determined whether a variable interest or interests give us a controlling financial interest in a variable interest entity. In determining whether we are the primary beneficiary of a variable interest entity, we consider a number of factors, including our ability to direct the activities that most significantly affect the entity's economic success, our contractual rights and responsibilities under the arrangement and the significance of the arrangement to each party. These considerations impact the way we account for our existing collaborative and joint venture relationships and may result in the future consolidation of companies or entities with which we have collaborative or other arrangements.
Valuation of Contingent Consideration Resulting from a Business Combination
For acquisitions completed after January 1, 2009, we record contingent consideration resulting from a business combination at its fair value on the acquisition date. Each reporting period thereafter, we revalue these obligations and record increases or decreases in their fair value as an adjustment to contingent consideration expense within the consolidated statement of income. Changes in the fair value of the contingent consideration obligations can result from adjustments to the discount rates and periods, updates in the assumed achievement or timing of any development milestones, or changes in the probability of certain clinical events and changes in the assumed probability associated with regulatory approval. Significant judgment is employed in determining the appropriateness of these assumptions as of the acquisition date and for each subsequent period. Accordingly, assumptions described above, could have a material impact on the amount of contingent consideration expense we record in any given period.
Valuation of Acquired Intangible Assets, including In Process Research and Development
We have acquired, and expect to continue to acquire, intangible assets through the acquisition of biotechnology companies or through the consolidation of variable interest entities. These intangible assets primarily consist of technology associated with human therapeutic products and in process research and development product candidates. When significant identifiable intangible assets are acquired, we generally engage an independent third-party valuation firm to assist in determining the fair values of these assets as of the acquisition date. Management will determine the fair value of less significant identifiable intangible assets acquired. Discounted cash flow models are typically used in these valuations, and these models require the use of significant estimates and assumptions including but not limited to:
| • | estimating the timing of and expected costs to complete the in process projects; | |
| • | projecting regulatory approvals; | |
| • | estimating future cash flows from product sales resulting from completed products and in process projects; and | |
| • | developing appropriate discount rates and probability rates by project. |
We believe the fair values assigned to the intangible assets acquired are based upon reasonable estimates and assumptions given available facts and circumstances as of the acquisition dates.
If these projects are not successfully developed, the sales and profitability of the company may be adversely affected in future periods. Additionally, the value of the acquired intangible assets may become impaired. We believe that the foregoing assumptions used in the IPR&D analysis were reasonable at the time of the respective acquisition. No assurance can be given, however, that the underlying assumptions used to estimate expected project sales, development costs or profitability, or the events associated with such projects, will transpire as estimated.
Prior to January 1, 2009, we measured acquired IPR&D in a business combination at fair value and expensed it on acquisition date if that technology lacked an alternative future use, or capitalized it as an intangible asset if certain criteria were met; however, effective January 1, 2009, if we are purchasing a business, the acquired IPR&D is measured at fair value, capitalized as an intangible asset and tested for impairment at least annually until commercialization, after which time the IPR&D is amortized over its estimated useful life. If we acquire an asset or group of assets, that do not meet the definition of a business under applicable accounting standards; then the
65
acquired IPR&D is expensed on its acquisition date. Future costs to develop these assets are recorded to expense as they are incurred if the technology lacks alternative future uses.
Inventory
Inventories are stated at the lower of cost or market with cost determined under the first-in, first-out (FIFO) method. Included in inventory are raw materials used in the production of pre-clinical and clinical products, which are expensed as research and development costs when consumed.
Capitalization of Inventory Costs
Our policy is to capitalize inventory costs associated with our products prior to regulatory approval, when, based on management's judgment, future commercialization is considered probable and the future economic benefit is expected to be realized. We consider numerous attributes in evaluating whether the costs to manufacture a particular product should be capitalized as an asset. We assess the regulatory approval process and where the particular product stands in relation to that approval process, including any known safety or efficacy concerns, potential labeling restrictions and other impediments to approval. We evaluate our anticipated research and development initiatives and constraints relating to the product and the indication in which it will be used. We consider our manufacturing environment including our supply chain in determining logistical constraints that could hamper approval or commercialization. We consider the shelf life of the product in relation to the expected timeline for approval and we consider patent related or contract issues that may prevent or delay commercialization. We are sensitive to the significant commitment of capital to scale up production and to launch commercialization strategies. We also base our judgment on the viability of commercialization, trends in the marketplace and market acceptance criteria. Finally, we consider the reimbursement strategies that may prevail with respect to the product and assess the economic benefit that we are likely to realize.
