UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
FORM 10-K
(Mark One)
☒ | ANNUAL REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended September 30, 2017.
☐ | TRANSITION REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number 000-21898
ARROWHEAD PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
Delaware |
| 46-0408024 |
(State of incorporation) |
| (I.R.S. Employer Identification No.) |
225 S. Lake Avenue, Suite 1050
Pasadena, California 91101
(626) 304-3400
(Address and telephone number of principal executive offices)
Securities registered under Section 12(b) of the Exchange Act:
Title of each class |
| Name of each exchange on which registered |
Common Stock, $0.001 par value |
| The NASDAQ Global Select Market |
Securities registered pursuant to Section 12(g) of the Exchange Act:
None
Indicate by a check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by a check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☒
Indicate by check mark whether the Registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See definitions of "large accelerated filer," "accelerated filer," "smaller reporting company" and "emerging growth company" in Rule 12b-2 of the Exchange Act.
Large accelerated filer ☐ |
| Accelerated filer ☒ |
| Non-accelerated filer ☐ |
| Smaller Reporting Company ☐ |
Emerging growth company ☐ |
|
|
|
|
|
|
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☒
The aggregate market value of issuer's voting and non-voting outstanding Common Stock held by non-affiliates was approximately $137 million based upon the closing stock price of issuer's Common Stock on March 31, 2017. Shares of common stock held by each officer and director and by each person who is known to own 10% or more of the outstanding Common Stock have been excluded in that such persons may be deemed to be affiliates of the Company. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
As of December 6, 2017, 74,828,280 shares of the issuer's Common Stock were issued and outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Definitive Proxy Statement to be filed for Arrowhead Pharmaceuticals Inc.'s 2017 Annual Meeting of Stockholders are incorporated by reference into Part III hereof.
PART I |
|
|
|
|
|
|
| ||
I TEM 1. |
| B USINESS |
| 1 |
I TEM 1A. |
| R ISK F ACTORS |
| 23 |
I TEM 1B. |
| U NRESOLVED S TAFF C OMMENTS |
| 34 |
I TEM 2. |
| P ROPERTIES |
| 34 |
I TEM 3. |
| L EGAL P ROCEEDINGS |
| 34 |
I TEM 4. |
| M INE S AFETY D ISCLOSURES |
| 34 |
|
|
| ||
PART II |
|
|
|
|
|
|
| ||
I TEM 5. |
| M ARKET FOR THE R EGISTRANT ' S C OMMON E QUITY , R ELATED S TOCKHOLDER M ATTERS AND I SSUER P URCHASES OF E QUITY S ECURITIES |
| 34 |
I TEM 6. |
| S ELECTED F INANCIAL D ATA |
| 37 |
I TEM 7. |
| M ANAGEMENT ' S D ISCUSSION AND A NALYSIS OF F INANCIAL C ONDITION AND R ESULTS OF O PERATIONS |
| 38 |
I TEM 7A. |
| Q UANTITATIVE AND Q UALITATIVE D ISCLOSURES A BOUT M ARKET R ISK |
| 52 |
I TEM 8. |
| F INANCIAL S TATEMENTS AND S UPPLEMENTARY D ATA |
| 52 |
I TEM 9. |
| C HANGES IN AND D ISAGREEMENTS WITH A CCOUNTANTS ON A CCOUNTING AND F INANCIAL D ISCLOSURE |
| 52 |
I TEM 9A. |
| C ONTROLS AND P ROCEDURES |
| 52 |
I TEM 9B. |
| O THER I NFORMATION |
| 53 |
|
|
| ||
PART III |
|
|
|
|
|
|
| ||
I TEM 10. |
| D IRECTORS , E XECUTIVE O FFICERS , AND C ORPORATE G OVERNANCE |
| 53 |
I TEM 11. |
| E XECUTIVE C OMPENSATION |
| 53 |
I TEM 12. |
| S ECURITY O WNERSHIP OF C ERTAIN B ENEFICIAL O WNERS AND M ANAGEMENT AND R ELATED S TOCKHOLDERS |
| 53 |
I TEM 13. |
| C ERTAIN R ELATIONSHIPS , R ELATED T RANSACTIONS AND D IRECTORS I NDEPENDENCE |
| 53 |
I TEM 14. |
| P RINCIPAL A CCOUNTANT F EES AND S ERVICES |
| 53 |
|
|
| ||
PART IV |
|
|
|
|
|
|
| ||
I TEM 15. |
| E XHIBITS AND F INANCIAL S TATEMENT S CHEDULES |
| 53 |
|
| |||
SIGNATURE |
| 56 | ||
|
| |||
INDEX TO FINANCIAL STATEMENTS AND SCHEDULES |
| F-1 | ||
FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains certain forward-looking statements within the meaning of Section 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934, and we intend that such forward-looking statements be subject to the safe harbors created thereby. For this purpose, any statements contained in this Annual Report on Form 10-K except for historical information may be deemed to be forward-looking statements. Without limiting the generality of the foregoing, words such as "may," "will," "expect," "believe," "anticipate," "intend," "could," "estimate," or "continue" or the negative or other variations thereof or comparable terminology are intended to identify forward-looking statements. In addition, any statements that refer to projections of our future financial performance, trends in our businesses, or other characterizations of future events or circumstances are forward-looking statements.
The forward-looking statements included herein are based on current expectations of our management based on available information and involve a number of risks and uncertainties, all of which are difficult or impossible to predict accurately and many of which are beyond our control. As such, our actual results may differ significantly from those expressed in any forward-looking statements. Factors that may cause or contribute to such differences include, but are not limited to, those discussed in more detail in Item 1 (Business) and Item 1A (Risk Factors) of Part I and Item 7 (Management's Discussion and Analysis of Financial Condition and Results of Operations) of Part II of this Annual Report on Form 10-K. Readers should carefully review these risks, as well as the additional risks described in other documents we file from time to time with the Securities and Exchange Commission. In light of the significant risks and uncertainties inherent in the forward-looking information included herein, the inclusion of such information should not be regarded as a representation by us or any other person that such results will be achieved, and readers are cautioned not to place undue reliance on such forward-looking information. Except as may be required by law, we disclaim any intent to revise the forward-looking statements contained herein to reflect events or circumstances after the date hereof or to reflect the occurrence of unanticipated events.
PART I
Unless otherwise noted, (1) the term "Arrowhead" refers to Arrowhead Pharmaceuticals, Inc., a Delaware corporation and its Subsidiaries, (2) the terms "Company," "we," "us," and "our," refer to the ongoing business operations of Arrowhead and its Subsidiaries, whether conducted through Arrowhead or a subsidiary of Arrowhead, (3) the term "Subsidiaries" refers collectively to Arrowhead Madison Inc. ("Arrowhead Madison"), Arrowhead Australia Pty Ltd ("Arrowhead Australia") and Ablaris Therapeutics, Inc. ("Ablaris"), (4) the term "Common Stock" refers to Arrowhead's Common Stock, (5) the term "Preferred Stock" refers to Arrowhead's Preferred Stock and (6) the term "Stockholder(s)" refers to the holders of Arrowhead Common Stock.
ITEM 1. | BUSINESS |
Description of Business
Arrowhead develops medicines that treat intractable diseases by silencing the genes that cause them. Using a broad portfolio of RNA chemistries and efficient modes of delivery, Arrowhead therapies trigger the RNA interference mechanism to induce rapid, deep and durable knockdown of target genes. RNA interference, or RNAi, is a mechanism present in living cells that inhibits the expression of a specific gene, thereby affecting the production of a specific protein. Deemed to be one of the most important recent discoveries in life science with the potential to transform medicine, the discoverers of RNAi were awarded a Nobel Prize in 2006 for their work. Arrowhead's RNAi-based therapeutics leverage this natural pathway of gene silencing.
In fiscal 2017, Arrowhead refocused its resources on therapeutics that exclusively utilize the company's Targeted RNAi Molecule (TRiM TM ) platform technology. Therapeutics built on the TRiM TM platform have demonstrated high levels of pharmacologic activity in multiple animal models spanning several therapeutic areas. TRiM TM enabled therapeutics offer several potential advantages over prior generation and competing technologies, including: simplified manufacturing and reduced costs; multiple routes of administration including subcutaneous injection and inhaled administration; ability to target multiple tissue types including liver, lung, and tumors; and the potential for improved safety and reduced risk of intracellular buildup, because there are less metabolites from smaller, simpler molecules.
As part of the refocusing of resources, Arrowhead announced in November 2016 that it would be discontinuing all clinical programs, that utilized the intravenously administered dynamic polyconjugate (DPC), or EX1, delivery vehicle. The decision to discontinue development of EX1-containing programs was based primarily on two factors. First, during discussions with regulatory agencies and outside experts, it became apparent that there would be substantial delays in all clinical programs that utilize EX1, while the Company further explored the cause of deaths in a non-clinical toxicology study in non-human primates exploring doses of EX1 higher than those planned to be used in humans. Second, the Company had made substantial advances in RNA chemistry and targeting, resulting in large potency gains for development programs utilizing the TRiM TM technology, making EX1 no longer necessary.
1
Pipeline Overview
Arrowhead is focused on developing innovative drugs for diseases with a genetic basis, typically characterized by the overproduction of one or more proteins. The depth and versatility of our RNAi technologies enable us to address conditions in virtually any therapeutic area and pursue disease targets that are not otherwise addressable by small molecules and biologics.
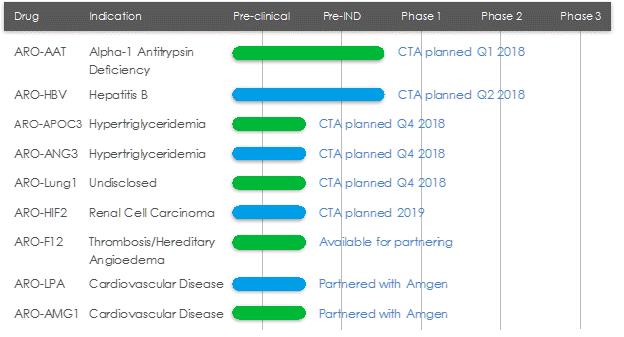
*Planned CTA filing time points discussed in figure above and in the paragraphs below are based on calendar quarters, not fiscal.
ARO-AAT
ARO-AAT is an RNAi therapeutic candidate for the treatment of liver disease associated with alpha-1 antitrypsin deficiency. ARO-AAT is designed to knock down the Alpha-1 antitrypsin (AAT) gene transcript and reduce the hepatic production of the mutant AAT protein. ARO-AAT is a next-generation subcutaneously administered compound that follows previous generation AAT compound ARC-AAT. A Clinical Trial Application (CTA) is planned for the first quarter of 2018.
Goal of ARO-AAT Treatment
The goal of ARO-AAT treatment is prevention and potential reversal of Z-AAT accumulation-related liver injury and fibrosis. Reduction of inflammatory Z-AAT protein, which has been clearly defined as the cause of progressive liver disease in AATD patients, is important as it is expected to halt the progression of liver disease and allow fibrotic tissue repair.
Alpha-1 Antitrypsin Deficiency (AATD)
AATD is a genetic disorder associated with liver disease in children and adults, and pulmonary disease in adults. AAT is a circulating glycoprotein protease inhibitor that is primarily synthesized and secreted by liver hepatocytes. Its physiologic function is the inhibition of neutrophil proteases to protect healthy tissues during inflammation and prevent tissue damage. The most common disease variant, the Z mutant, has a single amino acid substitution that results in improper folding of the protein. The mutant protein cannot be effectively secreted and accumulates in globules in the hepatocytes. This triggers continuous hepatocyte injury, leading to fibrosis, cirrhosis, and increased risk of hepatocellular carcinoma.
Current Treatments
Individuals with the homozygous PiZZ genotype have severe deficiency of functional AAT leading to pulmonary disease and hepatocyte injury and liver disease. Lung disease is frequently treated with AAT augmentation therapy. However, augmentation
2
therapy does nothing to treat liver disease, and there is no specific therapy for hepatic manifestations. There is a significant unmet need as liver transplant, with its attendant morbidity and mortality, is currently t he only available cure.
The Alpha-1 Project
Arrowhead has an agreement with The Alpha-1 Project (TAP), the venture philanthropy subsidiary of the Alpha-1 Foundation. TAP's mission is to support organizations in pursuit of cures and therapies for lung and liver disease caused by AATD. Under the terms of the agreement, TAP has partially funded development of ARO-AAT. In addition to the funding, TAP will make its scientific advisors available to Arrowhead, assist with patient recruitment for clinical trials with its Alpha-1 Foundation Patient Research Registry, and engage in other collaborative efforts that support development of ARO-AAT.
ARO-HBV
ARO-HBV is an RNAi therapeutic candidate for the treatment of chronic hepatitis B infection with the goal of achieving a functional cure. ARO-HBV is a next-generation subcutaneously administered compound that follows previous generation HBV compounds ARC-520 and ARC-521. A CTA is planned for the second quarter of 2018.
Goal of ARO-HBV Treatment
ARO-HBV is designed to silence the production of all HBV gene products with the goal of achieving a functional cure. The siRNAs target multiple components of HBV production including the pregenomic RNA that would be reverse transcribed to generate the viral DNA. The siRNAs intervene at the mRNA level, upstream of where NUCs act, and target the mRNAs that produce HBsAg proteins, the viral polymerase, the core protein that forms the capsid, the pre-genomic RNA, the HBeAg, and the hepatitis B X antigen (HBxAg). NUCs are effective at reducing production of viral particles, but are ineffective at controlling production of HBsAg and other HBV gene products. Arrowhead believes that a reduction in the production of HBsAg and other proteins that NUCs are ineffective at controlling is necessary to effective HBV therapy, because those proteins are thought to be major contributors to repression of the immune system and the persistence of liver disease secondary to HBV infection.
Chronic Hepatitis B Virus
According to the World Health Organization, 240 million people worldwide are chronically infected with hepatitis B virus, of which 500,000 to 1,000,000 people die each year from HBV-related liver disease. Chronic HBV infection is defined by the presence of hepatitis B surface antigen (HBsAg) for more than six months. In the immune tolerant phase of chronic infection, which can last for many years, the infected person typically produces very high levels of viral DNA and viral antigens. However, the infection is not cytotoxic and the carrier may have no symptoms of illness. Over time, the ongoing production of viral antigens causes inflammation and necrosis, leading to elevation of liver enzymes such as alanine and aspartate transaminases, hepatitis, fibrosis, and liver cancer (hepatocellular carcinoma, or HCC). If untreated, as many as 25% to 40% of chronic HBV carriers ultimately develop cirrhosis or HCC. Antiviral therapy is generally prescribed when liver enzymes become elevated.
We see the need for a next generation HBV treatment with a finite treatment period and an attractive dosing regimen, and that can be used at earlier stages of disease. We believe a novel therapeutic approach that can effectively treat or provide a functional cure (seroclearance of HBsAg and with or without development of excess patient antibodies against HBsAg) has the potential to take significant market share and may expand the available market to include patients that are currently untreated.
3
Current Treatments
The current standard of care for treatment of chronic HBV infection is a daily oral dose of nucleotide/nucleoside analogs (NUCs) or a regimen of interferon injections for approximately one year. NUCs are generally well tolerated, but patients may need lifetime treatment because viral replication often rebounds upon cessation of treatment. Interferon therapeutics can result in a functional cure in 10-20% of some patient types, but treatment is often associated with significant side effects, including severe flu-like symptoms, marrow suppression, and autoimmune disorders.
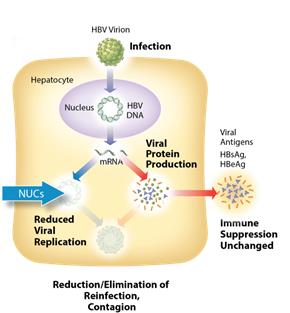
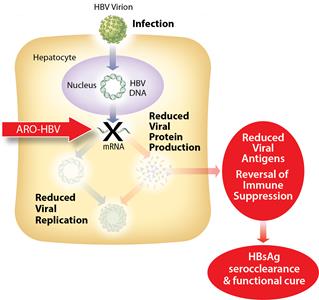
Figure 1: Mechanism of action NUCs Figure 2: Mechanism of action ARO-HBV
ARO-APOC3
ARO-APOC3 is designed to reduce production of Apolipoprotein C-III (apoC-III), a component of triglyceride rich lipoproteins (TRLs) including VLDL and chylomicrons and a key regulator of triglyceride metabolism. The company believes that knocking down the hepatic production of apoC-III may result in reduced VLDL synthesis and assembly, enhanced breakdown of TRLs, and better clearance of VLDL and chylomicron remnants. A CTA is planned for the fourth quarter of 2018.
Hypertriglyceridemia
Elevated triglyceride levels are an independent risk factor for cardiovascular disease. Severely elevated triglycerides (often over 2,000 mg/dL) in patients with familial chylomicronemia syndrome (FCS), a rare genetic disorder, can result in potentially fatal acute pancreatitis.
ARO-ANG3
ARO-ANG3 is designed to reduce production of angiopoietin-like protein 3 (ANGPTL3), a liver synthesized inhibitor of lipoprotein lipase and endothelial lipase. ANGPTL3 inhibition has been shown to lower serum LDL, serum and liver triglyceride and has genetic validation as a novel target for cardiovascular disease. A CTA is planned for the fourth quarter of 2018.
Hyperlipidemia and Hypertriglyceridemia
Hyperlipidemia and hypertriglyceridemia are risk factors for atherosclerotic coronary heart disease and cardiovascular events.
ARO-LUNG1
4
ARO-LUNG1 is Arrowhead's first therapeutic candidate to utilize the TRiM TM platform to target an undisclosed disease of the lung. A CTA is planned for the fourth qu arter of 2018.
ARO-HIF2
ARO-HIF2 is being developed for the treatment of clear cell renal cell carcinoma (ccRCC). ARO-HIF2 is designed to inhibit the production of HIF-2α, which has been linked to tumor progression and metastasis in ccRCC. Arrowhead believes it is an attractive target for intervention because over 90% of ccRCC tumors express a mutant form of the Von Hippel-Landau protein that is unable to degrade HIF-2α, leading to its accumulation during tumor hypoxia and promoting tumor growth. It is the first drug candidate to utilize the TRiM TM platform to target tumors. A CTA is planned for 2019.
ARO-F12
ARO-F12 is in development as a potential treatment for factor 12 (F12) mediated diseases such as hereditary angioedema (HAE) and thromboembolic disorders. Factor 12 initiates the intrinsic coagulation pathway and reducing its production using Arrowhead's RNAi technology may present opportunities in both disease areas.
Thrombosis and HAE
Thrombosis is the formation of blood clots that can obstruct blood flow. Broadly speaking, thrombosis can occur in veins or arteries and may cause serious repercussions if not treated. HAE is a rare genetic disorder with a prevalence of approximately 1/5,000-1/10,000. Patients with HAE can experience recurrent and dangerous acute inflammatory attacks in multiple tissues, with attacks of laryngeal edema being particularly serious and potentially fatal.
Partner-based Programs
ARO-LPA (AMG 890) and ARO-AMG1
ARO-LPA (AMG 890) is designed to reduce production of apolipoprotein A, a key component of lipoprotein(a), which has been genetically linked with increased risk of cardiovascular diseases, independent of cholesterol and LDL levels. Amgen acquired a worldwide, exclusive license in September 2016 to develop and commercialize ARO-LPA (AMG 890).
ARO-AMG1 is being developed against an undisclosed genetically-validated cardiovascular target under a license and collaboration agreement with Amgen.
Under the terms of the agreements taken together for ARO-LPA (AMG 890) and ARO-AMG1, the Company received $35 million in upfront payments, $21.5 million in the form of an equity investment by Amgen in the Company's Common Stock, and the Company is eligible to receive up to $617 million in option payments and development, regulatory and sales milestone payments. The Company is further eligible to receive single-digit royalties for sales of products under the ARO-AMG1 agreement and up to low double-digit royalties for sales of products under the ARO-LPA (AMG 890) agreement.
RNA Interference & the Benefits of RNAi Therapeutics
RNA interference (RNAi) is a mechanism present in living cells that inhibits the expression of a specific gene, thereby affecting the production of a specific protein. Deemed to be one of the most important recent discoveries in life science with the potential to transform medicine, the discoverers of RNAi were awarded a Nobel Prize in 2006 for their work. RNAi-based therapeutics may leverage this natural pathway of gene silencing to target and shut down specific disease-causing genes.
Small molecule and antibody drugs have proven effective at inhibiting certain cell surface, intracellular, and extracellular targets. However, other drug targets have proven difficult to inhibit with traditional drug-based and biologic therapeutics. Developing effective drugs for these targets would have the potential to address large underserved markets for the treatment of many diseases. Using the ability to specifically silence any gene, RNAi therapeutics may be able to address previously "undruggable" targets, unlocking the market potential of such targets.
5
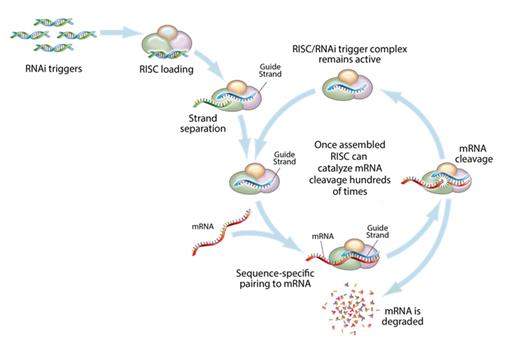
Figure 3: Mechanism of RNA interference
This figure depicts the mechanism by which gene silencing occurs. Double stranded RNAi triggers are introduced into a cell and get loaded into the RNA-induced silencing complex, (RISC). The strands are then separated, leaving an active RISC/RNAi trigger complex. This complex can then pair with and degrade the complementary messenger RNAs (mRNA), and stop the production of the target proteins. RNAi is a catalytic process, so each RNAi trigger can degrade mRNA hundreds of times, which results in a relatively long duration of effect for RNAi therapeutics.
Key Advantages of RNAi as a Therapeutic Modality
| • | Silences the expression of disease causing genes; |
| • | Potential to address any target in the transcriptome including previously "undruggable" targets; |
| • | Rapid lead identification; |
| • | High specificity; |
| • | Opportunity to use multiple RNA sequences in one drug product for synergistic silencing of related targets; and |
| • | RNAi therapeutics are uniquely suited for personalized medicine through target and cell specific delivery and gene knockdown. |
6
Targeted RNAi Molecule (TRiM TM ) Platform
Arrowhead's Targeted RNAi Molecule (TRiM TM ) platform utilizes ligand-mediated delivery and is designed to enable tissue-specific targeting while being structurally simple. Targeting has been core to Arrowhead's development philosophy and the TRiM TM platform builds on more than a decade of work on actively targeted drug delivery vehicles. Arrowhead scientists have discovered ways to progressively "TRiM" away extraneous features and chemistries and retain optimal pharmacologic activity.
The TRiM TM platform comprises a highly potent RNA trigger identified using Arrowhead's proprietary trigger selection rules and algorithms with the following components optimized, as needed, for each drug candidate: a high affinity targeting ligand; various linker and chemistries; structures that enhance pharmacokinetics; and highly potent RNAi triggers with sequence specific stabilization chemistries.
Therapeutics developed with the TRiM TM platform offer several advantages: simplified manufacturing and reduced costs; multiple routes of administration; and potential for improved safety because there are less metabolites from smaller molecules, thereby reducing the risk of intracellular buildup. At Arrowhead, we also believe that for RNAi to reach its true potential, it must target organs outside the liver. Arrowhead is leading this expansion with the TRiM TM platform, which has the potential to reach multiple tissues, including liver, lung, and tumor.
