UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2015
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission File Number 333-146542
AMPIO PHARMACEUTICALS, INC.
(Exact Name of Registrant as Specified in Its Charter)
| Delaware | 26-0179592 | |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification Number) |
373 Inverness Parkway Suite 200 Englewood, Colorado | 80112 | |
| (Address of principal executive offices) | (Zip Code) |
(720) 437-6500
(Registrant's telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
Title of each class | Name of each exchange on which registered | |
| Common Stock, par value $.0001 per share | The NYSE Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ¨ No x
Indicate by a check mark whether the Registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the Registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K x
Indicate by check mark whether the Registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definition of "large accelerated filer", "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act. (check one):
| Large Accelerated Filer | ¨ | Accelerated Filer | x | |||
| Non-Accelerated Filer | ¨ (Do not check if a smaller reporting company) | Smaller reporting company | ¨ | |||
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The aggregate market value of common stock held by non-affiliates of the Registrant as of June 30, 2015 was $108,000,000 based on the closing price of $2.37 as of that date.
Indicate the number of shares outstanding of each of the Registrant's classes of common stock, as of the latest practicable date: As of February 1, 2016, 52,016,432 shares of common stock were outstanding.
TABLE OF CONTENTS
| Page | ||||||
| PART I | ||||||
Item 1 | BUSINESS | 4 | ||||
Item 1A | RISK FACTORS | 16 | ||||
Item 1B | UNRESOLVED STAFF COMMENTS | 31 | ||||
Item 2 | PROPERTIES | 31 | ||||
Item 3 | LEGAL PROCEEDINGS | 31 | ||||
Item 4 | MINE SAFETY DISCLOSURES | 32 | ||||
| PART II | ||||||
Item 5 | MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES | 33 | ||||
Item 6 | SELECTED FINANCIAL DATA | 35 | ||||
Item 7 | MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS | 36 | ||||
Item 7A | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK | 48 | ||||
Item 8 | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA | 48 | ||||
Item 9 | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE | 48 | ||||
Item 9A | CONTROLS AND PROCEDURES | 48 | ||||
Item 9B | OTHER INFORMATION | 49 | ||||
| PART III | ||||||
Item 10 | DIRECTORS, EXECUTIVE OFFICERS, AND CORPORATE GOVERNANCE | 50 | ||||
Item 11 | EXECUTIVE COMPENSATION | 58 | ||||
Item 12 | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS | 67 | ||||
Item 13 | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE | 68 | ||||
Item 14 | PRINCIPAL ACCOUNTANT FEES AND SERVICES | 69 | ||||
| PART IV | ||||||
Item 15 | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES | 71 | ||||
SIGNATURES | 75 | |||||
Exhibit 23.1 | ||||||
Exhibit 31.1 | ||||||
Exhibit 31.2 | ||||||
Exhibit 32.1 | ||||||
This Report on Form 10-K refers to trademarks, such as Ampion, Optina, Zertane, Luoxis, Vyrix, RedoxSYS, MiOXSYS, ProstaScint, Primsol, Aytu and Rosewind, which are protected under applicable intellectual property laws and are our property or the property of our subsidiaries. This Form 10-K also contains trademarks, service marks, copyrights and trade names of other companies which are the property of their respective owners. Solely for convenience, our trademarks and tradenames referred to in this Form 10-K may appear without the ® or ™ symbols, but such references are not intended to indicate in any way that we will not assert, to the fullest extent under applicable law, our rights to these trademarks and tradenames.
Unless otherwise indicated or unless the context otherwise requires, references in this Form 10-K to the "Company," "Ampio," "we," "us," or "our" are to Ampio Pharmaceuticals, Inc. and its subsidiaries; references to "Life Sciences" are to DMI Life Sciences, Inc., our predecessor; and references to "BioSciences" are to DMI BioSciences, Inc.
2
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS AND INDUSTRY DATA
Forward Looking Statements
This Annual Report on Form 10-K, or Annual Report, includes forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, or the Exchange Act. All statements other than statements of historical facts contained in this Annual Report, including statements regarding our anticipated future clinical and regulatory events, future financial position, business strategy and plans and objectives of management for future operations, are forward-looking statements. Forward looking statements are generally written in the future tense and/or are preceded by words such as "may," "will," "should," "forecast," "could," "expect," "suggest," "believe," "estimate," "continue," "anticipate," "intend," "plan," or similar words, or the negatives of such terms or other variations on such terms or comparable terminology. Such forward-looking statements include, without limitation, statements regarding the anticipated start dates, durations and completion dates, as well as the potential future results, of our ongoing and future clinical trials, the anticipated designs of our future clinical trials, anticipated future regulatory submissions and events, the potential future commercialization of our product candidates, our anticipated future cash position and future events under our current and potential future collaborations. These forward-looking statements are subject to a number of risks, uncertainties and assumptions, including without limitation the risks described in "Risk Factors" in Part I, Item 1A of this Annual Report. These risks are not exhaustive. Other sections of this Annual Report include additional factors that could adversely impact our business and financial performance. Moreover, we operate in a very competitive and rapidly changing environment. New risk factors emerge from time to time and it is not possible for our management to predict all risk factors, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements. You should not rely upon forward-looking statements as predictions of future events. We cannot assure you that the events and circumstances reflected in the forward-looking statements will be achieved or occur and actual results could differ materially from those projected in the forward-looking statements. We assume no obligation to update or supplement forward-looking statements.