We expense previously capitalized costs related to pre-approval inventory upon a change in such judgment, due to, among other potential factors, a denial or delay of approval by necessary regulatory bodies. As of December 31, 2010 and 2009, the carrying value of our inventory did not include any costs associated with products that had not yet received regulatory approval.
There is a risk inherent in these judgments and any changes we make in these judgments may have a material impact on our results in future periods.
Obsolescence and Unmarketable Inventory
We periodically review our inventories for excess or obsolete inventory and write-down obsolete or otherwise unmarketable inventory to its estimated net realizable value. If the actual net realizable value is less than that estimated by us, or if it is determined that inventory utilization will further diminish based on estimates of demand, additional inventory write-downs may be required. Additionally, our products are subject to strict quality control and monitoring which we perform throughout the manufacturing process. In the event that certain batches or units of product no longer meet quality specifications or become obsolete due to expiration, we will record a charge to cost of sales to write-down any obsolete or otherwise unmarketable inventory to its estimated net realizable value. In all cases product inventory is carried at the lower of cost or its estimated net realizable value.
Impairment and Amortization of Long-lived Assets and Accounting for Goodwill
Long-lived Assets Other than Goodwill
Long-lived assets to be held and used, including property plant and equipment as well as intangible assets, are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of the assets may not be recoverable.
If such circumstances are determined to exist, an estimate of undiscounted future cash flows produced by the long-lived asset, including its eventual residual value, is compared to the carrying value to determine whether impairment exists. In the event that such cash flows are not expected to be sufficient to recover the carrying amount of the assets, the assets are written-down to their estimated fair values.
66
We also regularly evaluate our current manufacturing utilization strategy and assess alternatives. In June 2010, we decided to stop further validation of our large-scale manufacturing facility in Hillerød, Denmark following completion of the facility's operational qualification activities in the first half of 2011 as we continue to evaluate our current manufacturing utilization strategy. Recent manufacturing improvements have resulted in favorable production yields on TYSABRI, that along with slower than expected TYSABRI growth, have reduced our expected capacity requirements. As a result, we have decided to delay the start of manufacturing activities at this site until additional capacity is required by the business. If we decide to consolidate, co-locate or dispose of certain aspects of our business operations, for strategic or other operational reasons, we may dispose of or vacate one or more of our properties.
Our most significant intangible asset is the core technology related to our AVONEX product. We believe the economic benefit of our core technology is consumed as revenue is generated from our AVONEX product, which we refer to as the economic consumption amortization model. This amortization methodology involves calculating a ratio of actual current period sales to total anticipated sales for the life of the product and applying this ratio to the carrying amount of the intangible asset. An analysis of the anticipated product sales of AVONEX is performed at least annually during our long range planning cycle, and this analysis serves as the basis for the calculation of our economic consumption amortization model. Although we believe this process has allowed us to reliably determine the best estimate of the pattern in which we will consume the economic benefits of our core technology intangible asset, the model could result in deferring amortization charges to future periods in certain instances, due to continued sales of the product at a nominal level after patent expiration or otherwise. In order to ensure that amortization charges are not unreasonably deferred to future periods, we compare the amount of amortization determined under the economic consumption model against the minimum amount of amortization recalculated each year under the straight-line method. Amortization is then recorded based upon the higher of the amount of amortization determined under the economic consumption model or the minimum amortization amount determined under the straight-line method.
We completed our most recent long range planning cycle in the third quarter of 2010. This analysis is based upon certain assumptions that we evaluate on a periodic basis, such as the anticipated product sales of AVONEX and expected impact of competitor products and our own pipeline product candidates, as well as the issuance of new patents or the extension of existing patents. Based upon this analysis, we have continued to amortize this asset on the economic consumption model for the third and fourth quarters of 2010, and expect to apply the same model for the next two quarters.