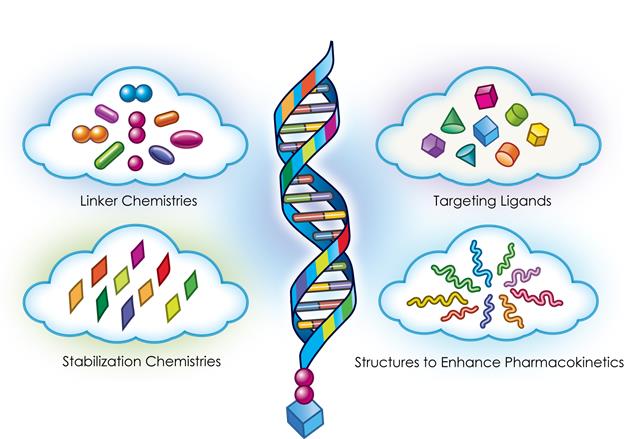
Figure 4: Targeted RNAi Molecule (TRiM TM ) Schematic
RNA Chemistries
The structure and chemistries of the oligonucleotide molecules used to trigger the RNAi mechanism can be tailored for optimal activity. Arrowhead's broad portfolio of RNA trigger structures and chemistries, including some proprietary structures, enable the company to optimize each drug candidate on a target-by-target basis and utilize the combination of structure and chemical modifications that yield the most potent RNAi trigger.
As a component of the TRiM TM platform, Arrowhead's design philosophy for RNA chemical modifications is to start with a structurally simple molecule and add only selective modification and stabilization chemistries as necessary to achieve the desired level
7
of target knockdown and dura tion of effect. The conceptual framework for the stabilization strategy starts with a more sophisticated RNAi trigger screening and selection process that identifies potent sequences rapidly in locations that others may miss. We pursue chemical stabilizati on strategies with a target duration of effect of 30-90 days and typically limit the use of strategies that produce longer activity because we anticipate that such strategies will increase long-term safety risks.
Intellectual Property and Key Agreements
The Company controls approximately 333 issued patents (including 179 directed to RNAi trigger molecules; 17 directed to targeting groups or targeting compounds; and 19 for hydrodynamic gene delivery), including European validations, and approximately 280 currently pending patent applications worldwide from 53 different patent families. The Company's patent applications have been filed throughout the world, including, in the United States, Argentina, ARIPO (Africa Regional Intellectual Property Organization), Australia, Brazil, Canada, Chile, China, Eurasian Patent Organization, Europe, GCC (Gulf Cooperation Council), Hong Kong, Israel, India, Indonesia, Iraq, Jordan, Japan, Republic of Korea, Lebanon, Mexico, New Zealand, OAPI (African Intellectual Property Organization), Peru, Philippines, Russian Federation, Saudi Arabia, Singapore, Thailand, Taiwan, Uruguay, Venezuela, Vietnam, and South Africa.
RNAi Triggers
The Company owns or has filed patent applications directed to RNAi trigger molecules, which serve as the foundation of Arrowhead's TRiM TM platform, and are targeted to reduce expression of several gene targets, including the following:
Patent Group | Estimated Year(s) of Expiration |
RNAi Triggers Gene Target | |
HBV | 2032, 2036, 2037 |
AAT | 2035 |
LPA | 2036 |
Factor 12 | 2036 |
HIF2α | 2034, 2036 |
RRM2 | 2031 |
APOC3 | 2035 |
α-ENaC | 2028 |
β-ENaC | 2031 |
β-Catenin | 2033 |
Cx43 | 2029 |
HCV | 2023 |
HIF1A | 2026 |
HRH1 | 2027 |
HSF1 | 2030, 2032 |
FRP-1 | 2026 |
KRAS | 2033 |
PDtype4 | 2026 |
PI4Kinase | 2028 |
SYK | 2027 |
TNF-α | 2027, 2028 |
Delivery Technologies
The delivery technology-related patent applications, which include components used in Arrowhead's TRiM TM platform, have been filed and issued in the United States, Australia, Canada, Europe, France, Germany, Italy, Spain, Switzerland, United Kingdom, India, Japan, Mexico, New Zealand, Philippines, Russia, South Korea, Singapore, and South Africa. The Company also controls a number of patents directed to hydrodynamic nucleic acid delivery, which issued in the United States, Australia and Europe (validated in Austria, Belgium, Switzerland, Germany, Denmark, Spain, Finland, France, the United Kingdom, Hungary, Ireland, Italy, Netherlands and Sweden). The approximate year of expiration for each of these various groups of patents are set forth below:
8
Patent Group | Estimated Year of Expiration |
Targeting ligands and other RNAi delivery technologies | |
Targeting groups ( Galactose derivative trimer-PK ) | 2031 |
Targeting groups (αvβ3 integrin) | 2035 |
Targeting groups (αvβ6 integrin) | 2037 |
Targeting groups (Galactose derivative ligands) | 2037 |
RNAi agent design (5′-phosphate mimic) | 2037 |
Physiologically labile linkers | 2036 |
Biologically cleavable linkers | 2036 |
Transferrin targeting | 2028 |
LDLR targeting | 2028 |
Peptide targeting (CPP-Arg) | 2028 |
Peptide targeting (YM3-10H) | 2032 |
| |
Hydrodynamic delivery | |
Second iteration | 2020 |
Third iteration | 2024 |
The RNAi and drug delivery patent landscapes are complex and rapidly evolving. As such, we may need to obtain additional patent licenses prior to commercialization of our candidates. You should review the factors identified in "Risk Factors" in Part I, Item 1A of this Annual Report on Form 10-K.
Non-Exclusively Licensed Patent Rights obtained from Roche
On October 21, 2011, Arrowhead acquired the RNAi therapeutics business of Hoffmann-La Roche, Inc. and F. Hoffmann-La Roche Ltd. (collectively, "Roche"). Roche built this business unit in a manner that only a large pharmaceutical company is capable of: backed by expansive capital resources, Roche systematically acquired technologies, licensed expansive intellectual property rights, attracted leading scientists, and developed new technologies internally. At a time when the markets were questioning whether RNAi could become a viable therapeutic modality, we saw great promise in the technology broadly and the quality of what Roche built specifically. The acquisition provided us with two primary sources of value:
| • | Broad freedom to operate with respect to key patents directed to the primary RNAi-trigger formats: canonical, UNA, meroduplex, and dicer substrate structures; and |
| • | A large team of scientists experienced in RNAi and oligonucleotide delivery. |
Pursuant to this acquisition, Roche assigned to Arrowhead its entire rights under certain licenses including: the License and Collaboration Agreement between Roche and Alnylam Pharmaceuticals, Inc. ("Alnylam") dated July 8, 2007 (the "Alnylam License"); the Non-Exclusive Patent License Agreement between Roche and MDRNA, Inc. dated February 12, 2009 ("MDRNA License"); and the Non-Exclusive License Agreement between Roche and City of Hope dated September 19, 2011 (the "COH License") (Collectively the "RNAi Licenses"). The RNAi Licenses provide the Company with non-exclusive, worldwide, perpetual, irrevocable, royalty-bearing rights and the right to sublicense a broad portfolio of intellectual property relating to the discovery, development, manufacture, characterization, and use of therapeutic products that function through the mechanism of RNA interference for specified targets.
Terms of the 2007 Alnylam License
The Alnylam License provides us with a non-exclusive, worldwide, perpetual, irrevocable, royalty-bearing right and sublicensable license under Alnylam's rights in certain intellectual property existing as of its effective date, to engage in discovery, development, commercialization and manufacturing activities, including to make, have made, use, offer for sale, sell and import certain licensed products in certain fields. The fields include the treatment or prophylaxis of indications comprising an RNAi compound complementary to, and function in mediating the RNAi of, a target known or believed to be primarily implicated in one or more primary therapeutic areas. The primary therapeutic areas are cancer, hepatic, metabolic disease and pulmonary disease. The hepatic therapeutic area specifically excludes targets of infectious pathogen.
The Alnylam License excludes access to intellectual property specifically related to "Blocked Targets." "Blocked Targets" are those targets that are subject to a contractual obligation of a pre-existing agreement between Alnylam and its alliance partners.
Under the Alnylam License, we may be obligated to pay development and sales milestone payments of up to the mid to upper double-digit millions of dollars for each licensed product that progresses through clinical trials in a particular indication, receives
9
marketing approval for that indication and is the subject of a first commercial sale. Additionally, we may be obligated to pay mid to high single-digit percentage royalties on sales of such products.
Core Patents relating to RNAi
The RNAi Licenses include patents relating to the general structure, architecture, and design of double-stranded oligonucleotide molecules, which engage RNA interference mechanisms in a cell. These rights include the "Tuschl II" patents, including issued U.S. Patent Nos. 7,056,704; 7,078,196; 7,078,196; 8,329,463; 8,362,231; 8,372,968; and 8,445,327; "Tuschl I" patents, including U.S. Patent Nos. 8,394,628 and 8,420,391; and allowed "Tuschl I" patent application, U.S. Publication No. 2011024446; "City of Hope" patents, including U.S. Patent No. 8,084,599; and "Kreutzer-Limmer" patents assigned to Alnylam, including U.S. Patent Nos. 7,829,693; 8,101,594; 8,119,608; 8,202,980; and 8,168,776.
Thomas Tuschl is the first named inventor on "Tuschl I" and "Tuschl II." "Tuschl I" patents refers to the patents arising from the patent application entitled "The Uses of 21-23 Sequence-Specific Mediators of Double-Stranded RNA Interference as a Tool to Study Gene Function and as a Gene-Specific Therapeutic." "Tuschl II" patents refer to the patents and patent applications arising from the patent application entitled "RNA Interference Mediating Small RNA Molecules." "City of Hope" is the first named assignee of certain core RNAi trigger patents. The second named assignee of these patents is Integrated DNA Technologies, Inc. Kreutzer-Limmer patents refer to the Alnylam patents and patent applications, relating to core siRNA IP, which includes inventors Roland Kreutzer and Stefan Limmer.
Chemical modifications of double-stranded oligonucleotides
The RNAi Licenses also include patents related to modifications of double-stranded oligonucleotides, including modifications to the base, sugar, or internucleoside linkage, nucleotide mimetics, and end modifications, which do not abolish the RNAi activity of the double-stranded oligonucleotides. Also included are patents relating to modified double-stranded oligonucleotides, such as meroduplexes described in U.S. Patent No. 9,074,205 assigned to Marina Biotech (f/k/a MDRNA, Inc.), and microRNAs described in U.S. Patent Nos. 7,582,744; 7,674,778, and 7,772,387 assigned to Alnylam, as well as U.S. Patent No. 8,314,227 related to unlocked nucleic acids (UNA). The ‘227 patent was assigned by Marina Biotech to Arcturus Therapeutics, Inc. but remains part of the MDRNA License. The RNAi Licenses also include rights from INEX/Tekmira relating to lipid-nucleic acid particles, and oligonucleotide modifications to improve pharmacokinetic activity including resistance to degradation, increased stability, and more specific targeting of cells from Alnylam and Ionis Pharmaceuticals, Inc. (f/k/a ISIS Pharmaceuticals, Inc.).
Manufacturing techniques for the double-stranded oligonucleotide molecules or chemical modifications
The RNAi Licenses also include patents relating to the synthesis and manufacture of double-stranded oligonucleotide molecules for use in RNA interference, as well as chemical modifications of such molecules, as described above. These include methods of synthesizing the double-stranded oligonucleotide molecules such as in the core "Tuschl I" allowed U.S. Application No. 12/897,749, the core "Tuschl II" U.S. Patent Nos. 7,056,704; 7,078,196; and 8,445,327; and Alnylam's U.S. Patent No. 8,168,776, as well as methods of making chemical modifications of the double-stranded oligonucleotides such as described in Alnylam's U.S. Patent No. 7,723,509. Patent applications are currently pending that further cover manufacturing techniques for double-stranded oligonucleotide molecules or chemical modifications.
Uses and Applications of Double-Stranded Oligonucleotide Molecules or Chemical modifications
The RNAi Licenses also include patents related to uses of the double-stranded oligonucleotides that function through the mechanism of RNA interference. These include for example, the core "Tuschl I" U.S. Patent No. 8,394,628 and "Tuschl II" U.S. Patent No. 8,329,463; Alnylam's U.S. Patent Nos. 7,763,590; 8,101,594, and 8,119,608, and City of Hope‘s U.S. Patent No. 8,084,599. Other more specific uses have been acquired and patent applications are currently pending that cover additional end uses and applications of double-stranded oligonucleotides functioning through RNA interference.
2012 License to Alnylam
In consideration for licenses obtained from Alnylam to certain RNAi intellectual property, in January 2012 we granted Alnylam a worldwide non-exclusive, sublicensable royalty-bearing license under our broad and target-specific DPC intellectual property rights to research, develop and commercialize RNAi-based products against a single undisclosed target in combination with DPC technology. Under the license to Alnylam, Alnylam may be obligated to pay us development and sales milestone payments of up to the low double-digit millions of dollars for each licensed product that progresses through clinical trials, receives marketing approval and is the subject of a first commercial sale. Additionally, Alnylam may be obligated to pay us low single-digit percentage royalties on sales of such products.
10
Acquisition of Assets from Novartis
On March 3, 2015, the Company entered into an Asset Purchase and Exclusive License Agreement (the "RNAi Purchase Agreement") with Novartis pursuant to which the Company acquired Novartis' RNAi assets and rights thereunder. Pursuant to the RNAi Purchase Agreement, the Company acquired or licensed certain patents and patent applications owned or controlled by Novartis related to RNAi therapeutics, assignment of Novartis's rights under a license from Alnylam (the "Alnylam-Novartis License"), rights to three pre-clinical RNAi candidates, and a license to certain Novartis assets (the "Licensed Novartis Assets"). The patents acquired from Novartis include multiple patent families covering delivery technologies and RNAi-trigger design rules and modifications. The Licensed Novartis Assets include an exclusive, worldwide right and license, solely in the RNAi field, with the right to grant sublicenses through multiple tiers under or with respect to certain patent rights and know how relating to delivery technologies and RNAi-trigger design rules and modifications. Under the assigned Alnylam-Novartis License, the Company acquired a worldwide, royalty-bearing, exclusive license with limited sublicensing rights to existing and future Alnylam intellectual property (including intellectual property that came under Alnylam's control on or before March 31, 2016), excluding intellectual property concerning delivery technology, to research, develop and commercialize 30 undisclosed gene targets.
We see the Roche and Novartis acquisitions as a powerful combination of intellectual property, R&D infrastructure, and RNAi experts. This foundation and substantial progress made by Arrowhead scientists over the last few years enable us to develop what we think are optimal RNAi therapeutics.
Cardiovascular Collaboration and License Agreements with Amgen
On September 28, 2016, the Company entered into two Collaboration and License agreements and a Common Stock Purchase Agreement with Amgen. Under the First Collaboration and License Agreement, Amgen received an option to a worldwide, exclusive license to ARO-AMG1 , an RNAi therapy for an undisclosed genetically validated cardiovascular target. Under the Second Collaboration and License, Amgen received a worldwide, exclusive license to Arrowhead's novel, RNAi ARO-LPA (AMG 890) program. The ARO-LPA (AMG-890) RNAi molecules are designed to reduce elevated lipoprotein(a), which is a genetically validated, independent risk factor for atherosclerotic cardiovascular disease. In both agreements, Amgen is wholly responsible for clinical development and commercialization. Under the terms of the agreements taken together, the Company has received $35 million in upfront payments, $21.5 million in the form of an equity investment by Amgen in the Company's Common Stock, and may receive up to $617 million in option payments and development, regulatory and sales milestone payments. The Company is further eligible to receive single-digit royalties for sales of products under the ARO-AMG1 agreement and up to low double-digit royalties for sales of products under the ARO-LPA (AMG 890) agreement.
Research and Development Facility
Arrowhead's research and development operations are located in Madison, Wisconsin. Substantially all of the Company's assets are located either in this facility or in our corporate headquarters in Pasadena. A summary of our research and development resources is provided below:
| • | Approximately 70 scientists currently; |
| • | State-of-the-art laboratories consisting of 60,000 total sq. ft.; |
| • | Complete small animal facility; |
| • | Primate colony housed at the Wisconsin National Primate Research Center, an affiliate of the University of Wisconsin; |
| • | In-house histopathology capabilities; |
| • | Animal models for cardio metabolic, viral, lung, and oncologic diseases; |
| • | Animal efficacy and safety assessment; |
| • | In-house drug manufacturing capabilities to produce first-in-human GMP (phase appropriate) material; |
| • | Polymer, peptide, oligonucleotide and small molecule synthesis and analytics capabilities (HPLC, NMR, MS, etc.); |
| • | Polymer, peptide and oligonucleotide PK, biodistribution, clearance methodologies; and |
| • | Conventional and confocal microscopy, flow cytometry, Luminex platform, qRT-PCR, clinical chemistry analytics. |
11
Research and Development Expenses
Research and development (R&D) expenses consist of costs incurred in discovering, developing and testing our clinical and preclinical candidates and platform technologies. R&D expenses also include costs related to clinical trials, including costs of contract research organizations to recruit patients and manage clinical trials. Other costs associated with clinical trials include manufacturing of clinical supplies, as well as good laboratory practice ("GLP") toxicology studies necessary to support clinical trials, both of which are outsourced to cGMP-compliant manufacturers and GLP-compliant laboratories. Total research and development expense for fiscal 2017 was $31.7 million, a decrease from $41.5 million in 2016 and a decrease from $47.3 million in 2015.
At September 30, 2017, we employed approximately 76 employees in an R&D function, primarily working from our facility in Madison, Wisconsin. These employees are engaged in various areas of research on Arrowhead candidate and platform development including synthesis and analytics, PK/biodistribution, formulation, CMC and analytics, tumor and extra-hepatic targeting, bioassays, live animal research, toxicology/histopathology, clinical and regulatory operations, and other areas. Salaries and payroll-related expenses for our R&D activities were $11.7 million in fiscal 2017, $13.9 million in fiscal 2016, and $11.6 million in fiscal 2015. Laboratory supplies including animal-related costs for in-vivo studies were $7.7 million, $4.3 million, and $3.1 million in fiscal 2017, 2016, and 2015, respectively.
Costs related to the manufacture of clinical supplies, GLP toxicology studies and other outsourced lab studies, as well as clinical trial costs were $21.3 million, $32.6 million, and $41.8 million in fiscal 2017, 2016, and 2015, respectively.
Facility-related costs, primarily rental costs for our leased laboratory in Madison, Wisconsin were $2.3 million, $1.3 million, and $1.0 million in fiscal 2017, 2016, and 2015, respectively. Other research and development expenses were $0.3 million, $3.2 million, and $1.4 million in fiscal 2017, 2016 and 2015, respectively. These expenses are primarily related to milestone payments, which can vary from period to period depending on the nature of our various license agreements, and the timing of reaching various development milestones requiring payment.
Government Regulation
Government authorities in the United States, at the federal, state, and local levels, and in other countries and jurisdictions, including the European Union, extensively regulate, among other things, the research, development, testing, product approval, manufacture, quality control, manufacturing changes, packaging, storage, recordkeeping, labeling, promotion, advertising, sales, distribution, marketing, and import and export of drugs and biologic products. All of our foreseeable product candidates are expected to be regulated as drugs. The processes for obtaining regulatory approval in the U.S. and in foreign countries and jurisdictions, along with compliance with applicable statutes and regulations and other regulatory authorities both pre- and post-commercialization, are a significant factor in the production and marketing of our products and our R&D activities and require the expenditure of substantial time and financial resources.
Review and Approval of Drugs in the United States
In the U.S., the FDA and other government entities regulate drugs under the Federal Food, Drug, and Cosmetic Act (the "FDCA"), the Public Health Service Act, and the regulations promulgated under those statutes, as well as other federal and state statutes and regulations. Failure to comply with applicable legal and regulatory requirements in the U.S. at any time during the product development process, approval process, or after approval, may subject us to a variety of administrative or judicial sanctions, such as a delay in approving or refusal by the FDA to approve pending applications, withdrawal of approvals, delay or suspension of clinical trials, issuance of warning letters and other types of regulatory letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil monetary penalties, refusals of or debarment from government contracts, exclusion from the federal healthcare programs, restitution, disgorgement of profits, civil or criminal investigations by the FDA, U.S. Department of Justice, State Attorneys General, and/or other agencies, False Claims Act suits and/or other litigation, and/or criminal prosecutions.
An applicant seeking approval to market and distribute a new drug in the U.S. must typically undertake the following:
(1) completion of pre-clinical laboratory tests, animal studies, and formulation studies in compliance with the FDA's GLP regulations;
(2) submission to the FDA of an Investigational New Drug Application ("IND") for human clinical testing, which must become effective without FDA objection before human clinical trials may begin;
(3) approval by an independent institutional review board ("IRB"), representing each clinical site before each clinical trial may be initiated;
12
(4) performance of adequate and well-controlled human clinical trials in accordance with the FDA's current good clinical practice ("cGCP") regulations, to establish the sa fety and effectiveness of the proposed drug product for each indication for which approval is sought;
(5) preparation and submission to the FDA of a New Drug Application ("NDA");
(6) satisfactory review of the NDA by an FDA advisory committee, where appropriate or if applicable,
(7) satisfactory completion of one or more FDA inspections of the manufacturing facility or facilities at which the drug product, and the active pharmaceutical ingredient or ingredients thereof, are produced to assess compliance with current good manufacturing practice ("cGMP") regulations and to assure that the facilities, methods, and controls are adequate to ensure the product's identity, strength, quality, and purity;
(8) payment of user fees, as applicable, and securing FDA approval of the NDA; and
(9) compliance with any post-approval requirements, such as any Risk Evaluation and Mitigation Strategies ("REMS") or post-approval studies required by the FDA.
Preclinical Studies and an IND
Preclinical studies can include in vitro and animal studies to assess the potential for adverse events and, in some cases, to establish a rationale for therapeutic use. The conduct of preclinical studies is subject to federal regulations and requirements, including GLP regulations. Other studies include laboratory evaluation of the purity, stability and physical form of the manufactured drug substance or active pharmaceutical ingredient and the physical properties, stability and reproducibility of the formulated drug or drug product. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and plans for clinical studies, among other things, to the FDA as part of an IND. Some preclinical testing, such as longer-term toxicity testing, animal tests of reproductive adverse events and carcinogenicity, may continue after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to a proposed clinical trial and places the trial on clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA allowing clinical trials to commence.
Following commencement of a clinical trial under an IND, the FDA may place a clinical hold on that trial. A clinical hold is an order issued by the FDA to the sponsor to delay a proposed clinical investigation or to suspend an ongoing investigation. A partial clinical hold is a delay or suspension of only part of the clinical work requested under the IND. For example, a specific protocol or part of a protocol is not allowed to proceed, while other protocols may do so. No more than 30 days after imposition of a clinical hold or partial clinical hold, the FDA will provide the sponsor a written explanation of the basis for the hold. Following issuance of a clinical hold or partial clinical hold, an investigation may only resume after the FDA has notified the sponsor that the investigation may proceed. The FDA will base that determination on information provided by the sponsor correcting the deficiencies previously cited or otherwise satisfying the FDA that the investigation can proceed.
Human Clinical Studies in Support of an NDA
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators in accordance with cGCP requirements, which include, among other things, the requirement that all research subjects provide their informed consent in writing before their participation in any clinical trial. Clinical trials are conducted under written study protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. In addition, an IRB representing each institution participating in the clinical trial must review and approve the plan for any clinical trial before it commences at that institution, and the IRB must conduct continuing review and reapprove the study at least annually. The IRB must review and approve, among other things, the study protocol and informed consent information to be provided to study subjects. An IRB must operate in compliance with FDA regulations. Information about certain clinical trials must be submitted within specific timeframes to the NIH for public dissemination on its ClinicalTrials.gov website.
Human clinical trials are typically conducted in three sequential phases, which may overlap or be combined:
Phase 1: The product candidate is initially introduced into healthy human subjects or patients with the target disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an early indication of its effectiveness.
13
Phase 2: The product candidate is administered to a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage.
Phase 3: The product candidate is administered to an expanded patient population, generally at geographically dispersed clinical trial sites, in well-controlled clinical trials to generate enough data to statistically evaluate the efficacy and safety of the product for approval, to establish the overall risk-benefit profile of the product, and to provide adequate information for the labeling of the product.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if serious adverse events occur. Phase 1, Phase 2, and Phase 3 clinical trials may not be completed successfully within any specified period, or at all. Furthermore, the FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution, or an institution it represents, if the clinical trial is not being conducted in accordance with the IRB's requirements or if the drug has been associated with unexpected serious harm to patients. The FDA will typically inspect one or more clinical sites in late-stage clinical trials to assure compliance with cGCP and the integrity of the clinical data submitted.