We obtained statistical data, market and product data, and forecasts used throughout this Form 10-K from market research, publicly available information and industry publications. While we believe that the statistical data, industry data and forecasts and market research are reliable, we have not independently verified the data, and we do not make any representation as to the accuracy of the information.
3
AMPIO PHARMACEUTICALS, INC.
PART I
| Item 1. | Business |
We are a biopharmaceutical company focused primarily on the development of therapies to treat prevalent inflammatory conditions for which there are limited treatment options.
Our product portfolio is primarily based on the work of Dr. David Bar-Or, the Director of Trauma Research LLC for both the Swedish Medical Center located in Englewood, CO and St. Anthony Hospital located in Lakewood, CO. For over two decades, while directing these two trauma research laboratories, Dr. Bar-Or and his staff have built a robust portfolio of product candidates focusing on inflammatory conditions. Our initial clinical programs were selected from Dr. Bar-Or's research based on certain criteria, particularly the ability to advance the candidates rapidly into late-stage clinical trials. The benchmarks used to build our pipeline were products with: (i) potential indications to address large underserved markets; (ii) strong intellectual property protection and the potential for market and data exclusivity; and (iii) a well-defined regulatory path to marketing approval.
We are primarily developing compounds that decrease inflammation by (i) inhibiting specific pro-inflammatory compounds by affecting specific pathways at the protein expression and at the transcription level; (ii) activating specific phosphatase or depleting available phosphate needed for the inflammation process; and (iii) decreasing vascular permeability.
We were also a majority stockholder in Aytu BioScience, Inc., or Aytu, at December 31, 2015, which was focused on acquiring, developing and commercializing products focused primarily on the urological disorders market, specifically sexual dysfunction, urological cancer, urinary tract infections and male infertility. On January, 4, 2016, we distributed a majority of our shares of common stock of Aytu to our shareholders on a pro rata basis. This transaction changed our ownership from 81.5% down to 8.6% of Aytu's outstanding shares on that date. See Note 1 – Basis of Presentation, Merger and Business Combination in the Notes to the Consolidated Financial Statements.
Corporate History
Our predecessor, DMI Life Sciences, Inc., or Life Sciences, was incorporated in Delaware in December 2008. In March 2010, Life Sciences was merged with a subsidiary of Chay Enterprises, Inc. As a result of this merger, Life Sciences stockholders became the controlling stockholders of Chay Enterprises. Following the merger, we reincorporated in Delaware as Ampio Pharmaceuticals, Inc. in March 2010.
In April 2015, Luoxis Diagnostics, Inc. and Vyrix Pharmaceuticals, Inc., each previously a subsidiary of ours, merged with and into Rosewind Corporation, or Rosewind. Following this transaction, we held 81.5% of the common stock of Rosewind, which changed its name to Aytu BioScience, Inc., or Aytu, in June 2015. In January 2016, we distributed a majority of our shares of common stock of Aytu to our shareholders on a pro rata basis. This transaction changed our ownership from 81.5% down to 8.6% of Aytu's outstanding shares on that date.
Our Product Pipeline
AMPION
Ampion for Osteoarthritis and Other Inflammatory Conditions
Ampion is a sub 5000 molecular weight fraction of commercial human serum albumin, or HSA. The primary constituent ingredient is aspartyl-alanyl diketopiperazine, or DA-DKP, an endogenous immunomodulatory molecule derived from the N-terminus of HSA. Based on Ampio's published in-vitro findings, DA-DKP appears to play a significant role in the homeostasis of inflammation. DA-DKP is believed to reduce inflammation by suppressing pro-inflammatory cytokine production in T-cells. Ampion also contains other known small molecules that confer anti-inflammatory effects to complement the activity of DA-DKP and derive in-vitro and in-vivo effects. We believe the non-steroidal, low molecular weight, anti-inflammatory biologic has the potential to be used in a wide variety of acute and chronic inflammatory conditions as well as immune-mediated diseases. We are currently developing Ampion as an intra-articular injection to treat pain due to osteoarthritis of the knee.