In addition, this analysis did not result in a significant change in the expected lifetime revenue of AVONEX. If there are any indications that the assumptions underlying our most recent analysis would be different than those utilized within our current estimates, our analysis would be updated and may result in a significant change in the anticipated lifetime revenue of AVONEX determined during our most recent annual review. For example, the occurrence of an adverse event, such as the invalidation of our AVONEX '755 Patent issued in September 2009, could substantially increase the amount of related amortization expense as compared to previous periods or our current expectations, which may result in a significant negative impact on our future results of operations.
We did not recognize an impairment charge related to our long-lived assets during 2010, 2009 and 2008.
Goodwill
Goodwill totaled approximately $1,146.3 million as of December 31, 2010, and relates largely to amounts that arose in connection with the merger of Biogen, Inc. and IDEC Pharmaceuticals Corporation. Our goodwill balances represent the difference between the purchase price and the fair value of the identifiable tangible and intangible net assets when accounted for using the purchase method of accounting.
We assess our goodwill balance within our single reporting unit annually and whenever events or changes in circumstances indicate the carrying value of goodwill may not be recoverable to determine whether any impairment in this asset may exist and, if so, the extent of such impairment. The provisions of this guidance require that we perform a two-step impairment test. In the first step, we compare the fair value of our reporting unit to its carrying value. If the carrying value of the net assets assigned to our reporting unit exceeds the fair value of our reporting unit, then the second step of the impairment test is performed in order to determine the implied fair value of our reporting unit's goodwill. If the carrying value of our reporting unit's goodwill exceeds its implied fair value, then the company records an impairment loss equal to the difference.
67
We completed our required annual impairment test in the fourth quarter of 2010, 2009 and 2008 and determined in each of those periods that the carrying value of goodwill was not impaired. In each year, the fair value of our reporting unit, which includes goodwill, was significantly in excess of the carry value of our reporting unit.
Investments, including Fair Value Measures and Impairments
We invest in various types of securities, including:
| • | short-term and long-term marketable securities, principally corporate notes, government securities including government sponsored enterprise mortgage-backed securities and credit card and auto loan asset-backed securities, in which our excess cash balances are invested; | |
| • | equity securities in certain publicly-traded biotechnology companies, some of which have collaborative agreements with us; | |
| • | equity securities of certain companies whose securities are not publicly traded and where fair value is not readily available; and | |
| • | investments in biotechnology oriented venture capital funds where fair value is not readily available. |
In accordance with the accounting standard for fair value measurements we have classified our financial assets and liabilities as Level 1, 2 or 3 within the fair value hierarchy. Fair values determined by Level 1 inputs utilize quoted prices (unadjusted) in active markets for identical assets or liabilities that we have the ability to access. Fair values determined by Level 2 inputs utilize data points that are observable such as quoted prices, interest rates and yield curves. Fair values determined by Level 3 inputs utilize unobservable data points for the asset or liability.
As noted in Note 6, Fair Value Measurements to our consolidated financial statements, a majority of our financial assets and liabilities have been classified as Level 2. These assets and liabilities have been initially valued at the transaction price and subsequently valued utilizing third party pricing services. The pricing services use many observable market inputs to determine value, including reportable trades, benchmark yields, credit spreads, broker/dealer quotes, bids, offers, current spot rates and other industry and economic events. We validate the prices provided by our third party pricing services by understanding the models used, obtaining market values from other pricing sources, analyzing pricing data in certain instances and confirming those securities trade in active markets.
We also have some investments classified as Level 3 whose fair value is initially measured at transaction prices and subsequently valued using the pricing of recent financing or by reviewing the underlying economic fundamentals and liquidation value of the companies. We apply judgments and estimates when we validate the prices provided by third parties. While we believe the valuation methodologies are appropriate, the use of valuation methodologies is highly judgmental and changes in methodologies can have a material impact on our results of operations.