Submission of an NDA to the FDA
Assuming successful completion of required clinical testing and other requirements, the results of the preclinical and clinical studies, together with detailed information relating to the product's chemistry, manufacture, controls and proposed labeling, among other things, are submitted to the FDA as part of an NDA requesting approval to market the drug product for one or more indications. Under federal law, the submission of most NDAs is additionally subject to an application user fee, currently exceeding $2.4 million for applications requiring clinical data, and the sponsor of an approved NDA is also subject to an annual program fee, currently exceeding $300,000. These fees are typically increased annually.
Under certain circumstances, the FDA will waive the application fee for the first human drug application that a small business, defined as a company with less than 500 employees, including employees of affiliates, submits for review. An affiliate is defined as a business entity that has a relationship with a second business entity if one business entity controls, or has the power to control, the other business entity, or a third party controls, or has the power to control, both entities. In addition, an application to market a prescription drug product that has received orphan designation is not subject to a prescription drug user fee unless the application includes an indication for other than the rare disease or condition for which the drug was designated.
The FDA conducts a preliminary review of an NDA within 60 days of its receipt and informs the sponsor by the 74th day after the FDA's receipt of the submission to determine whether the application is sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA has agreed to specified performance goals in the review process of NDAs. Most such applications are meant to be reviewed within ten months from the date of filing, and most applications for "priority review" products are meant to be reviewed within six months of filing. The review process may be extended by the FDA for three additional months to consider new information or clarification provided by the applicant to address an outstanding deficiency identified by the FDA following the original submission.
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA will typically inspect one or more clinical sites to assure compliance with cGCP.
The FDA also may require submission of an REMS plan to mitigate any identified or suspected serious risks. The REMS plan could include medication guides, physician communication plans, assessment plans, and elements to assure safe use, such as restricted distribution methods, patient registries, or other risk minimization tools.
The FDA is required to refer an application for a novel drug to an advisory committee or explain why such referral was not made. Typically, an advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
14
The FDA's Decision on an NDA
On the basis of the FDA's evaluation of the NDA and accompanying information, including the results of the inspection of the manufacturing facilities, the FDA may issue an approval letter or a complete response letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. If and when those deficiencies have been addressed to the FDA's satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
If the FDA approves a product, it may limit the approved indications for use for the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, be conducted to further assess the drug's safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution restrictions or other risk management mechanisms, including REMS, which can materially affect the potential market and profitability of the product. After approval, the FDA may seek to prevent or limit further marketing of a product based on the results of post-market studies or surveillance programs. Some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval.
The product may also be subject to official lot release, meaning that the manufacturer is required to perform certain tests on each lot of the product before it is released for distribution. If the product is subject to official release, the manufacturer must submit samples of each lot, together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the manufacturer's tests performed on the lot, to the FDA. The FDA may in addition perform certain confirmatory tests on lots of some products before releasing the lots for distribution. Finally, the FDA will conduct laboratory research related to the safety and effectiveness of drug products.
Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the U.S. and for which there is no reasonable expectation that the cost of developing and making available in the U.S. a drug for this type of disease or condition will be recovered from sales in the U.S. for that drug. Orphan drug designation entitles the applicant to incentives such as grant funding towards clinical study costs, tax advantages, and waivers of FDA user fees. Orphan drug designation must be requested before submitting an NDA, and both the drug and the disease or condition must meet certain criteria specified in the Orphan Drug Act and FDA's implementing regulations at 21 C.F.R. Part 316. The granting of an orphan drug designation does not alter the standard regulatory requirements and process for obtaining marketing approval. Safety and effectiveness of a drug must be established through adequate and well-controlled studies.
After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. If a product that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other application to market the same drug for the same indication, except in very limited circumstances, for seven years. Orphan drug exclusivity does not prevent the FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition.
Expedited Review and Accelerated Approval Programs
A sponsor may seek approval of its product candidate under programs designed to accelerate the FDA's review and approval of NDAs. For example, Fast Track Designation may be granted to a drug intended for treatment of a serious or life threatening disease or condition and data demonstrate its potential to address unmet medical needs for the disease or condition. The key benefits of Fast Track Designation are the eligibility for priority review, rolling review (submission of portions of an application before the complete marketing application is submitted), and accelerated approval, if relevant criteria are met. The FDA may grant the NDA a priority review designation, which sets the target date for FDA action on the application at six months after the FDA accepts the application for filing. Priority review is granted where there is evidence that the proposed product would be a significant improvement in the safety or effectiveness of the treatment, diagnosis, or prevention of a serious condition. Priority review designation does not change the scientific/medical standard for approval or the quality of evidence necessary to support approval.
The FDA may approve an NDA under the accelerated approval program if the drug treats a serious condition, provides a meaningful advantage over available therapies, and demonstrates an effect on either (1) a surrogate endpoint that is reasonably likely to predict clinical benefit, or (2) on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. Post-marketing studies or completion of
15
ongoing studies after marketing approval are generally required to verify the drug's clinical benefit in relationship to the surrogate endpoint or ultimate outcome in relationship to the clinical benefit.
In addition, the Food and Drug Administration Safety and Innovation Act of 2012, or FDASIA, established the Breakthrough Therapy designation. A sponsor may seek FDA designation of its product candidate as a breakthrough therapy if the drug is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. If a drug is designated as breakthrough therapy, FDA will provide more intensive guidance on the drug development program and expedite its review.
Post-Approval Requirements
Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims, are subject to prior FDA review and approval. There also are continuing, annual user fee requirements for any marketed products and the establishments at which such products are manufactured, as well as new application fees for supplemental applications with clinical data.
In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies, and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with cGMP requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events or problems with manufacturing processes of unanticipated severity or frequency, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
| • | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| • | fines, warning letters or holds on post-approval clinical trials; |
| • | refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product license approvals; |
| • | product seizure or detention, or refusal to permit the import or export of products; or |
| • | injunctions or the imposition of civil or criminal penalties. |
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability.
In addition, the distribution of prescription pharmaceutical products is subject to the Prescription Drug Marketing Act (PDMA), which regulates the distribution of drugs and drug samples at the federal level, and sets minimum standards for the registration and regulation of drug distributors by the states. Both the PDMA and state laws limit the distribution of prescription pharmaceutical product samples and impose requirements to ensure accountability in distribution.
Abbreviated New Drug Applications for Generic Drugs
In 1984, with passage of the Drug Price Competition and Patent Term Restoration Act of 1984 (commonly referred to as the "Hatch-Waxman Amendments") amending the FDCA, Congress authorized the FDA to approve generic drugs that are the same as drugs previously approved by the FDA under the NDA provisions of the statute. To obtain approval of a generic drug, an applicant must submit an abbreviated new drug application (ANDA) to the agency. In support of such applications, a generic manufacturer may rely on the preclinical and clinical testing previously conducted for a drug product previously approved under an NDA, known as the
16
reference listed drug, or RLD. To reference that information, however, the ANDA applicant must demonstrate, and the FDA must conclude, that the generic drug does, in fact, perform in the same way as the RLD it purports to copy. Specifically, in order f or an ANDA to be approved, the FDA must find that the generic version is identical to the RLD with respect to the active ingredients, the route of administration, the dosage form, and the strength of the drug.
At the same time, the FDA must also determine that the generic drug is "bioequivalent" to the innovator drug. Under the statute, a generic drug is bioequivalent to a RLD if "the rate and extent of absorption of the generic drug do not show a significant difference from the rate and extent of absorption of the RLD." Upon approval of an ANDA, the FDA indicates that the generic product is "therapeutically equivalent" to the RLD and it assigns a therapeutic equivalence rating to the approved generic drug in its publication "Approved Drug Products with Therapeutic Equivalence Evaluations," also referred to as the "Orange Book." Physicians and pharmacists consider the therapeutic equivalence rating to mean that a generic drug is fully substitutable for the RLD. In addition, by operation of certain state laws and numerous health insurance programs, the FDA's designation of a therapeutic equivalence rating often results in substitution of the generic drug without the knowledge or consent of either the prescribing physician or patient.
Under the Hatch-Waxman Amendments, the FDA may not approve an ANDA until any applicable period of nonpatent exclusivity for the RLD has expired. The FDCA provides a period of five years of data exclusivity for new drug containing a new chemical entity. In cases where such exclusivity has been granted, an ANDA may not be filed with the FDA until the expiration of five years unless the submission is accompanied by a Paragraph IV certification, in which case the applicant may submit its application four years following the original product approval. The FDCA also provides for a period of three years of exclusivity if the NDA includes reports of one or more new clinical investigations, other than bioavailability or bioequivalence studies, that were conducted by or for the applicant and are essential to the approval of the application. This three-year exclusivity period often protects changes to a previously approved drug product, such as a new dosage form, route of administration, combination or indication.
Hatch-Waxman Patent Certification and the 30 Month Stay
Upon approval of an NDA or a supplement thereto, NDA sponsors are required to list with the FDA each patent with claims that cover the applicant's product or a method of using the product. Each of the patents listed by the NDA sponsor is published in the Orange Book. When an ANDA applicant files its application with the FDA, the applicant is required to certify to the FDA concerning any patents listed for the reference product in the Orange Book, except for patents covering methods of use for which the ANDA applicant is not seeking approval.
Specifically, the applicant must certify with respect to each patent that:
| • | the required patent information has not been filed; |
| • | the listed patent has expired; |
| • | the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or |
| • | the listed patent is invalid, unenforceable or will not be infringed by the new product. |
A certification that the new product will not infringe the already approved product's listed patents or that such patents are invalid or unenforceable is called a Paragraph IV certification. If the applicant does not challenge the listed patents or indicate that it is not seeking approval of a patented method of use, the ANDA application will not be approved until all the listed patents claiming the referenced product have expired. If the ANDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days after the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit or a decision in the infringement case that is favorable to the ANDA applicant.
To the extent that a Section 505(b)(2) applicant is relying on studies conducted for an already approved product, the applicant is required to certify to the FDA concerning any patents listed for the approved product in the Orange Book to the same extent that an ANDA applicant would. As a result, approval of a 505(b)(2) NDA can be stalled until all the listed patents claiming the referenced product have expired, until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired, and, in the case of a Paragraph IV certification and subsequent patent infringement suit, until the earlier of 30 months, settlement of the lawsuit or a decision in the infringement case that is favorable to the Section 505(b)(2) applicant.
17
Pediatric Studies and Exclusivity
Under the Pediatric Research Equity Act of 2003, an NDA or supplement thereto must contain data that are adequate to assess the safety and effectiveness of the drug product for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. With the enactment of FDASIA, sponsors must also submit pediatric study plans prior to the assessment data. Those plans must contain an outline of the proposed pediatric study or studies the applicant plans to conduct, including study objectives and design, any deferral or waiver requests, and other information required by regulation. The applicant, the FDA, and the FDA's internal review committee must then review the information submitted, consult with each other, and agree upon a final plan. The FDA or the applicant may request an amendment to the plan at any time.
The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements. Additional requirements and procedures relating to deferral requests and requests for extension of deferrals are contained in FDASIA. Unless otherwise required by regulation, the pediatric data requirements do not apply to products with orphan designation.
Pediatric exclusivity is another type of non-patent marketing exclusivity in the United States and, if granted, provides for the attachment of an additional six months of marketing protection to the term of any existing regulatory exclusivity, including the non-patent and orphan exclusivity. This six-month exclusivity may be granted if an NDA sponsor submits pediatric data that fairly respond to a written request from the FDA for such data. The data do not need to show the product to be effective in the pediatric population studied; rather, if the clinical trial is deemed to fairly respond to the FDA's request, the additional protection is granted. If reports of requested pediatric studies are submitted to and accepted by the FDA within the statutory time limits, whatever statutory or regulatory periods of exclusivity or patent protection cover the product are extended by six months. This is not a patent term extension, but it effectively extends the regulatory period during which the FDA cannot accept or approve another application.
Patent Term Restoration and Extension
A patent claiming a new drug product may be eligible for a limited patent term extension under the Hatch-Waxman Amendments. Those Amendments permit a patent restoration of up to five years for patent term lost during product development and the FDA regulatory review. The restoration period granted is typically one-half the time between the effective date of an IND and the submission date of a NDA, plus the time between the submission date of a NDA and ultimate approval. Patent term restoration cannot be used to extend the remaining term of a patent past a total of 14 years from the product's approval date. Only one patent applicable to an approved drug product is eligible for the extension, and the application for the extension must be submitted prior to the expiration of the patent in question. The U.S. Patent and Trademark Office reviews and approves the application for any patent term extension or restoration in consultation with the FDA.
Legislative Developments
The 21st Century Cures Act (Cures Act), which was signed into law in December 2016, includes provisions to accelerate the development and delivery of new treatments. For example, the Cures Act requires the FDA to establish a program to evaluate the potential use of real world evidence to help to support the approval of a new indication for an approved drug and to help to support or satisfy post-approval study requirements, to issue guidance on adaptive and novel clinical trial designs for new drugs, and to establish a process for qualifying drug development tools used to support FDA approval for marketing or investigational use of a drug. The Cures Act also permits the FDA to rely on qualified data summaries to support the approval of a supplemental application for an already approved drug. The FDA is in the process of implementing the Cures Act requirements.
Review and Approval of Drug Products in the European Union
In order to market any product outside of the United States, a company must also comply with numerous and varying regulatory requirements of other countries and jurisdictions regarding quality, safety and efficacy and governing, among other things, clinical trials, marketing authorization, commercial sales and distribution of our products. Whether or not it obtains FDA approval for a product, the company would need to obtain the necessary approvals by the comparable foreign regulatory authorities before it can commence clinical trials or marketing of the product in those countries or jurisdictions. The approval process ultimately varies between countries and jurisdictions and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries and jurisdictions might differ from and be longer than that required to obtain FDA approval. Regulatory approval in one country or jurisdiction does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country or jurisdiction may negatively impact the regulatory process in others.
Pursuant to the EU Clinical Trial Directive 2001/20/EC, a system for the approval of clinical trials in the European Union has been implemented through national legislation of the member states. Under this system, an applicant must obtain approval from the competent national authority of a European Union member state in which the clinical trial is to be conducted. Furthermore, the applicant may only start a clinical trial after a competent ethics committee has issued a favorable opinion. Clinical trial applications
18
must be accompanied by an investigational medicinal product dossier with supporting information prescribed by the European Clinical Trials Directive and correspond ing national laws of the member states and further detailed in applicable guidance documents.
In 2014, a new Clinical Trial Regulation 536/2014, replacing the EU Clinical Trials Directive, was adopted. The new Regulation will become directly applicable in all European Union member states (without national implementation) once the relevant EU portal and database are fully functional. It is expected that this will occur in the second half of 2019. The new Regulation seeks to simplify and streamline the approval of clinical trials in the European Union, in particular through a harmonised electronic submission and assessment process for clinical trials conducted in multiple Member States. As part of the application process, the sponsor shall propose a reporting Member State, who will coordinate the validation and evaluation of the application. The reporting member state shall consult and coordinate with the other concerned member states. If an application is rejected, it can be amended and resubmitted through the EU portal. If an approval is issued, the sponsor can start the clinical trial in all concerned member states. However, a concerned member state can in limited circumstances declare an "opt-out" from an approval. In such a case, the clinical trial cannot be conducted in that member state. The Regulation also aims to streamline and simplify the rules on safety reporting, and introduces enhanced transparency requirements such as mandatory submission of a summary of the clinical trial results to the EU database. Information stored in the EU database will be made publicly available subject to transparency rules.
To obtain marketing approval of a drug under European Union regulatory systems, an applicant must submit a marketing authorization application (MAA) either under a centralized or decentralized procedure.
The centralized procedure provides for the grant of a single marketing authorization by the European Commission that is valid for all European Union member states. The centralized procedure is compulsory for specific products, including for medicines produced by certain biotechnological processes, products designated as orphan medicinal products, advanced therapy products and products with a new active substance indicated for the treatment of certain diseases (AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and viral diseases). For products with a new active substance indicated for the treatment of other diseases and products that are highly innovative or for which a centralized process is in the interest of patients, the centralized procedure may be optional.
Under the centralized procedure, the Committee for Medicinal Products for Human Use (CHMP) established at the European Medicines Agency (EMA) is responsible for conducting the initial assessment of a drug. The CHMP is also responsible for several post-authorization and maintenance activities, such as the assessment of modifications or extensions to an existing marketing authorization. Under the centralized procedure in the European Union, the maximum timeframe for the evaluation of an MAA is 210 days, excluding clock stops, when additional information or written or oral explanation is to be provided by the applicant in response to questions of the CHMP. Accelerated evaluation might be granted by the CHMP in exceptional cases, when a medicinal product is of major interest from the point of view of public health and in particular from the viewpoint of therapeutic innovation. In this circumstance, the EMA ensures that the opinion of the CHMP is given within 150 days.
The decentralized procedure is available to applicants who wish to market a product in various European Union member states where such product has not received marketing approval in any European Union member state before. The decentralized procedure provides for approval by one or more other, or concerned, member states of an assessment of an application performed by one member state designated by the applicant, known as the reference member state. Under this procedure, an applicant submits an application based on identical dossiers and related materials, including a draft summary of product characteristics, and draft labeling and package leaflet, to the reference member state and concerned member states. The reference member state prepares a draft assessment report and drafts of the related materials within 120 days after receipt of a valid application. Within 90 days of receiving the reference member state's assessment report and related materials, each concerned member state must decide whether to approve the assessment report and related materials.
If a member state cannot approve the assessment report and related materials on the grounds of potential serious risk to public health, the disputed points are subject to a dispute resolution mechanism and may eventually be referred to the European Commission, whose decision is binding on all member states.
19
Data and Market Exclusivity in the European Union
In the European Union, new chemical entities qualify for eight years of data exclusivity upon marketing authorization and an additional two years of market exclusivity. This data exclusivity, if granted, prevents regulatory authorities in the European Union from referencing the innovator's data to assess a generic (abbreviated) application for eight years, after which generic marketing authorization can be submitted, and the innovator's data may be referenced, but not approved for two years. The overall ten-year period will be extended to a maximum of eleven years if, during the first eight years of those ten years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies. Even if a compound is considered to be a new chemical entity and the sponsor is able to gain the prescribed period of data exclusivity, another company nevertheless could also market another version of the drug if such company can complete a full MAA with a complete database of pharmaceutical test, preclinical tests and clinical trials and obtain marketing approval of its product.
Other legislation regarding marketing, authorization and pricing of pharmaceutical products in the European Union
Other core legislation relating to the marketing, authorization and pricing of pharmaceutical products in the European Union includes the following:
| • | Directive 2001/83/EC, establishing the requirements and procedures governing the marketing authorization for medicinal products for human use, as well as the rules for the constant supervision of products following authorization. This Directive has been amended several times, most recently by Directive 2012/26/EU regarding pharmacovigilance, and the Falsified Medicines Directive 2011/62/EU. |
| • | Regulation (EC) 726/2004, as amended, establishing procedures for the authorization , supervision and pharmacovigilance of medicinal products for human and veterinary use and establishing the EMA. |
| • | Regulation (EC) 469/2009, establishing the requirements necessary to obtain a Supplementary Protection Certificate, which extends the period of patent protection applicable to medicinal products at the EU-level. |
| • | Directive 89/105/EEC, ensuring the transparency of measures taken by the European Union member states to set the prices and reimbursements of medicinal products. Specifically, while each member state has competence over the pricing and reimbursement of medicines for human use, they must also comply with this Directive, which establishes procedures to ensure that member state decisions and policies do not obstruct trade in medicinal products. The European Commission proposed to repeal and replace Directive 89/105/EEC, but this proposal was withdrawn in 2015. |
| • | Directive 2003/94/EC, laying down the principles of good manufacturing practice in respect of medicinal products and investigational medicinal products for human use (the "GMP Directive") . |
| • | Directive 2005/28/EC of April 8 2005, laying down principles and detailed guidelines for good clinical practice as regards investigational medicinal products for human use, as well as the requirements for authorisation of the manufacturing or importation of such products" (the "GCP Directive"). |
New Legislation and Regulations
From time to time, legislation is drafted, introduced and passed in the European Union, its member states and other states of Europe that could significantly change the statutory provisions governing the testing, approval, manufacturing, marketing, coverage and reimbursement of pharmaceutical products. In addition to new legislation, pharmaceutical regulations and policies are often revised or interpreted by the EMA and national agencies in ways that may significantly affect our business and our products.
On March 29 2017, the United Kingdom triggered Article 50 of the Treaty of the Functioning of the European Union in order to formally leave the European Union (the so-called "Brexit"). We cannot predict the regulatory implications of Brexit in the (i) enforcement of EU law in the UK before Brexit (which might be less stringent); and / or (ii) the post-Brexit UK regime on market authorizations.
Pharmaceutical Coverage, Pricing and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of products approved by the FDA and other government authorities. Sales of products will depend, in part, on the extent to which the costs of the products will be covered by third-party payors, including government health programs such as, in the United States, Medicare and Medicaid, commercial health insurers and managed care organizations. The process for determining whether a payor will provide coverage for a product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the product once coverage is approved. Third-party payors may limit coverage to specific products on an approved list, or formulary, which might not include all of the approved products for a particular indication.
20
In order to secure coverage and reimbursement for any product that might be approved for sale, a company may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of the product, in addition to the costs required to obtain FDA or other comparable regulatory approvals. A payor's decision to provide coverage for a drug product does not necessarily imply that an adequate reimbursement rate will be approved. Third-party reimbursement may not be sufficien t to maintain price levels high enough to realize an appropriate return on our investment in product development.
The containment of healthcare costs has become a priority of federal, state and foreign governments, and the prices of drugs have been a focus in this effort. Third-party payors are increasingly challenging the prices charged for medical products and services and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. If these third-party payors do not consider a product to be cost effective compared to other available therapies, they may not cover the product after approval as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow a company to sell its products at a profit. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost containment programs to limit the growth of government-paid health care costs, including price controls, risk sharing, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs. Adoption of such controls and measures, and tightening of restrictive policies in jurisdictions with existing controls and measures, could limit payments for pharmaceuticals. As a result, the marketability of any product which receives regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement.
In addition, an increasing emphasis on managed care in the United States has increased and will continue to increase the pressure on drug pricing. Coverage policies, third-party reimbursement rates and drug pricing regulation may change at any time. In particular, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, contains provisions that may reduce the profitability of drug products, including, for example, increased rebates for drugs sold to Medicaid programs, extension of Medicaid rebates to Medicaid managed care plans, mandatory discounts for certain Medicare Part D beneficiaries and annual fees based on pharmaceutical companies' share of sales to federal health care programs. Even if favorable coverage and reimbursement status is attained for one or more products that receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
In the European Union, pricing and reimbursement schemes vary widely from country to country. Some countries provide that drug products may be marketed only after a reimbursement price has been agreed. Some countries may require the completion of additional studies that compare the cost-effectiveness of a particular product candidate to currently available therapies. For example, the European Union provides options for its member states to restrict the range of drug products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. European Union member states may approve a specific price for a drug product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the drug product on the market. Other member states allow companies to fix their own prices for drug products, but monitor and control company profits. The downward pressure on health care costs in general, particularly prescription drugs, has become intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries, cross-border imports from low-priced markets exert competitive pressure that may reduce pricing within a country. Any country that has price controls or reimbursement limitations for drug products may not allow favorable reimbursement and pricing arrangements for any of our products.