Ampion is manufactured as the low molecular weight filtration product of commercial human serum albumin containing DA-DKP, N-acetyltryptophan, caprylate, and other small molecules either contained in HSA or added to HSA during the processing and production of commercial HSA products. DA-DKP, the primary constituent ingredient contained in Ampion, is a locally generated molecule formed as a physiological result of the cleavage and cyclization of the N-terminal aspartic acid and alanine residues of human albumin. The molecule was originally discovered in the blood and cerebrospinal fluid of patients several days after suffering severe closed head injuries. A high concentration of DA-DKP has also been detected in biofilms found on endotracheal tubes recovered from intubated patients and on implanted orthopedic plates and screws. Together these findings suggest a mechanism by which DA-DKP contributes to the ability to reduce the body's inflammatory response following insult or injury.
4
DA-DKP is believed to reduce inflammation through the activation of Ras-related protein 1, or Rap1. Rap1 interrupts the kinase cascade by regulating the amount of rapidly accelerated fibrosarcoma, or Raf, kinases available for interaction with Ras, inhibiting antigen-specific Ras activation. This decrease disrupts the mitogen-activation protein kinase, or MAPK, cascade and results in decreased immunoinflammatory cytokine gene transcription. The clinical results which are detailed below also suggest an effect other than anti-inflammatory properties are at work and imply more prolonged healing-like effects.
We have published several scientific papers on Ampion. Most recently in June 2015 we announced three peer-reviewed publications, "The Low Molecular Weight Fraction of Commercial Human Serum Albumin (LMWF5A-Ampion) Induces Morphologic and Transcriptional Changes of Bone Marrow-Derived Mesenchymal Stem Cells" and "Anti-Inflammatory Activity in the Low Molecular Weight Fraction of Commercial Human Serum Albumin (LMWF5A)" and "Inflammatory pathways in knee osteoarthritis: potential targets for treatment".
Market Opportunity
Osteoarthritis, or OA, is the most common form of arthritis, affecting over 100 million people in the United States with over 48 million people suffering from osteoarthritis of the knee. It is a progressive disorder of the joints involving degradation of the intra-articular cartilage, joint lining, ligaments, and bone. The incidence of developing osteoarthritis of the knee over a lifetime is approximately 45%. Certain risk factors in conjunction with natural wear and tear lead to the breakdown of cartilage. Osteoarthritis is caused by inflammation of the soft tissue and bony structures of the joint, which worsens over time and leads to progressive thinning of articular cartilage. Other progressive effects include narrowing of the joint space, synovial membrane thickening, osteophyte formation and increased density of subchondral bone. The global osteoarthritis of the knee market addresses moderate to moderately severe OA and is currently over $3.0 billion. We believe that if Ampion proves to be effective in the severely diseased patients, this market could grow substantially. The global demand for osteoarthritis of the knee treatment is expected to be fueled by aging demographics and increasing awareness of treatment options. Despite the size and growth of the osteoarthritis of the knee market, few adequate treatment options currently exist.
Competition
The currently available treatments for osteoarthritis of the knee include oral non-steroidal anti-inflammatory agents, opioids, pain patches, intra-articular, or IA, corticosteroids, and IA hyaluronic acid, or HA, injections. Despite wide availability and years of clinical use, none of these agents are adequately meeting the needs of the market. In May 2013, the American Academy of Orthopedic Surgeons, or AAOS, issued their second edition of clinical practice guidelines for the treatment of osteoarthritis of the knee. The AAOS was unable to recommend for or against the use of intra-articular corticosteroid injections as studies designed to indicate efficacy are inconclusive. Further, the AAOS was also unable to recommend for or against the use of acetaminophen, opioids, or pain patches as the efficacy studies in this area are also inconclusive. Most importantly, the AAOS does not recommend (with a strong ‘strength of recommendation') the use of hyaluronic acid injections as, in the AAOS' assessment, the clinical evidence does not support their use. This latest clinical practice guideline underscores a pervasive unmet need in the treatment of osteoarthritis of the knee given few accepted and available treatments. We believe Ampion is a novel treatment option that, if approved, would be the first non-steroidal, non-hyaluronic-based intra-articular treatment available for the treatment of osteoarthritis of the knee.
AIK Trial Results
In 2011 and 2012 we conducted our Phase I Ampion trial in Australia. The AIK study established that Ampion was safe for human use and showed efficacy treating patients with pain due to OA of the knee. The trial was conducted in Australia because the biologics legislation governing the Australian Therapeutic Goods Administration, or TGA, allowed us to move Ampion directly into human clinical trials as the TGA recognized that HSA has an already established safety profile in humans by virtue of its longstanding commercial use. The AIK trial was conducted in patients diagnosed with moderately-severe to severe osteoarthritis of the knee.