Impairment
We conduct periodic reviews to identify and evaluate each investment that has an unrealized loss, in accordance with the meaning of other-than-temporary impairment and its application to certain investments. An unrealized loss exists when the current fair value of an individual security is less than its amortized cost basis. Unrealized losses on available-for-sale debt securities that are determined to be temporary, and not related to credit loss, are recorded, net of tax, in accumulated other comprehensive income.
For available-for-sale debt securities with unrealized losses, management performs an analysis to assess whether we intend to sell or whether we would more likely than not be required to sell the security before the expected recovery of the amortized cost basis. Where we intend to sell a security, or may be required to do so, the security's decline in fair value is deemed to be other-than-temporary and the full amount of the unrealized loss is reflected within earnings as an impairment loss.
Regardless of our intent to sell a security, we perform additional analysis on all securities with unrealized losses to evaluate losses associated with the creditworthiness of the security. Credit losses are identified where we do not expect to receive cash flows sufficient to recover the amortized cost basis of a security and are reflected within earnings as an impairment loss.
68
For equity securities, when assessing whether a decline in fair value below our cost basis is other-than-temporary, we consider the fair market value of the security, the duration of the security's decline, and the financial condition of the issuer. We then consider our intent and ability to hold the equity security for a period of time sufficient to recover our carrying value. Where we have determined that we lack the intent and ability to hold an equity security to its expected recovery, the security's decline in fair value is deemed to be other-than-temporary and is reflected within earnings as an impairment loss.
Share-Based Compensation
We make certain assumptions in order to value and record expense for our share-based compensation arrangements. We review and evaluate our assumptions regularly and, as a result, we confirm or change the assumptions used to value share-based awards granted in future periods. Such changes may lead to a significant increase or decrease in the expense we recognize in connection with share-based payments.
In connection with valuing stock options and our employee stock purchase plan, we use the Black-Scholes options pricing model, which requires us to develop certain subjective assumptions. The key assumptions that most significantly affect the calculation include the expected volatility of our stock, the expected term of the award and the expected forfeiture rate associated with our stock option plan.
For each of our restricted stock programs, we make assumptions in accounting for these awards, principally related to the forfeiture rate.
For our time-vested and performance-vested restricted stock awards, each period end, we also develop an estimate of each performance factor in order to estimate the actual number of shares that will be earned. For our plan, the number of shares to be earned is based on company performance metrics, such as annual revenue and earnings per share. Thus, during the performance period, we estimate our full year revenue and earnings per share and then adjust the performance factor after the completion of the full year.
In addition, beginning in 2010, we granted certain employees restricted stock units which will vest based on stock price performance, referred to as market stock units, as well as performance-vested restricted stock units which will be settled in cash, rather than in shares, referred to as cash settled performance shares. These market stock units use a binomial model or Monte Carlo simulation to value each award at the grant date and include key assumptions such as the expected market price of our stock on the vest date and the expected number of shares to be vested under the terms of the award. The cash settled awards are "marked to market" at the end of each period, with fluctuations in value reported through earnings.
Income Taxes
We prepare and file income tax returns based on our interpretation of each jurisdiction's tax laws and regulations. In preparing our consolidated financial statements, we estimate our income tax liability in each of the jurisdictions in which we operate by estimating our actual current tax expense together with assessing temporary differences resulting from differing treatment of items for tax and financial reporting purposes. These differences result in deferred tax assets and liabilities, which are included in our consolidated balance sheets. Significant management judgment is required in assessing the realizability of our deferred tax assets. In performing this assessment, we consider whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. In making this determination, under the applicable financial accounting standards, we are allowed to consider the scheduled reversal of deferred tax liabilities, projected future taxable income, and the effects of tax planning strategies. Our estimates of future taxable income include, among other items, our estimates of future income tax deductions related to the exercise of stock options. In the event that actual results differ from our estimates, we adjust our estimates in future periods and we may need to establish a valuation allowance, which could materially impact our financial position and results of operations.