Healthcare Laws and Regulations
Healthcare providers, physicians and third-party payors will play a primary role in the recommendation and prescription of drug products that are granted marketing approval. Arrangements with healthcare providers, physicians, third-party payors and customers are subject to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute products for which we obtain marketing approval. Restrictions under applicable federal and state healthcare laws and regulations, include the following:
| • | the federal healthcare Anti-Kickback Statute prohibits, among other things, persons from soliciting, offering, receiving or providing any remuneration (in cash or in kind), directly or indirectly, to induce or reward either the referral of an individual for, or the purchase, lease, order or recommendation of, any item, facility or service for which payment may be made in whole or in part under a federal healthcare program such as Medicare and Medicaid; |
| • | the federal Foreign Corrupt Practices Act (FCPA) prohibits, among other things, U.S. corporations and persons acting on their behalf from offering, promising, authorizing or making payments to any foreign government official (including certain healthcare professionals in many countries), political party, or political candidate in an attempt to obtain or retain business or otherwise seek preferential treatment abroad; |
| • | the federal False Claims Act, which may be enforced by the U.S. Department of Justice or private whistlebloers to bring civil actions (qui tam actions) on behalf of the federal government, imposes civil penalties, as well as liability for damages and for attorneys' fees and costs, on individuals or entities for knowingly presenting, or causing to be presented, to the |
21
| federal government, claims for payment that are false or fraudulent, making a false statement material to a false or fraudulent claim, or improperly avoiding, decreasing, or concealing an obligation to pay money to the federal government; |
| • | the federal Health Insurance Portability and Accountability Act of 1996 (HIPAA) imposes criminal and civil liability for, among other conduct, executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters; |
| • | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act and its implementing regulations, also imposes criminal and civil liability and penalties on those who violate requirements, including mandatory contractual terms, intended to safeguard the privacy, security, transmission and use of individually identifiable health information; |
| • | the federal false statements statute relating to healthcare matters prohibits falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services; |
| • | the federal Physician Payment Sunshine Act requires manufacturers of drugs (among other products) to report to the Centers for Medicare and Medicaid Services within the U.S. Department of Health and Human Services information related to payments and other transfers of value to physicians and teaching hospitals, as well as physician ownership and investment interests in the reporting manufacturers; |
| • | similar state and foreign laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by nongovernmental third-party payors, including private insurers; |
| • | certain state laws require pharmaceutical companies to comply with voluntary compliance guidelines promulgated by a pharmaceutical industry association and relevant compliance guidance issues by HHS Office of Inspector General; bar drug manufacturers from offering or providing certain types of payments or gifts to physicians and other health care providers; and/or require disclosure of gifts or payments to physicians and other healthcare providers. |
Various state and foreign laws also govern the privacy and security of health information in some circumstances; many of these laws differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
Certain Financial Information
The financial information required in this Item 1 is included in Part II, Item 6 and Part IV, Item 15 of this Annual Report on Form 10-K.
Corporate Information
Arrowhead was originally incorporated in South Dakota in 1989 and was reincorporated in Delaware in 2000. In April 2016, Arrowhead changed its name from Arrowhead Research Corporation to Arrowhead Pharmaceuticals, Inc. The Company's principal executive offices are located at 225 South Lake Avenue, Suite 1050, Pasadena, California 91101, and its telephone number is (626) 304-3400. We also operate a research and development facility in Madison, Wisconsin. As of September 30, 2017, Arrowhead had 93 full-time employees.
Investor Information
Our website address is http://www.arrowheadpharmaceuticals.com Our reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended, including our annual reports on Form 10-K, our quarterly reports on Form 10-Q and our current reports on Form 8-K, and amendments to those reports, are accessible through our website, free of charge, as soon as reasonably practicable after these reports are filed electronically with, or otherwise furnished to, the SEC. These SEC reports can be accessed through the "Investors" section of our website.
You may read and copy any materials we file with the SEC at the SEC's Public Reference Room at 100 F Street, NE, Washington, DC 20549. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC also maintains an Internet website that contains reports, proxy and information statements, and other information regarding Arrowhead and other issuers that file electronically with the SEC. The SEC's Internet website address is http://www.sec.gov .
22
ITEM 1A. | RISK FACTORS |
You should carefully consider the risks discussed below and all of the other information contained in this report in evaluating us and an investment in our securities. If any of the following risks and uncertainties should occur, they could have a material adverse effect on our business, financial condition or results of operations. In that case, the trading price of our Common Stock could decline. Additionally, we note that we have accrued net losses annually since inception given the stage of our drug development. We urge you to consider our likelihood of success and prospects in light of the risks, expenses and difficulties frequently encountered by entities at similar stages of development.
Risks Related to Our Company
Risks Related to Our Discovery, Development, and Commercialization of Medicines
There are substantial risks inherent in attempting to commercialize our new drugs, and, as a result, we may not be able to successfully develop products for commercial use.
Our research and development efforts involve therapeutics based on RNA interference and our delivery systems, which are largely unproven technologies. Our scientists and engineers are working on developing technology in the early stages. However, such technology's commercial feasibility and acceptance are unknown. Scientific research and development requires significant amounts of capital and takes a long time to reach commercial viability, if it can be achieved at all. To date, our research and development projects have not produced commercially viable drugs, and may never do so. During the research and development process, we may experience technological barriers that we may be unable to overcome. Further, certain underlying premises in our development programs are not proven. For instance, the theory that knockdown of S-antigen in chronic hepatitis B patients will result in a functional cure is unproven. Similarly, the reduction of the production of mutant alpha-1 antitrypsin in the liver may not lead to a reduction of globules in the liver, and even if it leads to a reduction in such globules, this may not lead to other beneficial hepatic changes. It is also unknown at this time what changes in the liver may be required to gain regulatory approval and/or favorable reimbursement for a drug that reduces the production of mutant alpha-1 antitrypsin in the liver. Because of these and similar uncertainties, it is possible that no commercial products will be successfully developed. If we are unable to successfully develop commercial products, we will be unable to generate revenue or build a sustainable or profitable business.
There can be no assurance that our product candidates will obtain regulatory approval.
The sale of human therapeutic products in the U.S. and foreign jurisdictions is subject to extensive and time consuming regulatory approval which requires, among other things:
• | controlled research and human clinical testing; |
• | establishment of the safety and efficacy of the product; |
• | government review and approval of a submission containing manufacturing, pre-clinical and clinical data; and |
• | adherence to cGMP regulations during production and storage. |
The product candidates we currently have under development will require significant development, pre-clinical and clinical testing and investment of significant funds to gain regulatory approval before they can be commercialized. The results of our research and human clinical testing of our products may not meet regulatory requriements. Some of our product candidates, if approved, will require the completion of post-market studies. There can be no assurance that any of our products will be further developed and approved. The process of completing clinical testing and obtaining required approvals will take a number of years and require the use of substantial resources. Further, there can be no assurance that product candidates employing a new technology will be shown to be safe and effective in clinical trials or receive applicable regulatory approvals. If we fail to obtain regulatory approvals for any or all of our products, we will not be able to market such product and our operations may be adversely affected.
If testing of a particular product candidate does not yield successful results, then we will be unable to commercialize that product candidate.
We must demonstrate our product candidates' safety and efficacy in humans through extensive clinical testing. Our research and development programs are at an early stage of development. We may experience numerous unforeseen events during, or as a result of, the testing process that could delay or prevent commercialization of any products, including the following:
• | the results of pre-clinical studies may be inconclusive, or they may not be indicative of results that will be obtained in human clinical trials; |
23
• | safety and efficacy results attained in early human clinical trials may not be indicative of results that are obtained in later clinical trials; |
• | after reviewing test results, we may abandon projects that we might previously have believed to be promising; |
• | we or our regulators may suspend or terminate clinical trials because the participating subjects or patients are being exposed to unacceptable health risks; and |
• | our product candidates may not have the desired effects or may include undesirable side effects or other characteristics that preclude regulatory approval or limit their commercial use if approved. |
It may take us longer than we project to complete clinical trials, and we may not be able to complete them at all.
Although for planning purposes, we project the commencement, continuation and completion of our clinical trials, a number of factors, including scheduling conflicts with participating clinicians and clinical institutions, and difficulties in identifying or enrolling patients who meet trial eligibility criteria, may cause significant delays. We may not commence or complete clinical trials involving any of our product candidates as projected or may not conduct them successfully.
Even if our clinical studies are successful and we achieve regulatory approval, the approved product label may be more limited than we or analysts anticipate, which could limit the commercial opportunity for our product candidates.
At the time drugs are approved for commercialization, they are given a "product label" from the FDA or other regulatory body. In most countries this label sets forth the approved indication for marketing, and identifies potential safety concerns for prescribing physicians and patients. While we intend to seek as broad a product label as possible for our product candidates, we may receive a narrower label than is expected by either us or third parties, such as stockholders and securities analysts. For example, any approved products may only be indicated to treat refractory patients (i.e., those who have failed some other first-line therapy). Similarly, it is possible that only a specific sub-set of patients safely responds to one or more of our drug candidates. As a result, our product candidates, even if successful in clinical trials, could be approved only for a subset of patients. Additionally, safety considerations may result in contraindications that could further limit the scope of an approved product label. Any of these or other safety and efficacy considerations could limit the commercial opportunity for our product candidates.
Even if our product candidates are approved for commercialization, future regulatory reviews or inspections may result in the suspension or withdrawal of one or more of our products, closure of a facility or enforcement of substantial fines.
If regulatory approval to sell any of our product candidates is received, regulatory agencies will subject any marketed product(s), as well as the manufacturing facilities, to continual review and periodic inspection. If previously unknown problems with a product or manufacturing and laboratory facility are discovered, or we fail to comply with applicable regulatory approval requirements, a regulatory agency may impose restrictions on that product or on us. The agency may require the withdrawal of the product from the market, closure of the facility or enforcement of substantial fines.
We face potential product liability exposure, and if successful claims are brought against us, we may incur substantial liability for a product candidate and may have to limit its commercialization.
The use of our product candidates in clinical trials and the sale of any products for which we obtain marketing approval expose us to the risk of product liability claims. Product liability claims might be brought against us by clinical trial participants, consumers, health-care providers, pharmaceutical companies, or others selling our products. If we cannot successfully defend ourselves against these claims, we may incur substantial liabilities. Regardless of merit or eventual outcomes of such claims, product liability claims may result in:
• | decreased demand for our product candidates; |
• | impairment of our business reputation; |
• | withdrawal of clinical trial participants; |
• | costs of litigation; |
• | substantial monetary awards to patients or other claimants; and |
• | loss of revenues. |
|
|
|
|
Our insurance coverage may not be sufficient to reimburse us for all expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive and, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses.
24
The successful commercialization of our product candidates, if approved, will depend in part on the extent to which government authorities and health insurers establish adequate reimbursement levels and pricing policies.
Sales of any approved drug candidate will depend in part on the availability of coverage and reimbursement from third-party payers such as government insurance programs, including Medicare and Medicaid, private health insurers, health maintenance organizations and other health care related organizations, who are increasingly challenging the price of medical products and services. Accordingly, coverage and reimbursement may be uncertain. Adoption of any drug by the medical community may be limited if third-party payers will not offer coverage. Additionally, significant uncertainty exists as to the reimbursement status of newly approved drugs. Cost control initiatives may decrease coverage and payment levels for any drug and, in turn, the price that we will be able to charge and/or the volume of our sales. We are unable to predict all changes to the coverage or reimbursement methodologies that will be applied by private or government payers. Any denial of private or government payer coverage or inadequate reimbursement could harm our business and reduce our revenue. If we partner with third parties with respect to any of our product candidates, we may be reliant on that partner to obtain reimbursement from government and private payors for the drug, if approved, and any failure of that partner to establish adequate reimbursement could have a negative impact on our revenues and profitability.
In addition, both the federal and state governments in the United States and foreign governments continue to propose and pass new legislation, regulations, and policies affecting coverage and reimbursement rates, which are designed to contain or reduce the cost of health care. Further federal and state proposals and healthcare reforms are likely, which could limit the prices that can be charged for the product candidates that we develop and may further limit our commercial opportunity. There may be future changes that result in reductions in potential coverage and reimbursement levels for our product candidates, if approved and commercialized, and we cannot predict the scope of any future changes or the impact that those changes would have on our operations.
If future reimbursement for approved product candidates, if any, is substantially less than we project, or rebate obligations associated with them are substantially greater than we expect, our future net revenue and profitability could be materially diminished.
We may not enjoy the market exclusivity benefits of our orphan drug designation.
Although we may obtain orphan designations in the treatment of certain diseases our products are intended to treat, the designation may not be applicable to any particular product we might get approved and that product may not be the first product to receive approval for that indication. Under the Orphan Drug Act, the first product with an orphan designation receives market exclusivity, which prohibits FDA from approving the "same" drug for the same indication. The FDA has stated that drugs can be the "same" even when they are not identical, but has not provided guidance with respect to how it will determine "sameness" for RNAi drugs. It is possible that another RNAi drug could be approved for the treatment of a disease one of our orphan products is intended to treat before our product is approved, which means that we may not obtain orphan drug exclusivity and could also potentially be blocked from approval until the first product's orphan drug exclusivity period expires or we demonstrate, if we can, that our product is superior. Further, even if we obtain orphan drug exclusivity for a product, that exclusivity may not effectively protect the product from competition because different drugs can be approved for the same condition. Even after an orphan drug is approved and granted orphan drug exclusivity, the FDA can subsequently approve the same drug for the same condition if the FDA concludes that the later drug is safer, more effective or makes a major contribution to patient care.
Our success depends on the attraction and retention of senior management and scientists with relevant expertise.
Our future success depends to a significant extent on the continued services of our key employees, including our senior scientific, technical and managerial personnel. We do not maintain key person life insurance for any of our executives and we do not maintain employment agreements with many senior employees. Competition for qualified employees in the pharmaceutical industry is high, and our ability to execute our strategy will depend in part on our ability to continue to attract and retain qualified scientists and management. If we are unable to find, hire and retain qualified individuals, we will have difficulty implementing our business plan in a timely manner, or at all.
Risks Related to Our Intellectual Property
Our ability to protect our patents and other proprietary rights is uncertain, exposing us to the possible loss of competitive advantage.
We have licensed rights to pending patents and have filed and expect to continue to file patent applications. Researchers sponsored by us may also file patent applications that we may need to license. Such patent applications may not be available for licensing or may not be economically feasible to license. Certain of our patents may not be granted or may not contain claims of the necessary breadth because, for example, prior patents exist. If a particular patent is not granted, the value of the invention described in the patent would be diminished. Further, even if these patents are granted, they may be difficult to enforce. Even if ultimately successful, efforts to enforce our patent rights could be expensive, distracting for management, cause our patents to be invalidated or held unenforceable, and thus frustrate commercialization of products. Even if patents are issued and are enforceable, others may develop similar, superior or parallel technologies to any technology developed by us and not infringe on our patents. Our technology
25
may prove to infringe upon patents or rights owned by others. Patent prosecution and mai ntenance is expensive, and we may be forced to curtail prosecution or maintenance if our cash resources are limited. Thus, the patents held by or licensed to us may not afford us any meaningful competitive advantage. If we are unable to derive value from o ur licensed or owned intellectual property, the value of your investment may decline.
We are party to technology license agreements with third parties that require us to satisfy obligations to keep them effective and, if these agreements are terminated, our technology and our business would be seriously and adversely affected.
We are party to license agreements to incorporate third party proprietary technologies into our drug products under development. These license agreements require us to pay royalties and satisfy other conditions, including conditions in some cases related to the commercialization of the licensed technology. We may not be able to successfully incorporate these technologies into marketable products or, if we do, sales may not be sufficient to recover the amounts that we are obligated to pay to the licensors. If we fail to satisfy our obligations under these agreements, the terms of the licenses may be materially modified, such as by rendering currently exclusive licenses non-exclusive, or it may give our licensors the right to terminate their respective agreement with us, which would limit our ability to implement our current business plan and harm our business and financial condition.
We may be subject to patent infringement claims, which could result in substantial costs and liability and prevent us from commercializing our potential products.
Because the intellectual property landscape in the fields in which we participate is rapidly evolving and interdisciplinary, it is difficult to conclusively assess our freedom to operate without infringing on third party rights. However, we are currently aware of certain patent rights held by third parties that, if found to be valid and enforceable, could be alleged to render one or more of our drug candidates infringing. If a claim should be brought and is successful, we may be required to pay substantial damages, be forced to abandon any affected drug candidates and/or seek a license from the patent holder. In addition, any patent infringement claims brought against us, whether or not successful, may cause us to incur significant expenses and divert the attention of our management and key personnel from other business concerns. These could negatively affect our results of operations and prospects. We cannot be certain that patents owned or licensed by us will not be challenged, potentially successfully, by others.
In addition, if our product candidates infringe the intellectual property rights of third parties, these third parties may assert infringement claims against our customers, licensees, and other parties with whom we have business relationships and we may be required to indemnify those parties for any damages they suffer as a result of these claims. The claims may require us to initiate or defend protracted and costly litigation on behalf of customers, licensees, and other parties regardless of the merits of these claims. If any of these claims succeed, we may be forced to pay damages on behalf of those parties or may be required to obtain licenses for the products they use. If we cannot obtain all necessary licenses on commercially reasonable terms, we may be unable to continue selling such products.
We license patent rights from third-party owners and we rely on such owners to obtain, maintain and enforce the patents underlying such licenses.
We are a party to a number of licenses that give us rights to third-party intellectual property that is necessary or useful for our business. In particular, we have obtained licenses from, among others, Novartis and Alnylam. We also expect to enter into additional licenses to third-party intellectual property in the future.
Our success will depend in part on the ability of our licensors to obtain, maintain and enforce patent protection for our licensed intellectual property, in particular, those patents to which we have secured exclusive rights. Our licensors may not successfully prosecute the patent applications to which we are licensed. Even if patents are issued in respect of these patent applications, our licensors may fail to maintain these patents, may determine not to pursue litigation against other companies that are infringing these patents, or may pursue such litigation less aggressively than we would. Without protection for the intellectual property we license, other companies might be able to offer substantially identical products for sale, which could adversely affect our competitive business position and harm our business prospects.
Our technology licensed from various third parties may be subject to retained rights.
Our licensors often retain certain rights under their agreements with us, including the right to use the underlying technology for noncommercial academic and research use, to publish general scientific findings from research related to the technology, and to make customary scientific and scholarly disclosures of information relating to the technology. It is difficult to monitor whether our licensors limit their use of the technology to these uses, and we could incur substantial expenses to enforce our rights to our licensed technology in the event of misuse.
26
Confidentiality agreements with employees and others may not adequately prevent disclosure of trade secrets and other proprietary information.
In order to protect our proprietary technology and processes, we rely in part on confidentiality agreements with our collaborators, employees, consultants, outside scientific collaborators and sponsored researchers, and other advisors. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, others may independently discover trade secrets and proprietary information, and in such cases we could not assert any trade secret rights against such party. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
We may not be able to effectively secure first-tier technologies when competing against other companies or investors.
Our future success may require that we acquire patent rights and know-how to new or complimentary technologies. However, we compete with a substantial number of other companies that may also compete for technologies we desire. In addition, many venture capital firms and other institutional investors, as well as other pharmaceutical and biotech companies, invest in companies seeking to commercialize various types of emerging technologies. Many of these companies have greater financial, scientific and commercial resources than us. Therefore, we may not be able to secure the technologies we desire. Furthermore, should any commercial undertaking by us prove to be successful, there can be no assurance competitors with greater financial resources will not offer competitive products and/or technologies.
Risks Related to Our Business Model
Our business model assumes we will generate revenue by, among other activities, marketing or out-licensing the products we develop. Our drug candidates are in the early stages of development and because we have a short development history with both RNA interference and our delivery technologies, there is a limited amount of information about us upon which you can evaluate our business and prospects.
We have no approved drugs and thus have not begun to market or generate revenues from the commercialization of any products. We have only a limited history upon which one can evaluate our RNAi therapeutic business as our drug candidates are still at an early stage of development. Thus, we have limited experience and have not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the biopharmaceutical area. For example, to execute our business plan, we will need to successfully:
● | Execute product development activities using unproven technologies; |
● | Build, maintain, and protect a strong intellectual property portfolio; |
● | Demonstrate safety and efficacy of our drug candidates in multiple human clinical studies; |
● | Receive FDA approval and approval from similar foreign regulatory bodies; |
● | Gain market acceptance for the development and commercialization of any drugs we develop; |
● | Ensure our products are reimbursed by commercial and/or government payors at a rate that permits commercial viability; |
● | Develop and maintain successful strategic relationships with suppliers, distributors, and commercial licensing partners; |
● | Manage our spending and cash requirements as our expenses will increase in the near term if we add programs and additional preclinical and clinical trials; and |
● | Effectively market any products for which we obtain marketing approval. |
If we are unsuccessful in accomplishing these objectives, we may not be able to develop products, raise capital, expand our business or continue our operations.
We will need to establish additional relationships with strategic and development partners to fully develop our drug candidates and market any approved products.
During fiscal year 2016, we entered into two collaboration and license agreements with Amgen. Our business strategy includes obtaining additional collaborations with other pharmaceutical and biotech companies to support the development of our therapeutic siRNA and other drug candidates. We do not possess all of the financial and development resources necessary to develop and commercialize products that may result from our technologies. Unless we expand our product development capacity and enhance our internal marketing capability, we may need to make appropriate arrangements with strategic partners to develop and commercialize any drug candidates that may be approved. We may not be able to attract such partners, and even if we are able to enter into such partnerships, the terms may be less favorable than anticipated. Further, entering into partnership agreements may limit our
27
commercialization options and/or require us to share revenues and profits with our partners. If we do not find appropriate partners, or if our existing arrangements or future agreements are not successful, our ability to develop and commercialize products could be adversely affected. Even if we are able to find collabor ative partners, the overall success of the development and commercialization of product candidates in those programs will depend largely on the efforts of other parties and will be beyond our control. In addition, in the event we pursue our commercializati on strategy through collaboration or licenses to third parties, there are a variety of technical, business and legal risks, including:
● | We may not be able to control the amount and timing of resources that our collaborators may be willing or able to devote to the development or commercialization of our drug candidates or to their marketing and distribution; and |
● | Disputes may arise between us and our collaborators that result in the delay or termination of the research, development or commercialization of our drug candidates or that result in costly litigation or arbitration that diverts our management's resources. |
The occurrence of any of the above events or other related events could impair our ability to generate revenues and harm our business and financial condition.
We may lose a considerable amount of control over our intellectual property and may not receive anticipated revenues in strategic transactions, particularly where the consideration is contingent on the achievement of development or sales milestones.
Our business model has been to develop new technologies and to utilize the intellectual property created through the research and development process to develop commercially successful products. If the acquirers of our technologies fail to achieve performance milestones, we may not receive a significant portion of the total value of any sale, license or other strategic transaction.
We will need to achieve commercial acceptance of our drug candidates to generate revenues and achieve profitability.
Even if our research and development efforts yield technologically feasible applications, we may not successfully develop commercial products. Drug development takes years of study in human clinical trials prior to regulatory approval, and, even if we are successful, it may not be on a timely basis. During our drug development period, superior competitive technologies may be introduced which could diminish or extinguish the potential commercial uses for our drug candidates. Additionally, the degree to which the medical community and consumers will adopt any product we develop is uncertain. The rate and degree of market acceptance of our products will depend on a number of factors, including the establishment and demonstration in the medical community of the clinical efficacy and safety of our products, their potential advantage over alternative treatments, and the costs to patients and third-party payors, including insurance companies and Medicare. Recent efforts in the United States and abroad to reduce overall healthcare spending has put significant pressure on the price of prescription drugs and certain companies have been publicly criticized for the relatively high cost of their therapies. These pressures may force us to sell any approved drugs at a lower price than we or analysts may anticipate, or may result in lower levels of reimbursement and coverage from third parties.
We cannot predict whether significant commercial market acceptance for our products, if approved, will ever develop, and we cannot reliably estimate the projected size of any such potential market. Our revenue growth and achievement of profitability will depend substantially on our ability to introduce products that will be accepted by the medical community. If we are unable to cost-effectively achieve acceptance of our technology among the medical establishment and patients, or if the associated products do not achieve wide market acceptance, our business will be materially and adversely affected.
We rely on outside sources for various components and processes for our products.