We account for uncertain tax positions using a "more-likely-than-not" threshold for recognizing and resolving uncertain tax positions. We evaluate uncertain tax positions on a quarterly basis and consider various factors, that
69
include, but are not limited to, changes in tax law, the measurement of tax positions taken or expected to be taken in tax returns, the effective settlement of matters subject to audit, new audit activity and changes in facts or circumstances related to a tax position. We adjust the level of the liability to reflect any subsequent changes in the relevant facts surrounding the uncertain positions. Our liabilities for uncertain tax positions can be relieved only if the contingency becomes legally extinguished through either payment to the taxing authority or the expiration of the statute of limitations, the recognition of the benefits associated with the position meet the "more-likely-than-not" threshold or the liability becomes effectively settled through the examination process. We consider matters to be effectively settled once the taxing authority has completed all of its required or expected examination procedures, including all appeals and administrative reviews; we have no plans to appeal or litigate any aspect of the tax position; and we believe that it is highly unlikely that the taxing authority would examine or re-examine the related tax position. We also accrue for potential interest and penalties, related to unrecognized tax benefits in income tax expense.
As of December 31, 2010, undistributed foreign earnings and other basic differences of non-U.S. subsidiaries included in consolidated retained earnings aggregated approximately $2.4 billion. We intend to reinvest these earnings indefinitely in operations outside the U.S.; however, if we decide to repatriate funds in the future to execute our growth initiatives or to fund any other liquidity needs, the resultant tax consequences would negatively impact our results of operations. It is not practicable to estimate the amount of additional tax that might be payable if such earnings were remitted to the U.S.
Contingencies
We are currently involved in various claims and legal proceedings. On a quarterly basis, we review the status of each significant matter and assess its potential financial exposure. If the potential loss from any claim, asserted or unasserted, or legal proceeding is considered probable and the amount can be reasonably estimated, we accrue a liability for the estimated loss. Significant judgment is required in both the determination of probability and the determination as to whether an exposure is reasonably estimable. Because of uncertainties related to these matters, accruals are based only on the best information available at the time. As additional information becomes available, we reassess the potential liability related to pending claims and litigation and may revise our estimates. These revisions in the estimates of the potential liabilities could have a material impact on our consolidated results of operations and financial position.
Restructuring Charges
We have made estimates and judgments regarding the amount and timing of our restructuring expense and liability, including current and future period termination benefits and other exit costs to be incurred when related actions take place. We have also assessed the recoverability of certain long-lived assets employed in the business and in certain instances shortened the expected useful life of the assets based on changes in their expected use. When we determine that the useful lives of assets are shorter than we had originally estimated, we record additional depreciation to reflect the assets' new shorter useful lives. Severance and other related costs and asset-related charges are reflected within our consolidated statement of income as a component of total restructuring charges incurred. Actual results may differ from these estimates. For a more detailed description of our recent restructuring efforts, please read Note 3, Restructuring , to these consolidated financial statements.
New Accounting Standards
For a discussion of new accounting standards please read Note 1, Summary of Significant Accounting Principles to our consolidated financial statements included in this report.
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk |
We have operations or maintain distribution relationships in the U.S., Europe, Middle East, Canada, Central and South America, Australia, New Zealand, Japan, China, India and elsewhere in Asia in connection with the sale of AVONEX and TYSABRI and in Germany in connection with the sale of FUMADERM. We also receive royalty revenues based on worldwide product sales by our licensees and through Genentech on sales of RITUXAN in the
70
rest of world. As a result, our financial position, results of operations and cash flows can be affected by market fluctuations in foreign exchange rates, primarily with respect to the Euro, Canadian dollar, Swiss franc, Danish krone, Swedish krona, British pound, and Japanese yen.
We use foreign currency forward contracts to manage foreign currency risk but do not engage in currency speculation. The majority of our forward contracts are used to hedge certain forecasted revenue transactions denominated in foreign currencies. We also use forward contracts to mitigate the foreign currency exposure related to certain balance sheet items. We have not elected hedge accounting for the balance sheet related items.
The following quantitative information includes the impact of currency movements on forward contracts used in both programs. As of December 31, 2010 and 2009, a hypothetical adverse 10% movement in foreign exchange rates compared to the U.S. dollar across all maturities (for example, a strengthening of the Euro) would result in a hypothetical decrease in the fair value of forward contracts of approximately $65.5 million and $64.4 million, respectively. Our use of this methodology to quantify the market risk of such instruments should not be construed as an endorsement of its accuracy or the accuracy of the related assumptions. The quantitative information about market risk is limited because it does not take into account all foreign currency operating transactions.