We rely on third parties for various components and processes for our product candidates. We may not be able to achieve multiple sourcing because there may be no acceptable second source, other companies may choose not to work with us, or the component or process sought may be so new that a second source does not exist, or does not exist on acceptable terms. There may be a disruption or delay in the performance of our third-party contractors, suppliers or collaborators which is beyond our control. If such third parties are unable to satisfy their commitments to us, the development of our products would be adversely affected. Therefore, it is possible that our development plans will have to be slowed down or stopped completely at times due to our inability to obtain required raw materials, components, and outsourced processes at an acceptable cost, if at all, or to get a timely response from vendors.
We have limited manufacturing capability and must rely on third-party manufacturers to manufacture our clinical supplies and commercial products, if and when approved, and if they fail to meet their obligations, the development and commercialization of our products could be adversely affected.
We have limited manufacturing capabilities and experience. Our drug candidates are composed of multiple components and require specialized formulations for which scale-up and manufacturing could be difficult. We have limited experience in such scale-up and manufacturing requiring us to depend on a limited number of third parties, who may not be able to deliver in a timely manner, or at all. In order to develop products, apply for regulatory approvals, and commercialize our products, we will need to develop, contract
28
for, or otherwise arrange for the necessary manufacturing capabilities. Our internal manufacturing capabilities are limited to small-scale production of material for use in in vitro and in vivo experiments that is not required to be produ ced under cGMP standards beyond those applicable to active pharmaceutical ingredients used in phase 1 studies. There are a limited number of manufacturers that supply synthetic siRNAs. There are risks inherent in pharmaceutical manufacturing that could af fect the ability of our contract manufacturers to meet our delivery time requirements or provide adequate amounts of material to meet our needs. Included in these risks are synthesis and purification failures and contamination during the manufacturing proc ess, which could result in unusable product and cause delays in our development process, as well as additional expense to us.
Additionally, our product candidates have not yet been manufactured for commercial use. If any of our product candidates become approved for commercial sale, we will need to establish either internal or third-party manufacturing capacity. Manufacturing partner requirements may require us to fund capital improvements, perhaps on behalf of third parties, to support the scale-up of manufacturing and related activities. We may not be able to establish scaled manufacturing capacity for an approved product in a timely or economic manner, if at all. If we or our third party manufacturers are unable to provide commercial quantities of such an approved product, we will have to successfully transfer manufacturing technology to a different manufacturer. Engaging a new manufacturer for such an approved product could require us to conduct comparative studies or utilize other means to determine bioequivalence of the new and prior manufacturers' products, which could delay or prevent our ability to commercialize such an approved product. If we or any of these manufacturers is unable or unwilling to increase its manufacturing capacity or if we are unable to establish alternative arrangements on a timely basis or on acceptable terms, the development and commercialization of such an approved product may be delayed or there may be a shortage in supply. Any inability to manufacture our product candidates or future approved drugs in sufficient quantities when needed would seriously harm our business.
Manufacturers of our approved products, if any, must comply with cGMP requirements enforced by the FDA and foreign health authorities through facilities inspection programs. These requirements include quality control, quality assurance, and the maintenance of records and documentation. Manufacturers of our approved products, if any, may be unable to comply with these cGMP requirements and with other FDA, state, and foreign regulatory requirements. A failure to comply with these requirements may result in fines and civil penalties, suspension of production, suspension or delay in product approval, product seizure or recall, or withdrawal of product approval. If the safety of any quantities supplied is compromised due to a manufacturer's failure to adhere to applicable laws or for other reasons, we may not be able to obtain regulatory approval for or successfully commercialize our products, which would seriously harm our business.
We rely on third parties to conduct our clinical trials, and if they fail to fulfill their obligations, the development of our products may be adversely affected.
We rely on independent clinical investigators, contract research organizations and other third party service providers to assist us in managing, monitoring and otherwise carrying out our clinical trials. We contract with certain third-parties to provide certain services, including site selection, enrollment, monitoring and data management services. Although we depend heavily on these parties, we do not control them and therefore we cannot be assured that these third-parties will adequately perform all of their contractual obligations to us. If our third party service providers cannot adequately and timely fulfill their obligations to us, or if the quality and accuracy of our clinical trial data is compromised due to failure by such third parties to adhere to our protocols or regulatory requirements or if such third-parties otherwise fail to meet deadlines, our development plans may be delayed or terminated. Further, if clinical study results are compromised, then we may need to repeat the affected studies, which could result in significant additional costs and delays to us.
We face competition from various entities including large pharmaceutical companies, small biotech companies, private companies, and research institutions.
Many of our competitors have greater financial resources and may have more experience in research and development, manufacturing, managing clinical trials and/or regulatory compliance than we do. Our competitors may compete with us for lead clinical trial investigators, clinical trial site locations and patient enrollment. These competitors may also compete with us on recruiting scientific and management personnel. Because our products are in the early stages of development, along with many of the competing products, and given unpredictability inherent in drug development, it is difficult to predict which third parties may provide the most competition, and on what specific basis that competition may be based.
We may have difficulty expanding our operations successfully as we evolve from a company primarily involved in discovery and pre-clinical testing into one that develops and commercializes drugs.
We expect that as we increase the number of product candidates we are developing we will also need to expand our operations. This expected growth may place a strain on our administrative and operational infrastructure. As product candidates we develop enter and advance through clinical trials, we will need to expand our development, regulatory, manufacturing, marketing, and sales capabilities or contract with other organizations to provide these capabilities for us. As our operations expand due to our development
29
progress, we expect that we will need to manage additional relationships with various collaborators, suppliers, and other organizations. Our ability to manage our operations and future growth will require us to continue to improve our operational, f inancial and management controls, reporting systems and procedures. We may not be able to implement improvements to our management information and control systems in an efficient or timely manner and may discover deficiencies in existing systems and contro ls.
Our business and operations could suffer in the event of information technology system failures.
Our internal computer systems and those of our contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war, and telecommunication and electrical failures. Such events could cause interruption of our operations and loss of intellectual property. For example, the loss of pre-clinical trial data or data from completed or ongoing clinical trials for our product candidates could result in delays in our regulatory filings and development efforts and significantly increase our costs. To the extent that any disruption or security breach were to result in a loss of or damage to our data, or inappropriate disclosure of confidential, proprietary or private information, we could incur liability, we could lose valuable trade secret rights, and the development of our product candidates could be delayed.
If a natural or man-made disaster strikes our research and development facility or otherwise affects our business, it could delay our progress developing our product candidates.
We conduct research and development in a facility in Madison, Wisconsin. The facilities and the equipment we use are costly to replace and require substantial lead time to repair or replace. Our facilities may be harmed by natural or man-made disasters, including, without limitation, earthquakes, floods, fires and acts of terrorism; and if our facilities are affected by a disaster, our development efforts would be delayed. Significant delays in our development efforts could materially impact our ability to obtain regulatory approval and to commercialize our products. Any insurance we maintain against damage to our property and the disruption of our business due to disaster may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, or at all. In addition, our development activities could be harmed or delayed by a shutdown of the U.S. government, including the FDA.
Litigation claims may result in financial losses or harm our reputation and may divert management resources.
When the market price of a stock is volatile, holders of that stock have often initiated securities class action litigation against the company that issued the stock. Certain of our stockholders have recently brought such lawsuits against us, pursuant to which we could incur substantial costs. We cannot predict with certainty the eventual outcome of this or any other litigation, arbitration or third-party inquiry. We may not be successful in defending ourselves or asserting our rights in current or future lawsuits, investigations, or claims that have been or may be brought against us and, as a result, our business could be materially harmed. These lawsuits, arbitrations, investigations or claims may result in large judgments or settlements against us, any of which could have a negative effect on our financial performance and business. Additionally, lawsuits, arbitrations and investigations can be expensive to defend, whether or not the lawsuit, arbitration or investigation has merit, and the defense of these actions may divert the attention of our management and other resources that would otherwise be engaged in running our business.
Risks Related to Our Financial Condition
We have a history of net losses, and we expect to continue to incur net losses and may not achieve or maintain profitability.
We have incurred net losses since our inception, including net losses of $34.4 million for the fiscal year ended September 30, 2017. We expect that our operating losses will continue for the foreseeable future as we continue our drug development and discovery efforts. To achieve profitability, we must, either directly or through licensing and/or partnering relationships, meet certain milestones, successfully develop and obtain regulatory approval for one or more drug candidates and effectively manufacture, market and sell any drugs we successfully develop. Even if we successfully commercialize drug candidates that receive regulatory approval, we may not be able to realize revenues at a level that would allow us to achieve or sustain profitability.
Accordingly, we may never generate significant revenue and, even if we do generate significant revenue, we may never achieve profitability.
We will require substantial additional funds to complete our research and development activities.
Our business currently does not generate the cash that is necessary to finance our operations. Subject to the success of the research and development programs of our company and our partners, and potential licensing or partnering transactions, we will likely need to raise additional capital to:
● | Fund research and development infrastructure and activities relating to the development of our drug candidates, including pre-clinical and clinical trials and manufacturing to support these efforts; |
30
● | Fund our general and administrative infrastructure and activities; |
● | Pursue business development opportunities for our technologies; |
● | Add to and protect our intellectual property; and |
● | Retain our management and technical staff. |
Our future capital needs depend on many factors, including:
• | The scope, duration, and expenditures associated with our research and development; |
• | Regulatory requirements for our clinical trials; |
• | The extent to which our R&D and clinical efforts are successful; |
• | The outcome of potential partnering or licensing transactions, if any, and the extent to which our business development efforts result in the acquisition of new programs or technologies; |
• | Competing technological developments; |
• | Our intellectual property positions, if any, in our products; and |
• | The regulatory approval process and regulatory standards for our drug candidates. |
We will need to raise additional funds through public or private equity offerings, debt financings or additional strategic alliances and licensing arrangements in the future to continue our operations. We may not be able to obtain additional financing on terms favorable to us, if at all. General market conditions may make it very difficult for us to seek financing from the capital markets, and the terms of any financing may adversely affect the holdings or the rights of our stockholders. For example, if we raise additional funds by issuing equity securities, further dilution to our stockholders will result, which may substantially dilute the value of your investment. In addition, as a condition to providing additional funds to us, future investors may demand, and may be granted, rights superior to those of existing stockholders. Debt financing, if available, may involve restrictive covenants that could limit our flexibility in conducting future business activities and, in the event of insolvency, would be paid before holders of equity securities received any distribution of corporate assets. In order to raise additional funds through alliance, joint venture or licensing arrangements, we may be required to relinquish rights to our technologies or drug candidates, or grant licenses on terms that are not favorable to us. If adequate funds are not available, we may have to further delay, reduce or eliminate one or more of our planned activities. These actions would likely reduce the market price of our common stock.
If the estimates we make, or the assumptions on which we rely, in preparing our consolidated financial statements prove inaccurate, our actual results may vary from those reflected in our accruals.
Our consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (GAAP). The preparation of these consolidated financial statements requires us to make estimates and judgments that affect the reported amounts of our assets, liabilities, revenues and expenses, the amounts of charges accrued by us and related disclosure of contingent assets and liabilities. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. We cannot assure you, however, that our estimates, or the assumptions underlying them, will be correct.
The investment of our cash, cash equivalents and fixed income marketable securities is subject to risks which may cause losses and affect the liquidity of these investments.
At September 30, 2017, we had $40.8 million in fixed income marketable securities. These investments are in corporate bonds nearing maturity, but our investments may also include commercial paper, securities issued by the U.S. government obligations, certificates of deposit and money market funds meeting the criteria of our investment policy, which is focused on the preservation of our capital. These investments are subject to general credit, liquidity, and market and interest rate risks, particularly in the current economic environment. We may realize losses in the fair value of these investments or a complete loss of these investments, which would have a negative effect on our consolidated financial statements. In addition, should our investments cease paying or reduce the amount of interest paid to us, our interest income would suffer. The market risks associated with our investment portfolio may have an adverse effect on our results of operations, liquidity and financial condition.
Investment and Securities Risks
31
Our Board of Directors has adopted a rights agreement and has the authority to issue shares of "blank check" preferred stock, which may make an acquis ition of the Company by another company more difficult.
We have adopted and may in the future adopt certain measures that may have the effect of delaying, deferring or preventing a takeover or other change in control of the Company that a holder of our Common Stock might consider in its best interest. For example, on March 21, 2017, we entered into a rights agreement. The effect of this agreement could delay or prevent a third party from acquiring us or replacing members of our Board of Directors, or make more costly any attempt to acquire control of the Company, even if the acquisition or the board designees would be beneficial to our stockholders. Further, our Board of Directors, without further action by our stockholders, currently has the authority to issue up to 5,000,000 shares of preferred stock and to fix the rights (including voting rights), preferences and privileges of these shares ("blank check" preferred). Such preferred stock may have rights, including economic rights, senior to our Common Stock. These factors could also reduce the price that certain investors might be willing to pay for shares of our Common Stock and result in the market price being lower than it would be without these provisions.
We do not intend to declare cash dividends on our Common Stock.
We will not distribute cash to our stockholders unless and until we can develop sufficient funds from operations to meet our ongoing needs and implement our business plan. The time frame for that is unpredictable and investors should not expect dividends in the near future, if at all.
If securities or industry analysts do not publish research reports about our business or if they make adverse recommendations regarding an investment in our stock, our stock price and trading volume may decline.
The trading market for our Common Stock can be influenced by the research and reports that industry or securities analysts publish about our business. Currently, coverage of our Company by industry and securities analysts is limited. Investors have many investment opportunities and may limit their investments to companies that receive greater coverage from analysts. If additional industry or securities analysts do not commence coverage of the Company, the trading price of our stock could be negatively impacted. If one or more of the analysts downgrade our stock or comment negatively on our prospects, our stock price may decline. If one or more of these analysts cease to cover our industry or us or fail to publish reports about the Company regularly, our Common Stock could lose visibility in the financial markets, which could also cause our stock price or trading volume to decline. Further, incorrect judgments, estimates or assumptions made by research analysts may adversely affect our stock price, particularly if subsequent performance falls below the levels that were projected by the research analyst(s), even if we did not set or endorse such expectations. Any of these events could cause further volatility in our stock price and could result in substantial declines in the value of our stock.
The market for purchases and sales of our Common Stock may be limited, and the sale of a limited number of shares could cause the price to fall sharply.
Although our Common Stock is listed for trading on the NASDAQ Global Select Market, at various times our securities are relatively thinly traded. Investor trading patterns could serve to exacerbate the volatility of the price of our stock. For example, mandatory sales of our Common Stock by institutional holders could be triggered if an investment in our Common Stock no longer satisfies their investment standards and guidelines. It may be difficult to sell shares of our Common Stock quickly without significantly depressing the value of the stock. Unless we are successful in developing continued investor interest in our stock, sales of our stock could result in major fluctuations in the price of the stock.
Our Common Stock price has fluctuated significantly over the last several years and may continue to do so in the future, without regard to our results of operations and prospects.
Because we are early in the stage of our drug development, there are few objective metrics by which our progress may be measured. Consequently, we expect that the market price of our Common Stock will continue to fluctuate significantly. We may not generate substantial revenue from the license or sale of our technology for several years, if at all. In the absence of product revenue as a measure of our operating performance, we anticipate that investors and market analysts will assess our performance by considering factors such as:
● | Announcements of developments related to our business; |
● | Our ability to enter into or extend investigation phase, development phase, commercialization phase and other agreements with new and/or existing partners; |
● | Announcements regarding the status of any or all of our collaborations or products, including clinical trial results; |
● | Market perception and/or investor sentiment regarding our technology; |
● | Announcements of actions taken by regulatory authorities, such as the U.S. Food and Drug Administration; |
32
● | Announcements regarding developments in the RNA interference or biotechnology fields in general; |
● | Announcements regarding clinical trial results with our products or competitors' products; |
● | Market perception and/or announcements regarding other companies developing products in the field of biotechnology generally or specifically RNA interference; |
● | The issuance of competitive patents or disallowance or loss of our patent rights; |
● | The addition or departure of key executives; and |
● | Variations in our operating results. |
We will not have control over many of these factors but expect that they may influence our stock price. As a result, our stock price may be volatile and such volatility could result in the loss of all or part of your investment.
Stockholder equity interest may be substantially diluted in any additional equity issuances.
Our certificate of incorporation authorizes the issuance of 145,000,000 shares of Common Stock and 5,000,000 shares of Preferred Stock, on such terms and at such prices as our Board of Directors may determine. The following serves as a summary of share issuance activity during the fiscal year ended September 30, 2017:
| • | 2,670,989 shares of Common Stock pursuant to the conversion of certain Preferred Stock held by the Company's shareholders; |
| • | 1,745,810 shares of Common Stock to Amgen, Inc. pursuant to the Common Stock Purchase Agreement entered into with Amgen in September 2016; |
| • | 621,942 shares of Common Stock pursuant to the exercise of stock options, warrants and exchange rights and the vesting of restricted stock units; |
As of September 30, 2017, we had 74,785,426 shares of Common Stock issued and outstanding. The issuance of additional securities in financing transactions by us or through the exercise of options or warrants will dilute the equity interests of our existing stockholders, perhaps substantially, and could result in dilution in the tangible net book value of a share of our Common Stock, depending upon the price and other terms on which the additional shares are issued.
Risks Inherent in Our Industry
Drug development is time consuming, expensive and risky.
We are focused on technology related to new and improved pharmaceutical candidates. Product candidates that appear promising in the early phases of development, such as in animal and early human clinical trials, often fail to reach the market for a number of reasons, such as:
● | Clinical trial results may be unacceptable, even though preclinical trial results were promising; |
● | Inefficacy and/or harmful side effects in humans or animals; |
● | The necessary regulatory bodies, such as the U.S. Food and Drug Administration, may not approve our potential product for the intended use, or at all; and |
● | Manufacturing and distribution may be uneconomical. |
For example, any positive pre-clinical results in animals for our pre-clinical programs may not be replicated in human clinical studies. These programs may be also found to be unsafe in humans, particularly at higher doses needed to achieve the desired levels of efficacy. Also, the positive safety results from single dose human clinical studies may not be replicated in other human studies, including multiple dose studies. Clinical and pre-clinical study results are frequently susceptible to varying interpretations by scientists, medical personnel, regulatory personnel, statisticians and others, which often delays, limits, or prevents further clinical development or regulatory approvals of potential products. Clinical trials can take many years to complete, including the process of study design, clinical site selection and the recruitment of patients. As a result, we can experience significant delays in completing clinical studies, which can increase the cost of developing a drug candidate and shorten the time that an approved product may be protected by patents. If our drug candidates are not successful in human clinical trials, we may be forced to curtail or abandon certain development programs. If we experience significant delays in commencing or completing our clinical studies, we could suffer from significant cost overruns, which could negatively affect our capital resources and our ability to complete these studies.
33
The healthcare system is under significant financial pressure to reduce costs, which could reduce payment and reimbursement rates for drugs.
Throughout the world and particularly in the United States the healthcare system is under significant financial pressure to reduce costs. The price of pharmaceuticals has been a topic of considerable public discussion that could lead to price controls or other price-limiting strategies by payors that have the effect of lowering payment and reimbursement rates for drugs or otherwise making the commercialization of pharmaceuticals less profitable. These effects could reduce or eliminate our ability to return value to our shareholders.
Regulatory standards are subject to change over time, making it difficult to accurately predict the likelihood of marketing approval even when clinical trials meet their endpoints.
Regulatory standards are promulgated by various government entities and are subject to change based on factors such as scientific developments, public perceptions of risk, and political forces. Because clinical trials often take years to complete, it is sometimes possible for standards that exist during the conception and initiation of a clinical trial to change before the clinical trial is completed or reviewed by government regulators. While some government entities have safeguards intended to ensure standards agreed upon by sponsors and regulators at the outset of a clinical trial are applied during regulatory review processes, those safeguards generally permit regulators to apply more rigorous standards where regulators believe doing so is necessary. As such, there can be no assurance that regulatory standards that are appropriate at the outset of a clinical trial program will not become more rigorous during the regulatory approval process and could potentially result in a delayed approval or denial of marketing authorization.
ITEM 1B. | UNRESOLVED STAFF COMMENTS |
None.
ITEM 2. | PROPERTIES |
The Company does not own any real property. The following table summarizes the Company's leased facilities as of September 30, 2017.
| Office Space |
|
| Monthly Expenses |
|
| Primary Use |
| Lease Expiration |
|
| Lease Term |
| ||||
Pasadena, California |
| 8,500 sq. ft. |
|
| $ | 27,000 |
|
| Corp. Headqtrs. |
|
| September 2019 |
|
|
| 7 years |
|
Madison, Wisconsin |
| 60,000 sq. ft. |
|
| $ | 182,200 |
|
| Research Facility |
|
| September 2026 |
|
|
| 10 years |
|
ITEM 3. | LEGAL PROCEEDINGS |
Legal Proceedings are set forth in our financial statement schedules in Part IV, Item 15 of this Annual Report and are incorporated herein by reference. See Note 7 - Commitments and Contingencies of Notes to Consolidated Financial Statements of Part IV, Item 15. Exhibits and Financial Statement Schedules .
ITEM 4. | MINE SAFETY DISCLOSURES |
Not applicable.
PART II
ITEM 5. | MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Price Range of Common Stock
Our Common Stock is traded on the NASDAQ Global Select Market under the symbol "ARWR". The following table sets forth the high and low sales prices for a share of the Company's Common Stock during each period indicated.
34
| Fiscal Year Ended September 30, |
| |||||||||||||
| 2017 |
|
| 2016 |
| ||||||||||
| High |
|
| Low |
|
| High |
|
| Low |
| ||||
1st Quarter | $ | 7.74 |
|
| $ | 1.20 |
|
| $ | 6.45 |
|
| $ | 4.83 |
|
2nd Quarter |
| 2.45 |
|
|
| 1.42 |
|
|
| 6.05 |
|
|
| 3.07 |
|
3rd Quarter |
| 2.04 |
|
|
| 1.42 |
|
|
| 6.55 |
|
|
| 4.68 |
|
4th Quarter |
| 4.42 |
|
|
| 1.48 |
|
|
| 8.22 |
|
|
| 5.29 |
|
Shares Outstanding
At December 6, 2017, 74,828,280 shares of the Company's Common Stock were issued and outstanding, and were owned by 129 stockholders of record, based on information provided by the Company's transfer agent.
Dividends
The Company has never paid dividends on its Common Stock and does not anticipate that it will do so in the foreseeable future.
Securities Authorized for Issuance Under the Equity Compensation Plans
The disclosure required under this item related to equity compensation plans is incorporated by reference from Item 12 of Part III of this Annual Report on Form 10-K.
Sales of Unregistered Securities
All information under this Item has been previously reported on our Current Reports on Form 8-K.
Repurchases of Equity Securities
We did not repurchase any shares of our Common Stock during the years ended September 30, 2017, 2016 and 2015.
Performance Graph
The following performance graph shall not be deemed "soliciting material" or to be "filed" with the SEC, nor shall such information be incorporated by reference into any future filing under the Securities Act of 1933 or Securities Exchange Act of 1934, each as amended, except to the extent that we specifically incorporate it by reference into such filing. The graph compares the cumulative 5-year total return to shareholders on our Common Stock relative to the cumulative total returns of the NASDAQ Composite Index and the NASDAQ Biotechnology Index. We selected the NASDAQ Biotechnology Index because we believe the index reflects the market conditions within the industry in which we primarily operate. The comparison of total return on investment, defined as the change in year-end stock price plus reinvested dividends, for each of the periods assumes that $100 was invested on September 30, 2012, in each of our Common Stock, the NASDAQ Composite Index and the NASDAQ Biotechnology Index, with investment weighted on the basis of market capitalization.
The comparisons in the following graph are based on historical data and are not intended to forecast the possible future performance of our Common Stock.