Certain of our debt instruments are variable rate instruments and our interest expense associated with these instruments is, therefore, subject to changes in market interest rates. As of December 31, 2010 and 2009, a 100 basis-point adverse movement (increase in LIBOR) would increase annual interest expense by approximately $0.1 million and $0.2 million, respectively.
In addition, the fair value of our marketable securities is subject to change as a result of potential changes in market interest rates. The potential change in fair value for interest rate sensitive instruments has been assessed on a hypothetical 100 basis point adverse movement across all maturities. As of December 31, 2010 and 2009, we estimate that such hypothetical adverse 100 basis point movement would result in a hypothetical loss in fair value of approximately $10.5 million and $16.9 million, respectively, to our interest rate sensitive instruments.
The returns from cash, cash equivalents and marketable securities will vary as short-term interest rates change. A 100 basis-point adverse movement (decrease) in short-term interest rates would decrease interest income by approximately $11.4 million and $12.5 million as of December 31, 2010 and 2009, respectively.
We are exposed to equity price risks on the marketable portion of equity securities included in our portfolio of investments entered into for the promotion of business and strategic objectives. These investments are generally in small capitalization stocks in the biotechnology industry sector. We regularly review the market prices of these investments for impairment purposes. A hypothetical adverse 10% movement in market values would result in a hypothetical loss in fair value of approximately $4.5 million and $1.0 million as of December 31, 2010 and 2009, respectively.
| Item 8. | Consolidated Financial Statements and Supplementary Data |
The information required by this Item 8 is contained on pages F-1 through F-71 of this report and is incorporated herein by reference.
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
None.
| Item 9A. | Controls and Procedures |
Disclosure Controls and Procedures and Internal Control over Financial Reporting
Controls and Procedures
We have carried out an evaluation, under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended), as of December 31, 2010. Based upon that evaluation, our principal
71
executive officer and principal financial officer concluded that, as of the end of the period covered by this report, our disclosure controls and procedures are effective in ensuring that (a) the information required to be disclosed by us in the reports that we file or submit under the Securities Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC's rules and forms, and (b) such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate to allow timely decisions regarding required disclosure. In designing and evaluating our disclosure controls and procedures, our management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and our management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting during the quarter ended December 31, 2010 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Management's Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over our financial reporting. Internal control over financial reporting is defined in Rules 13a-15(f) and 15d-15(f) under the Securities Exchange Act as a process designed by, or under the supervision of, a company's principal executive and principal financial officers and effected by a company's board of directors, management and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles in the U.S. (U.S. GAAP). Our internal control over financial reporting includes those policies and procedures that:
| • | pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect our transactions and dispositions of our assets; | |
| • | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with U.S. GAAP, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and | |
| • | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on our financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2010. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control - Integrated Framework.
Based on our assessment, our management has concluded that, as of December 31, 2010, our internal control over financial reporting is effective based on those criteria.
The effectiveness of our internal control over financial reporting as of December 31, 2010 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report, which is included herein.
| Item 9B. | Other Information |
None.
72
PART III
| Item 10. | Directors, Executive Officers and Corporate Governance |
The information concerning our executive officers is set forth under the heading "Our Executive Officers" in Part I of this report. The text of our code of business conduct, which includes the code of ethics that applies to our principal executive officer, principal financial officer, principal accounting officer or controller, and persons performing similar functions, is posted on our website, www.biogenidec.com, under the "Board of Directors - Corporate Governance" subsection of the "About Us" section of the site. We intend to make all required disclosures regarding any amendments to, or waivers from, provisions of our code of business conduct, at the same location of our website. Our corporate governance principles (also posted on www.biogenidec.com) prohibit our Board of Directors from granting any waiver of the code of ethics for any of our directors or executive officers. We include our website address in this report only as an inactive textual reference and do not intend it to be an active link to our website.