35
36
ITEM 6. | SELECTED FINA NCIAL DATA |
The following selected financial data has been derived from our audited consolidated financial statements and should be read
in conjunction with the consolidated financial statements, the related notes thereto and the independent auditors' report thereon, and
"Management's Discussion and Analysis of Financial Condition and Results of Operations," which are included elsewhere in this
Form 10-K and in previously filed annual reports on Form 10-K of Arrowhead Pharmaceuticals, Inc.
|
| Year Ended September |
| |||||||||||||||||
|
| 2017 |
|
| 2016 |
|
| 2015 |
|
| 2014 |
|
| 2013 |
| |||||
OPERATING SUMMARY |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
REVENUE |
| $ | 31,407,709 |
|
| $ | 158,333 |
|
| $ | 382,000 |
|
| $ | 175,000 |
|
| $ | 290,266 |
|
OPERATING EXPENSES |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
| 31,690,298 |
|
|
| 41,454,452 |
|
|
| 47,267,361 |
|
|
| 23,138,050 |
|
|
| 8,705,627 |
|
Acquired in-process research and development |
|
| - |
|
|
| - |
|
|
| 10,142,786 |
|
|
| - |
|
|
| - |
|
Salaries and payroll-related costs |
|
| 17,292,883 |
|
|
| 19,461,656 |
|
|
| 16,554,008 |
|
|
| 12,829,355 |
|
|
| 6,667,669 |
|
General and administrative expenses |
|
| 6,838,402 |
|
|
| 9,940,737 |
|
|
| 7,931,184 |
|
|
| 5,894,008 |
|
|
| 3,488,864 |
|
Stock-based compensation |
|
| 7,891,595 |
|
|
| 11,595,816 |
|
|
| 10,232,897 |
|
|
| 5,696,173 |
|
|
| 1,536,271 |
|
Depreciation and amortization |
|
| 4,690,440 |
|
|
| 3,260,045 |
|
|
| 2,336,207 |
|
|
| 1,345,655 |
|
|
| 1,751,412 |
|
Impairment expense |
|
| - |
|
|
| 2,050,817 |
|
|
| - |
|
|
| 2,172,387 |
|
|
| 1,308,047 |
|
Contingent consideration - fair value adjustments |
|
| - |
|
|
| (5,862,464 | ) |
|
| 1,891,533 |
|
|
| 2,375,658 |
|
|
| 1,421,652 |
|
TOTAL OPERATING EXPENSES (a) |
|
| 68,403,618 |
|
|
| 81,901,059 |
|
|
| 96,355,976 |
|
|
| 53,451,286 |
|
|
| 24,879,542 |
|
OPERATING LOSS |
|
| (36,995,909 | ) |
|
| (81,742,726 | ) |
|
| (95,973,976 | ) |
|
| (53,276,286 | ) |
|
| (24,589,276 | ) |
LOSS FROM CONTINUING OPERATIONS |
|
| (34,380,295 | ) |
|
| (81,723,002 | ) |
|
| (91,940,882 | ) |
|
| (58,725,412 | ) |
|
| (31,703,079 | ) |
Income (loss) from discontinued operations |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| (354 | ) |
NET LOSS |
|
| (34,380,295 | ) |
|
| (81,723,002 | ) |
|
| (91,940,882 | ) |
|
| (58,725,412 | ) |
|
| (31,703,433 | ) |
Net (gain) loss attributable to non-controlling interests |
|
| - |
|
|
| - |
|
|
| - |
|
|
| 95,222 |
|
|
| 560,144 |
|
NET LOSS ATTRIBUTABLE TO ARROWHEAD |
| $ | (34,380,295 | ) |
| $ | (81,723,002 | ) |
| $ | (91,940,882 | ) |
| $ | (58,630,190 | ) |
| $ | (31,143,289 | ) |
EARNINGS PER SHARE (BASIC AND DILUTED): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net Income (Loss) attributable to Arrowhead common shareholders |
| $ | (0.47 | ) |
| $ | (1.34 | ) |
| $ | (1.60 | ) |
| $ | (1.25 | ) |
| $ | (1.30 | ) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average shares outstanding – basic and diluted |
|
| 73,898,598 |
|
|
| 61,050,880 |
|
|
| 57,358,442 |
|
|
| 46,933,030 |
|
|
| 24,002,224 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CASH DIVDEND PAID PER COMMON SHARE |
| $ | - |
|
| $ | - |
|
| $ | - |
|
| $ | - |
|
| $ | - |
|
|
| September 30, |
| |||||||||||||||||
|
| 2017 |
|
| 2016 |
|
| 2015 |
|
| 2014 |
|
| 2013 |
| |||||
FINANCIAL POSITION SUMMARY |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CASH AND CASH EQUIVALENTS (b) |
| $ | 24,838,567 |
|
| $ | 85,366,448 |
|
| $ | 81,214,354 |
|
| $ | 132,510,610 |
|
| $ | 19,114,444 |
|
SHORT- AND LONG-TERM INVESTMENTS (b) |
|
| 40,769,539 |
|
|
| - |
|
|
| 17,539,902 |
|
|
| 44,741,378 |
|
|
| 10,732,414 |
|
TOTAL ASSETS (c) |
|
| 104,022,280 |
|
|
| 128,176,505 |
|
|
| 132,267,914 |
|
|
| 182,816,756 |
|
|
| 37,329,631 |
|
CAPITAL LEASE OBLIGATIONS |
|
| - |
|
|
| - |
|
|
| 758,340 |
|
|
| 972,331 |
|
|
| 1,282,458 |
|
OTHER LONG-TERM OBLIGATIONS |
|
| 4,454,070 |
|
|
| 7,508,452 |
|
|
| 6,204,917 |
|
|
| 4,226,137 |
|
|
| 1,808,709 |
|
| (a) | The decrease in our Total Operating Expenses during the year ended September 30, 2017 is primarily due to the Company's decision to discontinue its previous clinical trial candidates: ARC-520, ARC-AAT and ARC-521. However, we expect research and development expenses to increase as our next generation candidates progress toward the clinic. |
| (b) | The Company's Cash and Cash Equivalents decreased from September 30, 2016 to September 30, 2017 due primarily to the Company's $40.8 million investments in fixed-income debt securities, as well as cash used in research and development expenditures. |
| (c) | The Company's Total Assets decreased from September 30, 2016 to September 30, 2017 due primarly to cash used in research and development expenditures. |
37
ITEM 7. | MANAGE MENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERA TIONS |
Description of Business
Unless otherwise noted, (1) the term "Arrowhead" refers to Arrowhead Pharmaceuticals, Inc., a Delaware corporation, (2) the terms "Company," "we," "us," and "our," refer to the ongoing business operations of Arrowhead and its Subsidiaries, whether conducted through Arrowhead or a subsidiary of Arrowhead, (3) the term "Subsidiaries" refers collectively to Arrowhead Madison Inc. ("Arrowhead Madison"), Arrowhead Australia Pty Ltd ("Arrowhead Australia") and Ablaris Therapeutics, Inc. ("Ablaris"), (4) the term "Common Stock" refers to Arrowhead's Common Stock, (5) the term "Preferred Stock" refers to Arrowhead's Preferred Stock and (6) the term "Stockholder(s)" refers to the holders of Arrowhead Common Stock.
Overview
Arrowhead Pharmaceuticals, Inc. develops medicines that treat intractable diseases by silencing the genes that cause them. Using a broad portfolio of RNA chemistries and efficient modes of delivery, Arrowhead therapies trigger the RNA interference mechanism to induce rapid, deep and durable knockdown of target genes. RNA interference, or RNAi, is a mechanism present in living cells that inhibits the expression of a specific gene, thereby affecting the production of a specific protein. Deemed to be one of the most important recent discoveries in life science with the potential to transform medicine, the discoverers of RNAi were awarded a Nobel Prize in 2006 for their work. Arrowhead's RNAi-based therapeutics leverage this natural pathway of gene silencing. The company's pipeline includes ARO-HBV for chronic hepatitis B virus, ARO-AAT for liver disease associated with alpha-1 antitrypsin deficiency (AATD), ARO-APOC3 and ARO-ANG3 for hypertriglyceridemia, ARO-Lung1 for an undisclosed pulmonary target, ARO-HIF2 for renal cell carcinoma, ARO-F12 for hereditary angioedema and thromboembolic disorders, and ARO-AMG1 for an undisclosed genetically validated cardiovascular target under a license and collaboration agreement with Amgen, Inc., a Delaware corporation ("Amgen"). ARO-LPA (AMG 890) for cardiovascular disease was out-licensed to Amgen in 2016.
Arrowhead operates a lab facility in Madison, Wisconsin, where the Company's research and development activities, including the development of RNAi therapeutics, are based. The Company's principal executive offices are located in Pasadena, California.
In fiscal 2017, Arrowhead refocused its resources on therapeutics that exclusively utilize the company's Targeted RNAi Molecule (TRiM TM ) platform technology. Therapeutics built on the TRiM TM platform have demonstrated high levels of pharmacologic activity in multiple animal models spanning several therapeutic areas. TRiM TM enabled therapeutics offer several potential advantages over prior generation and competing technologies, including: simplified manufacturing and reduced costs; multiple routes of administration including subcutaneous injection and inhaled administration; the ability to target multiple tissue types including liver, lung, and tumors; and the potential for improved safety and reduced risk of intracellular buildup, because there are less metabolites from smaller, simpler molecules.
As part of the refocusing of resources, Arrowhead announced in November 2016 that it would be discontinuing all clinical programs that utilized the intravenously administered DPC, or EX1, delivery vehicle. The decision to discontinue development of EX1-containing programs was based primarily on two factors. First, during discussions with regulatory agencies and outside experts, it became apparent that there would be substantial delays in all clinical programs that utilize EX1, while the Company further explored the cause of deaths in a non-clinical toxicology study in non-human primates exploring doses of EX1 higher than those planned to be used in humans. Second, the Company had made substantial advances in RNA chemistry and targeting resulting in large potency gains for development programs utilizing the TRiM TM technology, making EX1 no longer necessary.
As part of an R&D day in September 2017, the Company introduced its new TRiM TM platform and made the following announcements regarding its pipeline candidates:
| - | ARO-AAT, Arrowhead's second generation subcutaneously administered clinical candidate for the treatment of alpha-1 antitrypsin deficiency liver disease, achieved up to 92% knockdown in monkeys, thought to be near complete suppression of hepatic production of the alpha-1 antitrypsin protein. In non-GLP rat and monkey exploratory toxicology studies, no changes in clinical chemistries or histopathology suggestive of organ toxicity were observed at doses up to 300 mg/kg (100x expected human dose). |
| - | ARO-HBV, Arrowhead's third generation subcutaneously administered clinical candidate for the treatment of chronic hepatitis B virus infection, achieved up to 99.9% knockdown of hepatitis B surface antigen (HBsAg), e-antigen (HBeAg), and HBV DNA in rodent models. In a non-GLP rat exploratory toxicology study, no changes in clinical chemistries or histopathology changes suggestive of organ toxicity were observed at doses up to 300 mg/kg (75-100x expected human dose). |
38
| - | Arrowhead has expanded its cardiovascular disease portfolio utilizing the TRiM™ platform. ARO-APOC3, targeting apolipoprotein C-III, and ARO-ANG3, targeting angio poietin-like protein 3 (ANGPTL3) will be added to ARO-LPA (AMG 890) and ARO-AMG1, which are both partnered with Amgen. ARO-APOC3 and ARO-ANG3 will both be developed for the treatment of hypertriglyceridemia. |
| - | ARO-Lung1, the first generation candidate against an undisclosed gene target in the lung, reached almost 90% target knockdown following inhaled administration in rodents. |
| - | The ARO-HIF2 candidate targeting renal cell carcinoma achieved 85% target gene knockdown in a rodent tumor model. |
On September 28, 2016, the Company entered into two Collaboration and License agreements and a Common Stock Purchase Agreement with Amgen. Under one of the license agreements (the "First Collaboration and License Agreement" or "ARO-AMG1 Agreement"), Amgen has received an option to a worldwide, exclusive license for ARO-AMG1 , an RNAi therapy for an undisclosed genetically validated cardiovascular target. Under the other license agreement (the "Second Collaboration and License Agreement" or "ARO-LPA (AMG 890) Agreement"), Amgen has received a worldwide, exclusive license to Arrowhead's novel, RNAi ARO-LPA (AMG 890) program. The ARO-LPA (AMG-890) RNAi molecules are designed to reduce elevated lipoprotein(a), which is a genetically validated, independent risk factor for atherosclerotic cardiovascular disease. In both agreements, Amgen is wholly responsible for clinical development and commercialization. Under the terms of the agreements taken together, the Company has received $35 million in upfront payments and $21.5 million in the form of an equity investment by Amgen in the Company's Common Stock, and could receive up to $617 million in option payments and development, regulatory and sales milestone payments. The Company is further eligible to receive single-digit royalties for sales of products under the ARO-AMG1 agreement and up to low double-digit royalties for sales of products under the ARO-LPA (AMG 890) agreement.
The Company continues to develop other clinical candidates for future clinical trials. Clinical candidates are tested internally and through GLP toxicology studies at outside laboratories. Drug materials for such studies and clinical trials are contracted to third-party manufacturers when cGMP production is required. The Company engages third-party contract research organizations (CROs) to manage clinical trials and works cooperatively with such organizations on all aspects of clinical trial management, including plan design, patient recruiting, and follow up. These outside costs, relating to the preparation for and administration of clinical trials, are referred to as "program costs". If the clinical candidates progress through human testing, program costs will increase.
Net losses were $34.4 million, $81.7 million and $91.9 million during the years ended September 30, 2017, 2016 and 2015, respectively. Diluted losses per share were $0.47, $1.34 and $1.60 during the years ended September 30, 2017, 2016 and 2015, respectively. An increase in revenue in 2017 from the collaborations with Amgen is the driver of the reductions in the net losses and diluted losses per share, as discussed further below.
The Company strengthened its liquidity and financial position through an equity offering completed in August 2016, which generated $43.2 million of net cash proceeds for the Company. Additionally, the Company received $56.5 million in upfront payments and equity investments from Amgen. These cash proceeds secured the funding needed to continue to advance our preclinical candidates. The Company had $24.8 million of cash and cash equivalents, $40.8 million in short-term investments and $104.0 million of total assets as of September 30, 2017, as compared to $85.4 million, $0 million and $128.2 million as of September 30, 2016, respectively. Based upon the Company's current cash and short-term investment resources and operating plan, the Company expects to have sufficient liquidity to fund operations for at least the next twelve months.
Critical Accounting Policies and Estimates
Management makes certain judgments and uses certain estimates and assumptions when applying GAAP in the preparation of our Consolidated Financial Statements. We evaluate our estimates and judgments on an ongoing basis and base our estimates on historical experience and on assumptions that we believe to be reasonable under the circumstances. Our experience and assumptions form the basis for our judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may vary from what we anticipate and different assumptions or estimates about the future could change our reported results. We believe the following accounting policies are the most critical to us, in that they require our most difficult, subjective or complex judgments in the preparation of our consolidated financial statements. For further information, see Note 1, Organization and Significant Accounting Policies , to our Consolidated Financial Statements, which outlines our application of significant accounting policies.
Revenue Recognition
Revenue from product sales is recorded when persuasive evidence of an arrangement exists, title has passed and delivery has occurred, a price is fixed and determinable, and collection is reasonably assured.
39
The Company may generate revenue from technolo gy licenses, collaborative research and development arrangements, research grants and product sales. Revenue under technology licenses and collaborative agreements typically consists of nonrefundable and/or guaranteed technology license fees, collaborative research funding, manufacturing and development services and various milestone and future product royalty or profit-sharing payments. These agreements are generally referred to as "multiple element arrangements".
The Company applies the accounting standard on revenue recognition for multiple element arrangements. The fair value of deliverables under the arrangement may be derived using a best estimate of selling price if vendor specific objective evidence and third-party evidence is not available. Deliverables under the arrangement will be separate units of accounting if a delivered item has value to the customer on a standalone basis and if the arrangement includes a general right of return for the delivered item, delivery or performance of the undelivered item is considered probable and substantially in the Company's control.
The Company recognizes upfront license payments as revenue upon delivery of the license only if the license has standalone value from any undelivered performance obligations and that value can be determined. The undelivered performance obligations typically include manufacturing or development services or research and/or steering committee services. If the fair value of the undelivered performance obligations can be determined, then these obligations would be accounted for separately. If the license is not considered to have standalone value, then the license and other undelivered performance obligations would be accounted for as a single unit of accounting. In this case, the license payments and payments for performance obligations are recognized as revenue over the estimated period of when the performance obligations are performed or deferred indefinitely until the undelivered performance obligation is determined.
Whenever the Company determines that an arrangement should be accounted for as a single unit of accounting, the Company determines the period over which the performance obligations will be performed and revenue will be recognized. Revenue is recognized using a proportional performance or straight-line method. The proportional performance method is used when the level of effort required to complete performance obligations under an arrangement can be reasonably estimated. The amount of revenue recognized under the proportional performance method is determined by multiplying the total payments under the contract, excluding royalties and payments contingent upon achievement of milestones, by the ratio of the level of effort performed to date to the estimated total level of effort required to complete performance obligations under the arrangement. If the Company cannot reasonably estimate the level of effort to complete performance obligations under an arrangement, the Company recognizes revenue under the arrangement on a straight-line basis over the period the Company is expected to complete its performance obligations. Significant management judgment is required in determining the level of effort required under an arrangement and the period over which the Company is expected to complete its performance obligations under an arrangement.
Many of the Company's collaboration agreements entitle the Company to additional payments upon the achievement of development, regulatory and sales performance-based milestones. If the achievement of a milestone is considered probable at the inception of the collaboration, the related milestone payment is included with other collaboration consideration, such as upfront fees and research funding, in the Company's revenue calculation. Typically, these milestones are not considered probable at the inception of the collaboration. As such, milestones will typically be recognized in one of two ways depending on the timing of when the milestone is achieved. If the milestone is achieved during the performance period, then the Company will only recognize revenue to the extent of the proportional performance achieved at that date, or the proportion of the straight-line basis achieved at that date, and the remainder will be recorded as deferred revenue to be amortized over the remaining performance period. If the milestone is achieved after the performance period has completed and all performance obligations have been delivered, then the Company will recognize the milestone payment as revenue in its entirety in the period the milestone was achieved.
Deferred revenue will be classified as part of Current or Long-Term Liabilities in the accompanying Consolidated Balance Sheets based on the Company's estimate of the portion of the performance obligations regarding that revenue will be completed within the next 12 months divided by the total performance period estimate. This estimate is based on the Company's current operating plan and, if the Company's operating plan should change in the future, the Company may recognize a different amount of deferred revenue over the next 12-month period.
Impairment of Long-lived Assets
We review long-lived assets for impairment whenever events or changes in business circumstances indicate that the carrying amount of assets may not be fully recoverable or that our assumptions about the useful lives of these assets are no longer appropriate. If impairment is indicated, recoverability is measured by a comparison of the carrying amount of an asset to the estimated undiscounted future cash flows expected to be generated by the asset. If the carrying amount of an asset exceeds its estimated future cash flows, an impairment charge is recognized in the amount by which the carrying amount of the asset exceeds the fair value of the asset.
Impairment of Intangible assets
40
Intangible assets consist of a license agreement and patents acquired in conjunction with a business or asset acquisition. Intangible assets are monitored for potential impairment whenever events or circums tances indicate that the carrying amount may not be recoverable, and are also reviewed annually to determine whether any impairment is necessary. Based on ASU 2012-02, the annual review of intangible assets is performed via a two-step process. First, a qua litative assessment is performed to determine if it is more likely than not that the intangible asset is impaired. If required, a quantitative assessment is performed and, if necessary, impairment is recorded.
Stock-Based Compensation
We account for stock-based compensation arrangements in accordance with FASB ASC 718, which requires the measurement and recognition of compensation expense for all share-based payment awards to be based on estimated fair values. The Company uses the Black-Scholes option valuation model to estimate the fair value of its stock options at the date of grant. The Black-Scholes option valuation model requires the input of subjective assumptions to calculate the value of stock options. For restricted stock units, the value of the award is based on the Company's stock price at the grant date. For performance-based restricted stock unit awards, the value of the award is based on the Company's stock price at the grant date. The Company uses historical data and other information to estimate the expected price volatility for stock option awards and the expected forfeiture rate for all awards. Expense is recognized over the vesting period for all awards, and commences at the grant date for time-based awards and upon the Company's determination that the achievement of such performance conditions is probable for performance-based awards. This determination requires significant judgement by management.
Derivative Assets and Liabilities
We account for warrants and other derivative financial instruments as either equity or assets/liabilities based upon the characteristics and provisions of each instrument. Warrants classified as equity are recorded as additional paid-in capital on our Consolidated Balance Sheet and no further adjustments to their valuation are made. Some of our warrants were determined to be ineligible for equity classification because of provisions that may result in an adjustment to their exercise price. Warrants classified as derivative liabilities and other derivative financial instruments that require separate accounting as assets or liabilities are recorded on our Consolidated Balance Sheet at their fair value on the date of issuance and are revalued on each subsequent balance sheet date until such instruments are exercised or expire, with any changes in the fair value between reporting periods recorded as other income or expense. We estimate the fair value of these assets/liabilities using option pricing models that are based on the individual characteristics of the warrants or instruments on the valuation date, as well as assumptions for expected volatility, expected life and risk-free interest rate. Changes in the assumptions used could have a material impact on the resulting fair value. The primary input affecting the value of our derivatives liabilities is the Company's stock price.
Contingent Consideration
The consideration for our acquisitions often includes future payments that are contingent upon the occurrence of a particular event. For example, milestone payments might be based on progress of clinical development, the achievement of various regulatory approvals or future sales milestones, and royalty payments might be based on drug product sales levels. The Company records a contingent consideration obligation for such contingent payments at fair value on the acquisition date. The Company estimates the fair value of contingent consideration obligations through valuation models designed to estimate the probability of the occurrence of such contingent payments based on various assumptions and incorporating estimated success rates. Estimated payments are discounted using present value techniques to arrive at estimated fair value at the balance sheet date. Changes in the fair value of our contingent consideration obligations are recognized within our Consolidated Statements of Operations. Changes in the fair value of the contingent consideration obligations can result from changes to one or multiple inputs, including adjustments to the discount rates, changes in the amount or timing of expected expenditures associated with product development, changes in the amount or timing of cash flows from products upon commercialization, changes in the assumed achievement or timing of any development milestones, changes in the probability of certain clinical events and changes in the assumed probability associated with regulatory approval. These fair value measurements are based on significant inputs not observable in the market. Substantial judgment is employed in determining the appropriateness of these assumptions as of the acquisition date and for each subsequent period. Accordingly, changes in assumptions could have a material impact on the amount of contingent consideration expense the Company records in any given period.
Results of Operations
The following data summarizes our results of operations for the following periods indicated:
|
| Year ended September 30, |
| |||||||||
|
| 2017 |
|
| 2016 |
|
| 2015 |
| |||
Revenue |
| $ | 31,407,709 |
|
| $ | 158,333 |
|
| $ | 382,000 |
|
Operating Loss |
|
| (36,995,909 | ) |
|
| (81,742,726 | ) |
|
| (95,973,976 | ) |
Loss from Continuing Operations |
|
| (34,380,295 | ) |
|
| (81,723,002 | ) |
|
| (91,940,882 | ) |
41
Net Loss |
|
| (34,380,295 | ) |
|
| (81,723,002 | ) |
|
| (91,940,882 | ) |
Earnings per Share (Basic and Diluted) |
| $ | (0.47 | ) |
| $ | (1.34 | ) |
| $ | (1.60 | ) |
The increase in our Revenue during the year ended September 30, 2017 was driven by the upfront payments received from Amgen that we are recognizing as revenue as performance is completed for the ARO-LPA (AMG 890) and ARO-AMG1 agreements. These payments were also the key driver of the decrease in our Operating Loss, Net Loss and Loss per Share. Research and Development expenses also decreased in fiscal 2017 due to the discontinuation of our previous clinical programs in November 2016.
Results of Operations Comparison for 2017 and 2016
Revenues
Total revenue was $31,407,709 for the year ended September 30, 2017 and $158,333 for the year ended September 30, 2016. Revenue in the current period is primarily related to the upfront payments received from Amgen that we are recognizing as performance is completed for the ARO-LPA (AMG 890) and ARO-AMG1 agreements
Under the terms of the ARO-LPA (AMG 890) Agreement, the Company has granted a worldwide, exclusive license to ARO-LPA (AMG 890) . The collaboration between the Company and Amgen is governed by a joint research committee comprised of an equal number of representatives from each party; however, Amgen has the final decision making authority regarding ARO-LPA (AMG 890) in this committee. The Company is also responsible for assisting Amgen in the oversight of certain development and manufacturing activities, most of which are to be covered at Amgen's cost. The Company has determined that the significant deliverables under the ARO-LPA (AMG 890) Agreement include the license and the oversight of certain of the development and manufacturing activities. The Company also determined that, pursuant to the accounting guidance governing revenue recognition on multiple element arrangements, the license and collective undelivered activities and services do not have standalone value due to the specialized nature of the activities and services to be provided by the Company. Therefore, the deliverables are not separable and, accordingly, the license and undelivered services are being treated as a single unit of accounting. The Company will recognize revenue on a straight-line basis from November 18, 2016 (the Hart-Scott-Rodino clearance date) through October 31, 2017, which is the date where the significant development and manufacturing related deliverables were completed. The Company received the upfront payment of $30 million due under this agreement in November 2016. The initial $30 million payment was recorded as Deferred Revenue, and $27.3 million of this was amortized into Revenue during the year ended September 30, 2017.
Under the terms of the ARO-AMG1 Agreement, the Company has granted an option to a worldwide, exclusive license to ARO-AMG1, an undisclosed genetically validated cardiovascular target. The collaboration between the Company and Amgen is governed by a joint steering committee comprised of an equal number of representatives from each party. The Company is also responsible for developing, optimizing and manufacturing the candidate through certain preclinical efficacy and toxicology studies to determine whether the candidate the Company has developed meets the required criteria as defined in the agreement (the "Arrowhead Deliverable"). If this is achieved, Amgen will then have the option to an exclusive license for the intellectual property generated through the Company's development efforts, and will likely assume all development, regulatory and commercialization efforts for the candidate upon the option exercise. The Company has determined that the significant deliverables under the ARO-AMG1 Agreement include the license, the joint research committee and the development and manufacturing activities toward achieving the Arrowhead Deliverable. The Company also determined that, pursuant to the accounting guidance governing revenue recognition on multiple element arrangements, the license and collective undelivered activities and services do not have standalone value due to the specialized nature of the activities and services to be provided by the Company. Therefore, the deliverables are not separable and, accordingly, the license and undelivered services are being treated as a single unit of accounting. The Company will recognize revenue on a straight-line basis from October 1, 2016, through September 30, 2018. The due date for achieving the Arrowhead Deliverable is September 28, 2018. The Company received the upfront payment of $5 million due under this agreement in September 2016. The initial $5 million payment was recorded as Deferred Revenue, and $2.5 million of this was amortized into Revenue during the year ended September 30, 2017.
In January 2017, the Company also entered into a separate services agreement with Amgen to provide certain services related to process development, manufacturing, materials supply, discovery studies, and other consulting services related to ARO-LPA (AMG 890). During the year ended September 30, 2017, work orders under this services agreement generated approximately $1.5 million of Revenue.
Operating Expenses
The analysis below details the operating expenses and discusses the expenditures of the Company within the major expense categories. Certain reclassifications have been made to prior-period operating expense categories to conform to the current period presentation. For purposes of comparison, the amounts for the years ended September 30, 2017 and 2016 are shown in the tables below.
42
Research a nd Development Expenses
R&D expenses are related to the Company's on-going research and development efforts, primarily related to program costs, composed primarily of outsourced costs related to the manufacturing of clinical supplies, toxicity/efficacy studies and clinical trial expenses. Internal costs primarily relate to operations at our research and development facility in Madison, Wisconsin, including facility costs and laboratory-related expenses. The following table provides details of research and development expense for the periods indicated:
(in thousands)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
|
| Twelve Months Ended |
|
| % of Expense |
|
| Twelve Months Ended |
|
| % of Expense |
|
| Increase (Decrease) |
| |||||||||
|
| September 30, 2017 |
|
| Category |
|
| September 30, 2016 |
|
| Category |
|
| $ |
|
| % |
| ||||||
Laboratory supplies & services |
| $ | 5,303 |
|
|
| 17 | % |
| $ | 2,706 |
|
|
| 7 | % |
| $ | 2,597 |
|
|
| 96 | % |
In vivo studies |
|
| 2,410 |
|
|
| 8 | % |
|
| 1,611 |
|
|
| 4 | % |
|
| 799 |
|
|
| 50 | % |
Outside labs & contract services |
|
| 495 |
|
|
| 2 | % |
|
| 155 |
|
|
| 0 | % |
|
| 340 |
|
|
| 219 | % |
Toxicity/efficacy studies |
|
| 1,768 |
|
|
| 6 | % |
|
| 7,766 |
|
|
| 19 | % |
|
| (5,998 | ) |
|
| -77 | % |
Drug manufacturing |
|
| 9,812 |
|
|
| 31 | % |
|
| 9,855 |
|
|
| 24 | % |
|
| (43 | ) |
|
| 0 | % |
Clinical trials |
|
| 9,258 |
|
|
| 29 | % |
|
| 14,800 |
|
|
| 36 | % |
|
| (5,542 | ) |
|
| -37 | % |
License, royalty & milestones |
|
| 2 |
|
|
| 0 | % |
|
| 3,054 |
|
|
| 7 | % |
|
| (3,052 | ) |
|
| -100 | % |
Facilities and related |
|
| 2,337 |
|
|
| 7 | % |
|
| 1,315 |
|
|
| 3 | % |
|
| 1,022 |
|
|
| 78 | % |
Other research expenses |
|
| 305 |
|
|
| 1 | % |
|
| 192 |
|
|
| 1 | % |
|
| 113 |
|
|
| 59 | % |
Total |
| $ | 31,690 |
|
|
| 100 | % |
| $ | 41,454 |
|
|
| 100 | % |
| $ | (9,764 | ) |
|
| -24 | % |
Laboratory supplies and services expense increased $2,597,000 from $2,706,000 during the year ended September 30, 2016 to $5,303,000 during the current period. The increase in laboratory supplies and services is a result of additional supply purchases necessary to support the expansion of the Company's preclinical pipeline as well as the development of the subcutaneous versions of its new drug candidates.
In vivo studies expense increased $799,000 from $1,611,000 during the year ended September 30, 2016 to 2,410,000 during the current period. In vivo expense can vary depending on the stage of preclinical candidates, the nature and amount of testing required and the cost variation of different in vivo testing models. The increase in in vivo studies in the current period is a result of additional discovery studies being conducted for the Company's subcutaneous candidates.
Outside labs and contract services expense increased $340,000 from $155,000 during the year ended September 30, 2016 to $495,000 during the current period. The increase in outside labs and contract services in the current period is a result of additional discovery work being conducted for the Company's subcutaneous candidates.
Toxicity/efficacy studies expense decreased $5,998,000 from $7,766,000 during the year ended September 30, 2016 to $1,768,000 during the current period. This category includes IND-enabling toxicology studies as well as post-IND toxicology studies, such as long-term toxicology studies, and other efficacy studies. The decrease primarily relates to toxicology studies related to one of our discontinued drug candidates, ARC-521. We anticipate this expense to increase as we prepare to enter clinical trials with our new subcutaneous drug candidates.
Drug manufacturing expense was relatively consistent at $9,855,000 during the year ended September 30, 2016 to $9,812,000 during the current period. The current period expense relates to campaigns for our ARO-HBV and ARO-AAT programs, and the previous period related to campaigns for our previous generation candidates. We anticipate this expense to increase as we prepare to enter clinical trials with our new subcutaneous drug candidates.
Clinical trials expense decreased $5,542,000 from $14,800,000 during the year ended September 30, 2016 to $9,258,000 during the current period. The decrease is primarily due to the discontinuation of our previous clinical candidates, and the close out of those studies. We anticipate this expense to increase as we prepare to enter clinical trials with our new subcutaneous drug candidates.
License, royalty and milestones expense decreased $3,052,000 from $3,054,000 during the year ended September 30, 2016 to $2,000 during the current period. This category can include milestone payments which can vary from period to period depending on the nature of our various license agreements and the timing of reaching various development milestones requiring payment. We reached milestones related to our clinical candidates that required a $3 million accrual in fiscal 2016.
43
Facilities expense increased $1,022,000 from $1,315,000 during the year ended September 30, 2016 to $2,337,000 during the current period. The increase relates to increased rental cost s for our lease for a larger facility in Madison, Wisconsin, which we began occupying in October 2016.
Other research expense increased $113,000 from $192,000 during the year ended September 30, 2016 to $305,000 during the current period. The increase primarily relates to additional miscellaneous supplies purchased to support efforts at our larger facility in Madison, Wisconsin.
Salary and Payroll-Related Expenses
The Company employs scientific, technical and administrative staff at its corporate offices and its research facilities. Salaries and payroll-related expense consists of salaries, bonuses, payroll taxes and related benefits. Salary and payroll-related expenses include two major categories, based on the primary activities of each employee: general and administrative (G&A) compensation expense and research and development (R&D) compensation expense. The following table provides detail of salary and payroll-related expenses for the periods indicated:
(in thousands)
|
| Twelve months |
|
| % of |
|
| Twelve months |
|
| % of |
|
|
|
|
|
|
|
|
| ||||
|
| Ended |
|
| Expense |
|
| Ended |
|
| Expense |
|
| Increase (Decrease) |
| |||||||||
|
| September 30, 2017 |
|
| Category |
|
| September 30, 2016 |
|
| Category |
|
| $ |
|
| % |
| ||||||
R&D - compensation-related |
| $ | 11,722 |
|
|
| 68 | % |
| $ | 13,883 |
|
|
| 71 | % |
| $ | (2,161 | ) |
|
| -16 | % |
G&A - compensation-related |
|
| 5,571 |
|
|
| 32 | % |
|
| 5,579 |
|
|
| 29 | % |
|
| (8 | ) |
|
| 0 | % |
Total |
| $ | 17,293 |
|
|
| 100 | % |
| $ | 19,462 |
|
|
| 100 | % |
| $ | (2,169 | ) |
|
| -11 | % |
R&D compensation expense decreased $2,161,000 from $13,883,000 during the year ended September 30, 2016 to $11,722,000 during the current period. The decrease is primarily due to the reduction in force in November 2016 associated with the discontinuation of our previous clinical candidates.
G&A compensation expense was consistent at $5,579,000 during the year ended September 30, 2016 to $5,571,000 during the current period.
General & Administrative Expenses
The following table provides details of our general and administrative expenses for the periods indicated:
(in thousands)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
|
| Twelve Months Ended |
|
| % of Expense |
|
| Twelve Months Ended |
|
| % of Expense |
|
| Increase (Decrease) |
| |||||||||
|
| September 30, 2017 |
|
| Category |
|
| September 30, 2016 |
|
| Category |
|
| $ |
|
|
| % |
| |||||
Professional/outside services |
| $ | 2,915 |
|
|
| 43 | % |
| $ | 4,201 |
|
|
| 42 | % |
| $ | (1,286 | ) |
|
| -31 | % |
Patent expense |
|
| 1,154 |
|
|
| 17 | % |
|
| 1,529 |
|
|
| 15 | % |
|
| (375 | ) |
|
| -25 | % |
Facilities and related |
|
| 327 |
|
|
| 5 | % |
|
| 320 |
|
|
| 3 | % |
|
| 7 |
|
|
| 2 | % |
Travel |
|
| 713 |
|
|
| 10 | % |
|
| 864 |
|
|
| 9 | % |
|
| (151 | ) |
|
| -17 | % |
Business insurance |
|
| 591 |
|
|
| 9 | % |
|
| 632 |
|
|
| 6 | % |
|
| (41 | ) |
|
| -6 | % |
Communication and Technology |
|
| 426 |
|
|
| 6 | % |
|
| 686 |
|
|
| 7 | % |
|
| (260 | ) |
|
| -38 | % |
Office expenses |
|
| 563 |
|
|
| 8 | % |
|
| 303 |
|
|
| 3 | % |
|
| 260 |
|
|
| 86 | % |
Other |
|
| 149 |
|
|
| 2 | % |
|
| 1,406 |
|
|
| 14 | % |
|
| (1,257 | ) |
|
| -89 | % |
Total |
| $ | 6,838 |
|
|
| 100 | % |
| $ | 9,941 |
|
|
| 100 | % |
| $ | (3,103 | ) |
|
| -31 | % |
Professional/outside services include legal, accounting, consulting and other outside services retained by the Company. All periods include normally recurring legal and audit expenses related to SEC compliance and other corporate matters. Professional/outside services expense decreased $1,286,000 from $4,201,000 during the year ended September 30, 2016 to $2,915,000 during the current period. The decrease primarily related to higher legal fees in the previous periods related to litigation cases.
44
Patent expense decreased $375,000 from $1,529,000 during the year ended September 30, 2016 to $1,154,000 during the current period. The Company continues to invest in patent protection for its product candidates and other RNAi technology through patent fi lings in numerous countries. The Company expects to extend and maintain protection for its current portfolios, as appropriate, and file new patent applications as technologies are developed and improved. Expenses can vary from period to period as patents proceed through their prosecution life cycle.
Facilities-related expense was consistent at $320,000 during the year ended September 30, 2016 and $327,000 in the current period. Facilities expense represents the expense associated with our corporate headquarters in Pasadena.
Travel expense decreased $151,000 from $864,000 during the year ended September 30, 2016 to $713,000 during the current period. Travel expense decreased due to the discontinuation of our clinical trials in November 2016 and a reduction in R&D headcount. We anticipate this expense to increase in the near term as our next generation candidates approach the clinic.
Business insurance expense decreased $41,000 from $632,000 during the year ended September 30, 2016 to $591,000 during the current period. Business insurance costs consist of directors and officers insurance, property insurance, corporate liability insurance, as well as insurance related to our previous clinical programs.
Communication and technology expense decreased $260,000 from $686,000 during the year ended September 30, 2016 to $426,000 during the current period. This category includes costs associated with the Company's IT infrastructure. The decrease was primarily due to several IT consulting projects completed during fiscal 2016.
Office expense increased $260,000 from $303,000 during the year ended September 30, 2016 to $563,000 during the current period. These expenses relate to conferences/training, office supplies, miscellaneous administrative expenses, and expenses related to office expansions at our R&D facility in Madison, Wisconsin and our corporate headquarters in Pasadena, California. The increase is primarily related to moving expenses for the Company's move to its new facility in Madison, Wisconsin.
Other expense decreased $1,257,000 from $1,406,000 during the year ended September 30, 2016 to $149,000 during the current period. This category consists primarily of conference attendance fees, franchise and property tax expenses and marketing expenses. The decrease in other expense relates to litigation in the fiscal 2016 periods that was settled.
Stock-based compensation expense
Stock-based compensation expense, a noncash expense, decreased $3,704,221 from $11,595,816 during the year ended September 30, 2016 to $7,891,595 during the current period. Stock-based compensation expense is based upon the valuation of stock options and restricted stock units granted to employees, directors, and certain consultants. Many variables affect the amount expensed, including the Company's stock price on the date of the grant, as well as other assumptions. The decrease in the expense in each period is primarily related to the decrease in the Company's stock price, which is a key input in deriving the valuations of the awards.
Depreciation and amortization expense
Depreciation and amortization expense, a noncash expense, increased $1,430,095 from $3,260,045 during the year ended September 30, 2016 to $4,690,440 during the current period. The majority of depreciation and amortization expense relates to depreciation on lab equipment. In addition, the Company records depreciation on leasehold improvements at its Madison, Wisconsin research facility and its Pasadena, California corporate headquarters. The increase in depreciation and amortization expense is primarily due to the depreciation on leasehold improvements at the Company's new Madison, Wisconsin research facility in the current period.
Impairment expense
Impairment expense, a noncash expense, was $2,050,817 during the year ended September 30, 2016 and $0 during the current period. During the previous period, the Company recognized an impairment expense of $1.1 million related to leasehold improvements at its previous research facility in Madison, Wisconsin. This amount represented the entire net book value remaining for the leasehold improvements associated with the previous facility, and was recognized during the year ended September 30, 2016 as the Company moved into a larger research facility. During the previous period, the Company also recognized a $0.9 million impairment expense related to acquired in-process research and development assets that were acquired in the acquisition of the Roche RNAi business. In November 2016, the Company announced the discontinuation of its clinical trial efforts for ARC-520, ARC-AAT and ARC-521. Given this development, the Company assessed the fair value of this indefinite-lived intangible asset to be $0 at September 30, 2016.
Contingent Consideration – Fair Value Adjustments
45
Contingent Consideration – Fair Value Adjustments was ($5,862,464) during the year ended September 30, 2016 and $0 during the current period. Contingent consideration resulting from the acquisition of Roche's RNAi business is calculated by modeling res earch and development activities for clinical candidates, forecasting timelines to market, and using "peak sales" estimate modeling, cash flows and potential milestone and royalty payments. The modeling assumes certain success rates and discount factors r elated to riskiness of projects and the time value of money to calculate a net present value of future consideration payments to Roche. Each reporting period, the Company re-evaluates its contingent consideration, and if material, makes adjustments to the recorded liability. In November 2016, the Company announced the discontinuation of its clinical trial efforts for ARC-520, ARC-AAT and ARC-521. Given this development, the Company assessed the fair value of its contingent consideration obligation to be $0 at September 30, 2016 and September 30, 2017.
Other Income / Expense
Other income / expense was income of $22,124 during the year ended September 30, 2016 as compared to income of $2,618,014 during the current period. The largest component of other income / expense in the current period was $1.3 million in other income due to an insurance settlement related to one of the Company's recent litigation cases. The settlement amount was received in fiscal 2017. The other significant component of other income / expense is related to the change in the value of derivative liabilities related to certain warrants with a price adjustment feature, which requires derivative accounting. The change in value of derivative liabilities was an expense of approximately $0.3 million in 2016 and an income of approximately $0.9 million in the current period. The fluctuations in each period were primarily driven by changes in the Company's stock price, which had a corresponding impact on the valuation of the underlying warrant liability.
Results of Operations Comparison for 2016 and 2015
Revenues
Total revenue was $158,333 for the year ended September 30, 2016 and $382,000 for the year ended September 30, 2015. Revenue during these periods is primarily related to licensed technology. In addition, the Company had collaboration revenue of $160,000 during the year ended September 30, 2015.
Operating Expenses
The analysis below details the operating expenses and discusses the expenditures of the Company within the major expense categories. Certain reclassifications have been made to prior-period operating expense categories to conform to the current period presentation. For purposes of comparison, the amounts for the years ended September 30, 2016 and 2015 are shown in the tables below.
Research and Development Expenses
R&D expenses are related to the Company's on-going research and development efforts, primarily related to program costs, composed primarily of outsourced costs related to the manufacturing of clinical supplies, toxicity/efficacy studies and clinical trial expenses. Internal costs primarily relate to operations at our research and development facility in Madison, Wisconsin, including facility costs and laboratory-related expenses. The following table provides details of research and development expense for the periods indicated:
(in thousands)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
|
| Twelve Months Ended |
|
| % of Expense |
|
| Twelve Months Ended |
|
| % of Expense |
|
| Increase (Decrease) |
| |||||||||
|
| September 30, 2016 |
|
| Category |
|
| September 30, 2015 |
|
| Category |
|
| $ |
|
|
| % |
| |||||
Laboratory supplies & services |
| $ | 2,706 |
|
|
| 7 | % |
| $ | 2,531 |
|
|
| 5 | % |
| $ | 175 |
|
|
| 7 | % |
In vivo studies |
|
| 1,611 |
|
|
| 4 | % |
|
| 556 |
|
|
| 1 | % |
|
| 1,055 |
|
|
| 190 | % |
Outside labs & contract services |
|
| 155 |
|
|
| 0 | % |
|
| 489 |
|
|
| 1 | % |
|
| (334 | ) |
|
| -68 | % |
Toxicity/efficacy studies |
|
| 7,766 |
|
|
| 19 | % |
|
| 6,572 |
|
|
| 14 | % |
|
| 1,194 |
|
|
| 18 | % |
Drug manufacturing |
|
| 9,855 |
|
|
| 25 | % |
|
| 21,431 |
|
|
| 46 | % |
|
| (11,576 | ) |
|
| -54 | % |
Clinical trials |
|
| 14,800 |
|
|
| 36 | % |
|
| 13,329 |
|
|
| 28 | % |
|
| 1,471 |
|
|
| 11 | % |
License, royalty & milestones |
|
| 3,054 |
|
|
| 6 | % |
|
| 1,065 |
|
|
| 2 | % |
|
| 1,989 |
|
|
| 187 | % |
Facilities and related |
|
| 1,315 |
|
|
| 3 | % |
|
| 977 |
|
|
| 2 | % |
|
| 338 |
|
|
| 35 | % |
Other research expenses |
|
| 192 |
|
|
| 1 | % |
|
| 317 |
|
|
| 1 | % |
|
| (125 | ) |
|
| -39 | % |
Total |
| $ | 41,454 |
|
|
| 100 | % |
| $ | 47,267 |
|
|
| 100 | % |
| $ | (5,813 | ) |
|
| -12 | % |
46
Laboratory supplies and services expense increased $175,000 from $2,531,000 during the year ended September 30, 2015 to $2,706,000 during the year ended September 30, 2016. The increase in laboratory supplies and services is a result of additional supply purchases necessary to support the expansion of the Company's preclinical pipeline as well as the development of the subcutaneous versions of its drug candidates.
In vivo studies expense increased $1,055,000 from $556,000 during the year ended September 30, 2015 to $1,611,000 during the year ended September 30, 2016. In vivo expense can vary depending on the stage of preclinical candidates, the nature and amount of testing required and the varying costs of different in vivo testing models. The expense in both periods relates to studies in connection with the development of new clinical candidates, and the increase in fiscal year 2016 was primarily driven by studies for ARO-LPA (AMG 890) before it was licensed to Amgen.
Outside labs and contract services expense decreased $334,000 from $489,000 during the year ended September 30, 2015 to $155,000 during the year ended September 30, 2016. The decrease primarily relates to reduced contracted labor services and more functions being performed in house.
Toxicity/efficacy studies expense increased $1,194,000 from $6,572,000 during the year ended September 30, 2015 to $7,766,000 during the year ended September 30, 2016. This category includes IND-enabling toxicology studies, post-IND toxicology studies, such as long-term toxicology studies, and other efficacy studies. The increase primarily relates to toxicology studies related to ARC-521.
Drug manufacturing expense decreased $11,576,000 from $21,431,000 during the year ended September 30, 2015 to $9,855,000 during the year ended September 30, 2016. The decrease primarily relates to the substantial completion of our ARC-520 Phase 2b manufacturing campaign during fiscal 2015. Drug manufacturing expense for the year ended September 30, 2016 primarily relates to manufacturing for ARC-521 clinical trials.
Clinical trials expense increased $1,471,000 from $13,329,000 during the year ended September 30, 2015 to $14,800,000 during the year ended September 30, 2016. In both periods, the primary driver of the expenses was related to ARC-520 Phase 2b trials. We also incurred costs in fiscal 2016 related to our clinical trials for ARC-AAT and ARC-521.
License, royalty and milestones expense increased $1,989,000 from $1,065,000 during the year ended September 30, 2015 to $3,054,000 during the year ended September 30, 2016. This category can include milestone payments which can vary from period to period depending on the nature of our various license agreements and the timing of reaching various development milestones requiring payment. We reached milestones related to our clinical candidates that required a $3 million payment in fiscal 2016 and a $1 million payment in fiscal 2015.
Facilities expense increased $338,000 from $977,000 during the year ended September 30, 2015 to $1,315,000 during the year ended September 30, 2016. The increase relates to rent for our additional research and development facility in Middleton, Wisconsin and increased repairs and maintenance costs for our lab equipment.
Other research expense decreased $125,000 from $317,000 during the year ended September 30, 2015 to $192,000 during the year ended September 30, 2016. The decrease primarily relates to costs associated with a collaboration agreement to identify muscle targeting peptide molecules, for which the Company has been reimbursed from its collaboration partner.