The response to the remainder of this item is incorporated by reference from the discussion responsive thereto in the sections entitled " Proposal 1 - Election of Directors," "Corporate Governance," "Stock Ownership - Section 16(a) Beneficial Ownership Reporting Compliance " and " Miscellaneous - Stockholder Proposals " contained in the proxy statement for our 2011 annual meeting of stockholders.
| Item 11. | Executive Compensation |
The response to this item is incorporated by reference from the discussion responsive thereto in the sections entitled " Executive Compensation and Related Information " and "Corporate Governance" contained in the proxy statement for our 2011 annual meeting of stockholders.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The response to this item is incorporated by reference from the discussion responsive thereto in the sections entitled "Stock Ownership" and "Equity Compensation Plan Information" contained in the proxy statement for our 2011 annual meeting of stockholders.
| Item 13. | Certain Relationships and Related Transactions, and Director Independence |
The response to this item is incorporated by reference from the discussion responsive thereto in the sections entitled " Certain Relationships and Related Person Transactions " and "Corporate Goverance " contained in the proxy statement for our 2011 annual meeting of stockholders.
| Item 14. | Principal Accountant Fees and Services |
The response to this item is incorporated by reference from the discussion responsive thereto in the section entitled " Proposal 2 - Ratification of the Selection of our Independent Registered Public Accounting Firm " contained in the proxy statement for our 2011 annual meeting of stockholders.
73
PART IV
| Item 15. | Exhibits, Financial Statement Schedules |
a. (1) Consolidated Financial Statements:
The following financial statements are filed as part of this report:
| Financial Statements | Page Number | |||
Consolidated Statements of Income | F-2 | |||
Consolidated Balance Sheets | F-3 | |||
Consolidated Statements of Cash Flows | F-4 | |||
Consolidated Statements of Equity | F-5 | |||
Notes to Consolidated Financial Statements | F-8 | |||
Report of Independent Registered Public Accounting Firm | F-71 | |||
(2) Financial Statement Schedules
Schedules are omitted because they are not applicable, or are not required, or because the information is included in the consolidated financial statements and notes thereto.
(3) Exhibits
The exhibits listed on the Exhibit Index beginning on page A-1, which is incorporated herein by reference, are filed or furnished as part of this report or are incorporated into this report by reference.
74
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
BIOGEN IDEC INC.
| By: | /s/ George A. Scangos |
George A. Scangos
Chief Executive Officer
Date: February 4, 2011
Pursuant to the requirements the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Name | Capacity | Date | ||||
/s/ George A. Scangos George A. Scangos | Director and Chief Executive Officer (principal executive officer) | February 4, 2011 | ||||
/s/ Paul J. Clancy Paul J. Clancy | Executive Vice President, Finance and Chief Financial Officer (principal financial officer) | February 4, 2011 | ||||
/s/ Robert E. Gagnon Robert E. Gagnon | Vice President, Finance, Chief Accounting Officer and Controller (principal accounting officer) | February 4, 2011 | ||||
/s/ William D. Young William D. Young | Director and Chairman of the Board of Directors | February 3, 2011 | ||||
Alexander J. Denner | Director | |||||
/s/ Caroline D. Dorsa Caroline D. Dorsa | Director | February 3, 2011 | ||||
/s/ Nancy L. Leaming Nancy L. Leaming | Director | February 3, 2011 | ||||
/s/ Richard C. Mulligan Richard C. Mulligan | Director | February 3, 2011 | ||||
/s/ Robert W. Pangia Robert W. Pangia | Director | February 4, 2011 | ||||
/s/ Stelios Papadopoulos Stelios Papadopoulos | Director | February 3, 2011 | ||||
75
| Name | Capacity | Date | ||||
/s/ Brian S. Posner Brian S. Posner | Director | February 3, 2011 | ||||
/s/ Eric K. Rowinsky Eric K. Rowinsky | Director | February 3, 2011 | ||||
/s/ Lynn Schenk Lynn Schenk | Director | February 3, 2011 | ||||
/s/ Stephen A. Sherwin Stephen A. Sherwin | Director | January 30, 2011 | ||||
76
BIOGEN
IDEC INC. AND SUBSIDIARIES
CONSOLIDATED FINANCIAL STATEMENTS