47
Salary and Payroll-Related Expenses
The Company employs scientific, technical and administrative staff at its corporate offices and its research facilities. Salaries and payroll-related expense consists of salaries, bonuses, payroll taxes and related benefits. Salary and payroll-related expenses include two major categories, based on the primary activities of each employee: general and administrative (G&A) compensation expense and research and development (R&D) compensation expense. The following table provides detail of salary and payroll-related expenses for the periods indicated:
(in thousands)
|
| Twelve Months |
|
|
|
|
| Twelve Months |
|
|
|
|
|
|
|
|
|
|
|
| ||||
|
| Ended |
|
| % of |
|
| Ended |
|
| % of |
|
| Increase (Decrease) |
| |||||||||
|
| September 30, 2016 |
|
| Expense Category |
|
| September 30, 2015 |
|
| Expense Category |
|
| $ |
|
| % |
| ||||||
R&D - compensation-related |
| $ | 13,883 |
|
|
| 71 | % |
| $ | 11,605 |
|
|
| 70 | % |
| $ | 2,278 |
|
|
| 20 | % |
G&A - compensation-related |
|
| 5,579 |
|
|
| 29 | % |
|
| 4,949 |
|
|
| 30 | % |
|
| 630 |
|
|
| 13 | % |
Total |
| $ | 19,462 |
|
|
| 100 | % |
| $ | 16,554 |
|
|
| 100 | % |
| $ | 2,908 |
|
|
| 18 | % |
R&D compensation expense increased $2,278,000 from $11,605,000 during the year ended September 30, 2015 to $13,883,000 during the year ended September 30, 2016. An increase in headcount accounted for the majority of the change in compensation-related expense.
G&A compensation expense increased $630,000 from $4,949,000 during the year ended September 30, 2015 to $5,579,000 during the year ended September 30, 2016. Annual merit increases accounted for the majority of the change in compensation-related expense.
General & Administrative Expenses
The following table provides details of our general and administrative expenses for the periods indicated:
(in thousands)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
|
| Twelve Months Ended |
|
| % of Expense |
|
| Twelve Months Ended |
|
| % of Expense |
|
| Increase (Decrease) |
| |||||||||
|
| September 30, 2016 |
|
| Category |
|
| September 30, 2015 |
|
| Category |
|
| $ |
|
|
|
|
| |||||
Professional/outside services |
| $ | 4,201 |
|
|
| 43 | % |
| $ | 3,989 |
|
|
| 50 | % |
| $ | 212 |
|
|
| 5 | % |
Patent expense |
|
| 1,529 |
|
|
| 15 | % |
|
| 950 |
|
|
| 12 | % |
|
| 579 |
|
|
| 61 | % |
Facilities and related |
|
| 320 |
|
|
| 3 | % |
|
| 308 |
|
|
| 4 | % |
|
| 12 |
|
|
| 4 | % |
Travel |
|
| 864 |
|
|
| 9 | % |
|
| 841 |
|
|
| 11 | % |
|
| 23 |
|
|
| 3 | % |
Business insurance |
|
| 632 |
|
|
| 6 | % |
|
| 523 |
|
|
| 7 | % |
|
| 109 |
|
|
| 21 | % |
Communication and Technology |
|
| 686 |
|
|
| 7 | % |
|
| 691 |
|
|
| 9 | % |
|
| (5 | ) |
|
| -1 | % |
Office expenses |
|
| 303 |
|
|
| 3 | % |
|
| 270 |
|
|
| 3 | % |
|
| 33 |
|
|
| 12 | % |
Other |
|
| 1,406 |
|
|
| 14 | % |
|
| 359 |
|
|
| 4 | % |
|
| 1,047 |
|
|
| 292 | % |
Total |
| $ | 9,941 |
|
|
| 100 | % |
| $ | 7,931 |
|
|
| 100 | % |
| $ | 2,010 |
|
|
| 25 | % |
Professional/outside services include legal, accounting, consulting and other outside services retained by the Company. All periods include normally recurring legal and audit expenses related to SEC compliance and other corporate matters. Professional/outside services expense increased $212,000 from $3,989,000 during the year ended September 30, 2015 to $4,201,000 during the year ended September 30, 2016. The increase in professional fees primarily related to higher legal fees related to previous litigation events.
Patent expense increased $579,000 from $950,000 during the year ended September 30, 2015 to $1,529,000 during the year ended September 30, 2016. Patent expense costs increased due to additional prosecution requirements associated with new patents acquired through the Novartis asset acquisition. The Company continues to invest in patent protection for its product candidates and other RNAi technology through patent filings in numerous countries. The Company expects to extend and maintain protection for its current portfolios, as appropriate, and file new patent applications as technologies are developed and improved. Expenses can vary from period to period as patents proceed through their prosecution life cycle.
48
Facilities-related expense was consistent at $308,000 during t he year ended September 30, 2015 and $320,000 in the year ended September 30, 2016. Facilities expense represents the expense associated with our corporate headquarters in Pasadena, California.
Travel expense was consistent at $841,000 during the year ended September 30, 2015 and $864,000 during the year ended September 30, 2016. Travel expense is incurred to support our R&D function, primarily our GMP manufacturing campaign and our clinical trials, as well as other corporate and business development related travel.
Business insurance expense increased $109,000 from $523,000 during the year ended September 30, 2015 to $632,000 during the year ended September 30, 2016. Business insurance costs increased primarily due to added coverage related to the Company's clinical trials, as well as increases in other corporate liability insurance.
Communication and technology expense was consistent at $691,000 during the year ended September 30, 2015 and $686,000 during the year ended September 30, 2016. This category covers IT equipment and services for our personnel.
Office expense increased $33,000 from $270,000 during the year ended September 30, 2015 to $303,000 during the year ended September 30, 2016. These expenses relate to conferences/training, office supplies, miscellaneous administrative expenses, and expenses related to office expansions at our R&D facility in Madison, Wisconsin and our corporate headquarters in Pasadena, California. Conference trainings and seminar expenses were increased in the current period.
Other expense increased $1,047,000 from $359,000 during the year ended September 30, 2015 to $1,406,000 during the year ended September 30, 2016. The increase in the current period pertains to litigation as discussed in Note 7 – Commitments and Contingencies o f Notes to Consolidated Financial Statements of Part IV, Item 15. Exhibits and Financial Statement Schedules . This category also consists primarily of franchise and property tax expenses and marketing.
Acquired in-process research and development – Novartis pre-clinical candidates
Acquired in-process research and development expense related to the Novartis pre-clinical candidates was $10,142,786 for fiscal year 2015 and $0 for the year ended September 30, 2016. This expense pertains to the acquisition of the Novartis RNAi assets. The value of the purchase price allocated to certain preclinical candidates was expensed during the period, while certain patents and a third-party license were capitalized as intangible assets.
Stock-based compensation expense
Stock-based compensation expense, a noncash expense, increased $1,362,919 from $10,232,897 during the year ended September 30, 2015 to $11,595,816 during the year ended September 30, 2016. Stock-based compensation expense is based upon the valuation of stock options and restricted stock units granted to employees, directors, and certain consultants. Many variables affect the amount expensed, including the Company's stock price on the date of the grant, as well as other assumptions. The increase in the current period was primarily due to certain performance based awards that were deemed probable of achievement during the period.
Depreciation and amortization expense
Depreciation and amortization expense, a noncash expense, increased $923,838 from $2,336,207 during the year ended September 30, 2015 to $3,260,045 during the year ended September 30, 2016. The majority of depreciation and amortization expense relates to depreciation on lab equipment. In addition, the Company records depreciation on leasehold improvements at its Madison, Wisconsin research facility and its Pasadena, California corporate headquarters. The increase in depreciation and amortization expense is primarily due to the amortization of the intangible assets acquired in the Novartis RNAi asset acquisition.
Impairment expense
Impairment expense, a noncash expense, was $2,050,817 in the year ended September 30, 2016 and $0 during the year ended September 30, 2015. During the year ended September 30, 2016, the Company recognized an impairment expense of $1.1 million related to leasehold improvements at its previous research facility in Madison, Wisconsin. This amount represented the entire net book value remaining for the leasehold improvements associated with the previous facility, and was recognized during the year ended September 30, 2016 as the Company moved into a larger research facility. During the year ended September 30, 2016, the Company also recognized a $0.9 million impairment expense related to acquired in-process research and development assets that were acquired in the acquisition of the Roche RNAi business. In November 2016, the Company announced the discontinuation of its clinical trial efforts for ARC-520, ARC-AAT and ARC-521. Given this development, the Company assessed the fair value of this indefinite-lived intangible asset to be $0 at September 30, 2016.
Contingent Consideration – Fair Value Adjustments
49
Contingent Consideration – Fair Value Adjustments decreased $7,753,997 from $1,891,5 33 during the year ended September 30, 2015 to ($5,862,464) during the year ended September 30, 2016. Contingent consideration resulting from the acquisition of Roche's RNAi business is calculated by modeling research and development activities for clinic al candidates, forecasting timelines to market, and using "peak sales" estimate modeling, cash flows and potential milestone and royalty payments. The modeling assumes certain success rates, and discount factors related to riskiness of projects and the ti me value of money to calculate a net present value of future consideration payments to Roche. Each reporting period, the Company re-evaluates its contingent consideration, and if material, makes adjustments to the recorded liability. In November 2016, th e Company announced the discontinuation of its clinical trial efforts for ARC-520, ARC-AAT and ARC-521. Given this development, the Company assessed the fair value of its contingent consideration obligation to be $0 at September 30, 2016.
Other Income / Expense
Other income / expense was income of $4,035,494 during the year ended September 30, 2015 as compared to income of $22,124 during the year ended September 30, 2016. The largest component of other income / expense is related to the change in the value of derivative liabilities related to certain warrants with a price adjustment feature, which requires derivative accounting. The change in value of derivative liabilities was a reduction of approximately $2.9 million in 2015 and an increase of approximately $0.3 million in the year ended September 30, 2016. The fluctuations in each period were primarily driven by changes in the Company's stock price, which had a corresponding impact to the valuation of the underlying warrant liability.
Liquidity and Cash Resources
Arrowhead has historically financed its operations through the sale of its equity securities. Research and development activities have required significant capital investment since the Company's inception, and are expected to continue to require significant cash expenditure in fiscal year 2017 and beyond .
At September 30, 201 7 , the Company had cash on hand of approximately $24.8 million as compared to $85.4 million at September 30, 2016. Excess cash invested in fixed income securities was $40.8 million at September 30, 2017, compared to $0 million at September 30, 2016. The Company believes its current financial resources are sufficient to fund its operations through at least the next twelve months.
A summary of cash flows for the years ended September 30, 2017, 2016, and 2015 is as follows:
|
| Year ended September 30, |
| |||||||||
|
| 2017 |
|
| 2016 |
|
| 2015 |
| |||
Cash Flow from: |
|
|
|
|
|
|
|
|
|
|
|
|
Operating Activities |
| $ | (23,938,972 | ) |
| $ | (64,427,486 | ) |
| $ | (65,707,615 | ) |
Investing Activities |
|
| (48,644,218 | ) |
|
| 13,447,763 |
|
|
| 14,120,838 |
|
Financing Activities |
|
| 12,055,309 |
|
|
| 55,131,817 |
|
|
| 290,521 |
|
Net increase (decrease) in cash |
|
| (60,527,881 | ) |
|
| 4,152,094 |
|
|
| (51,296,256 | ) |
Cash at beginning of period |
|
| 85,366,448 |
|
|
| 81,214,354 |
|
|
| 132,510,610 |
|
Cash at end of period |
| $ | 24,838,567 |
|
| $ | 85,366,448 |
|
| $ | 81,214,354 |
|
During the year ended September 30, 2017, the Company used $23.9 million in cash from operating activities, primarily driven by $53.9 million of cash used for the on-going expenses of its research and development programs and general and administrative expenses, partially offset by the $30 million upfront payment received from Amgen. Cash used in investing activities was $48.6 million, which was primarily related to investments in short-term fixed-income securities of $45.0 million and $7.9 million of capital expenditures primarily for leasehold improvements on the Company's Madison, Wisconsin research facility and lab equipment purchases. Cash generated by financing activities of $12.1 million was driven by the $12.5 million equity investment received from Amgen, and was partially offset by cash paid for employee taxes on net share settlements of restricted stock units that vested during the period.
During the year ended September 30, 2016, the Company used $64.4 million in cash from operating activities, which represents the on-going expenses of its research and development programs and corporate overhead. Cash outlays were primarily composed of the following: research and development costs were $42.6 million, salary and payroll-related expenses were $16.4 million and general and administrative costs were $9.9 million. These expenditures were partially offset by $5 million received under the First Collaboration and License Agreement with Amgen. Cash provided by investing activities was $13.4 million, primarily related to the maturity of certain marketable securities of $17.3 million partially offset by capital expenditures of $3.9 million. Cash provided by
50
financing activities of $55.1 million primarily includes an equity financing in August 2016, which yielded net proceeds of $43.2 million to the Company, and $9 million of equity investments from the Company's Common Stock Purchase Agreement with Amge n.
During the year ended September 30, 2015, the Company used $65.7 million in cash from operating activities, which represents the on-going expenses of its research and development programs and corporate overhead. Cash outlays were primarily composed of the following: research and development costs were $41.2 million, salary and payroll-related expenses were $16.6 million, and general and administrative costs were $7.9 million. Cash provided by investing activities was $14.1 million, primarily related to the maturity of certain marketable securities partially offset by $10.0 million of cash used to acquire the Novartis assets discussed above. Capital expenditures were $ 2.0 million. Cash provided by financing activities of $0.3 million primarily includes the exercise of warrants and stock options during the year ended September 30, 2015.
Contractual Obligations
In the table below, we set forth our enforceable and legally binding obligations and future commitments at September 30, 2017 for the categories shown, as well as obligations related to contracts in such categories that we are likely to continue. Some of the figures that we include in this table are based on management's estimates and assumptions about these obligations, including their duration, the possibility of renewal, anticipated actions by third parties and other factors. Because these estimates and assumptions are necessarily subjective, the obligations we will actually pay in future periods may vary from those reflected in the table. The following table does not include any future obligations that may be owed under existing license agreements, as the certainty of achieving the relevant milestones that would trigger these payments is currently unknown.
|
| Payments due by Period |
| |||||||||||||||||
|
| Total |
|
| Less than 1 Year |
|
| 1-3 Years |
|
| 3-5 Years |
|
| More than 5 Years |
| |||||
Long-Term Debt |
|
| 2,325,018 |
|
|
| - |
|
|
| 464,078 |
|
|
| 534,748 |
|
|
| 1,326,192 |
|
Capital Leases |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
Operating Leases |
|
| 10,848,066 |
|
|
| 1,531,234 |
|
|
| 2,479,840 |
|
|
| 2,167,664 |
|
|
| 4,669,328 |
|
Purchase Obligations |
|
| 11,400,000 |
|
|
| 11,400,000 |
|
|
| - |
|
|
| - |
|
|
| - |
|
Other Long-Term Liabilities |
|
| 2,129,052 |
|
|
| - |
|
|
| 519,495 |
|
|
| 424,320 |
|
|
| 1,185,237 |
|
Total |
| $ | 26,702,136 |
|
| $ | 12,931,234 |
|
| $ | 3,463,413 |
|
| $ | 3,126,732 |
|
| $ | 7,180,757 |
|
Off-Balance Sheet Arrangements
As of September 30, 2017, we did not have any off-balance sheet arrangements, as defined in Item 303(a)(4)(ii) of SEC Regulation S-K.
Recent Accounting Pronouncements
See Note 1 to our Consolidated Financial Statements of this annual report on Form 10-K for a description of recent accounting pronouncements applicable to our business.
51
ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
We are exposed to market risk related to changes in interest rates, which could adversely affect the value of our interest rate-sensitive assets and liabilities. We do not hold any instruments for trading purposes and investment criteria are governed by the Company's Investment Policy. As of September 30, 2017 and 2016, we had cash and cash equivalents of $24.8 million and $85.4 million, respectively, and investments of $40.8 million and $0, respectively. At times, we have invested our cash reserves in corporate bonds typically with maturities of less than 2 years, and we have historically classified these investments as held-to-maturity. Due to the relatively short-term nature of the investments that we hold, we do not believe that the results of operations or cash flows would be affected to any significant degree by a sudden change in market interest rates relative to our investment portfolio. Our liability instrument sensitive to changes in interest rates is our derivative liability with its fair value determined using an option pricing model, which uses interest rate as an input. However, any change associated with this valuation would result in a noncash expense and would not significantly impact our operations.
ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
The information required by this item is included in Item 15 of this Annual Report on Form 10-K.
ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE. |
None.
ITEM 9A. | CONTROLS AND PROCEDURES. |
Our Chief Executive Officer and our Chief Financial Officer, after evaluating our "disclosure controls and procedures" (as defined in Securities Exchange Act of 1934 (the "Exchange Act") Rules 13a-15(e) and 15d-15(e)) as of the end of the period covered by this Annual Report on Form 10-K (the "Evaluation Date") have concluded that as of the Evaluation Date, our disclosure controls and procedures are effective to ensure that information we are required to disclose in reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in Securities and Exchange Commission rules and forms, and to ensure that information required to be disclosed by us in such reports is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, where appropriate, to allow timely decisions regarding required disclosure.
No change in the Company's internal controls over financial reporting (as defined in Rule 13a-15(f) and 15d-15(f) of the Exchange Act) occurred during the Company's most recent fiscal quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Management's Annual Report on Internal Control over Financial Reporting
Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Our internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles in the United States. This process includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures are being made only in accordance with authorizations of our management and directors; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on our financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of the internal control over financial reporting to future periods are subject to risk that the internal control may become inadequate because of changes in conditions, or that the degree of compliance with policies or procedures may deteriorate.
Management's Assessment of the Effectiveness of our Internal Control over Financial Reporting
The Company's management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rule 13a-15(f) under the Exchange Act). Management conducted an assessment of the effectiveness of the Company's
52
inte rnal control over financial reporting based on the criteria set forth in Internal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on the Company's assessment, management has conclu ded that its internal control over financial reporting was effective as of September 30, 2017 to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements in accordance with GAAP. The Company' s independent registered public accounting firm, Rose, Snyder and Jacobs LLP, has issued an audit report on the Company's internal control over financial reporting, which appears in Item 15 of this Form 10-K.
Changes in Internal Control Over Financial Reporting
There was no change in our internal control over financial reporting that occurred during the fourth quarter of the year ended September 30, 2017 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
ITEM 9B. | OTHER INFORMATION |
None.
PART III
ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE. |
The information called for by this Item will be incorporated by reference from our Definitive Proxy Statement to be filed for our 2018 Annual Meeting of Stockholders, which proxy statement will be filed no later than January 28, 2018.
ITEM 11. | EXECUTIVE COMPENSATION |
The information called for by this Item will be incorporated by reference from our Definitive Proxy Statement to be filed for our 2018 Annual Meeting of Stockholders, which proxy statement will be filed no later than January 28, 2018.
ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS. |
The information called for by this Item will be incorporated by reference from our Definitive Proxy Statement to be filed for our 2018 Annual Meeting of Stockholders, which proxy statement will be filed no later than January 28, 2018.
ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE. |
The information called for by this Item will be incorporated by reference from our Definitive Proxy Statement to be filed for our 2018 Annual Meeting of Stockholders, which proxy statement will be filed no later than January 28, 2018.
ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The information called for by this Item will be incorporated by reference from our Definitive Proxy Statement to be filed for our 2018 Annual Meeting of Stockholders, which proxy statement will be filed no later than January 28, 2018.
PART IV
ITEM 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES |
The following documents are filed as part of this Annual Report on Form 10-K:
| (1) | Financial Statements. |
See Index to Financial Statements and Schedule on page F-1.
| (2) | Financial Statement Schedules. |
See Index to Financial Statements and Schedule on page F-1. All other schedules are omitted as the required information is not present or is not present in amounts sufficient to require submission of the schedule, or because the information required is included in the consolidated financial statements or notes thereto.
53
| (3) | Exhibits. |
The following exhibits are filed (or incorporated by reference herein) as part of this Annual Report on Form 10-K:
Exhibit Number |
| Description |
| Incorporated by Reference Herein | ||
|
| Form |
| Date | ||
2.1 |
|
Stock and Asset Purchase Agreement between Arrowhead Research Corporation and Roche entities, dated October 21, 2011† |
|
Annual Report on Form 10-K for the fiscal year ended September 30, 2011, as Exhibit 2.1 |
|
December 20, 2011 |
|
|
|
|
|
|
|
2.2 |
| Asset Purchase and Exclusive License Agreement between Arrowhead Research Corporation and Novartis Institutes for BioMedical Research, Inc., dated March 3, 2015† |
| Quarterly Report on Form 10-Q, as Exhibit 2.1 |
| May 11, 2015 |
|
|
|
|
|
|
|
3.1 |
| Amended and Restated Certificate of Incorporation of Arrowhead Research Corporation, a Delaware corporation, filed with the Secretary of State of the State of Delaware on April 5, 2016 |
| Current Report on Form 8-K as Exhibit 3.3 |
| April 6, 2016 |
|
|
|
|
|
|
|
3.2 |
| Amended and Restated Bylaws of Arrowhead Pharmaceuticals, Inc. |
| Current Report on form 8-K as Exhibit 3.4 |
| April 6, 2016 |
|
|
|
|
|
|
|
3.3 |
| Certificate of Designation, Preferences, and Rights of Series D Junior Participating Preferred Stock of Arrowhead Pharmaceuticals, Inc. |
| Current Report on form 8-K as Exhibit 3.1 |
| March 23, 2017 |
|
|
|
|
|
|
|
4.1 |
| Form of Warrant to Purchase Common Stock expiring May 2017 |
| Current Report on Form 8-K, as Exhibit 4.1 |
| May 30, 2007 |
|
|
|
|
|
|
|
4.2 |
| Form of Warrant to Purchase Shares of Common Stock expiring December 12, 2017 |
| Current Report on Form 8-K, as exhibit 4.2 |
| December 12, 2012 |
|
|
|
|
|
|
|
4.3 |
| Form of Warrant to Purchase Shares of Common Stock expiring January 30, 2018 |
| Current Report on Form 8-K, as exhibit 4.2 |
| January 30, 2013 |
|
|
|
|
|
|
|
4.4 |
| Form of Series C Preferred Stock Certificate |
| Annual Report on Form 10-K for the fiscal year ended September 30, 2013, as Exhibit 4.19 |
| December 18, 2013 |
|
|
|
|
|
|
|
4.5 |
| Form of Common Stock Certificate of Arrowhead Pharmaceuticals, Inc. |
| Current Report on Form 8-K, as Exhibit 4.1 |
| April 6, 2016 |
|
|
|
|
|
|
|
4.6 |
| Form of Indenture |
| Registration Statement on Form S-3 (File No. 333-214315) |
| October 28, 2016 |
|
|
|
|
|
|
|
4.7 |
| Rights Agreement dated as of March 21, 2017, between the Company and Computershare Trust Company, N.A., as rights agent, which includes as Exhibit B the Form of Rights Certificate |
| Current Report on Form 8-K, as Exhibit 4.1 |
| March 23, 2017 |
|
|
|
|
|
|
|
10.1** |
| Arrowhead Research Corporation (fka InterActive, Inc.) 2000 Stock Option Plan |
| Schedule 14C, as Exhibit D |
| December 22, 2000 |
|
|
|
|
|
|
|
10.2** |
| Arrowhead Research Corporation 2004 Equity Incentive Plan, as amended |
| Schedule 14C, as Annex B |
| January 12, 2012 |
|
|
|
|
|
|
|
10.3** |
| Arrowhead Research Corporation 2013 Incentive Plan |
| Schedule 14C, as Annex A |
| December 20, 2013 |
|
|
|
|
|
|
|
10.4** |
| Form of Stock Option Agreement for use with the 2013 Incentive Plan |
| Current Report on Form 8-K, as Exhibit 10.1 |
| February 12, 2014 |
|
|
|
|
|
|
|
54
Exhibit Number |
| Description |
| Incorporated by Reference Herein | ||
|
| Form |
| Date | ||
10.5** |
| Form of Restricted Stock Unit Agreement for use with the 2013 Incentive Plan | ||||