SPRING Pivotal Trial Results
In the second half of 2013 we announced results of our first pivotal trial, the SPRING study, of Ampion for the treatment of pain due to osteoarthritis of the knee. The results of this study establish the safety and efficacy of Ampion for reduction of pain due to OA at 12 weeks after a single intra-articular injection in the knee. The SPRING study was a U.S. multicenter, randomized, double-blind, vehicle controlled trial. Three hundred twenty-nine patients were randomized to receive one of two doses (4 mL or 10 mL) of Ampion or corresponding saline control via intra-articular injection. Both doses of Ampion, 4 mL and 10 mL, showed a statistically significant reduction in pain compared to control, and there were no significant differences between the efficacy of the two Ampion doses. As such, the lowest required dose, 4 mL, was selected as the optimal dose. Patients who received Ampion experienced, on average, greater than a 40% reduction in pain from baseline at 12 weeks. Patients who received Ampion also showed a significant improvement in function and quality of life (quality of life was assessed using the Patient Global Assessment, or PGA) compared to patients who
5
received saline control at 12 weeks. Furthermore, the trial included severely diseased patients (defined as Kellgren-Lawrence IV) and those patients who received Ampion had a significantly greater reduction in pain than those patients who received saline control. Ampion was well tolerated with minimal adverse events, or AEs, reported equally across Ampion and saline groups in the study. There were no drug-related serious adverse events, or SAEs.
STEP Trial
In early 2014 we announced the STEP study clinical trial of Ampion for the treatment of pain due to osteoarthritis of the knee. The STEP study was a randomized, vehicle controlled, double-blind study in which 538 patients with osteoarthritis knee pain were randomized to receive either a 4 mL single injection of Ampion or saline control. A deviation of temperature protocols occurred during the drug distribution process of the STEP Study, which interfered with efficacy analysis. There were minimal adverse events reported and there were no drug-related SAEs in the STEP study.
STRUT Trial
In mid-2014 we announced the beginning of a Phase I multiple injection study, the STRUT study, at a single site for patients with pain due to mostly severe or very severe osteoarthritis of the knee. Patients showed a 65% improvement in pain and a 74% improvement in function from baseline at one-month post-injection. No drug-related SAEs were reported. Following these results, we initiated the randomized, double-blind, vehicle controlled (Phase II) portion of the multiple injection STRUT study.
In 2015, we announced the results from the Phase II STRUT study, which showed that patients who received Ampion demonstrated a significant improvement in pain when compared to patients who received saline control. Patients who received Ampion demonstrated, on average, a 64% reduction in pain at 20 weeks compared to baseline. The safety profile of Ampion in this trial was highly favorable, with no treatment-related SAEs. The results of this study establish the safety and efficacy of Ampion for reduction of pain at 20 weeks after a series of three IA injections administered two weeks apart in the knee of patients with OA.
STRIDE Trial
In late 2014 we enrolled 329 patients in the vehicle controlled, multiple injection, multi-center STRIDE study. Enrollment in this study differed from previous trials in both disease severity and patient Body Mass Index, or BMI. In the STRIDE study 68% of patients had severe osteoarthritis (Kellgren-Lawrence IV), compared to 23% in the SPRING study. Patients in this study were also significantly heavier and had a larger BMI than in any previous trials. In mid-2015 we announced that, although patients showed a marked reduction in pain from baseline to 20 weeks when treated with Ampion, the study failed to reach its primary endpoint.
PIVOT Trial
Together with the U.S. Food and Drug Administration, or the FDA, we agreed on a Special Protocol Assessment, an agreement on the trial design and size to form the primary basis of efficacy for a Biologics License Application, or BLA, filing for the confirmatory pivotal trial, the PIVOT study. The PIVOT trial is a randomized, vehicle controlled, double-blind study, planned to have approximately 484 patients with osteoarthritis knee pain who are randomized to receive either a 4mL single injection of Ampion or saline control. The primary objective of this study is to evaluate the efficacy of 4 mL Ampion versus 4 mL placebo intra-articular injection in improving knee pain, when administered to patients suffering from OA of the knee. The study started in October 2015 and in January 2016 we announced enrollment for this confirmatory study was approximately 80% complete. We expect the study to be completed by mid-2016.
Clinical Development Pathway
Upon conclusion of the AIK trial, pre-clinical and clinical data were presented to the blood products division of the Center for Biologics Evaluation and Research, or CBER, of the FDA for guidance toward an Ampion novel biologic BLA filing. The FDA provides novel biologics twelve years of market exclusivity against would-be "biosimilar" competitors. The FDA granted an active Investigational New Drug, or IND, for Ampion for the treatment of pain due to osteoarthritis of the knee in March 2013.
We met with the FDA in late 2013 and the FDA confirmed the SPRING study is the first of two pivotal clinical trials required to demonstrate efficacy in a BLA. In collaboration with the FDA we confirmed the trial design of the PIVOT study and the FDA granted us a Special Protocol Assessment, or SPA, an agreement on the design and size of the clinical trial intended to form the primary basis of an efficacy claim in the BLA, for the PIVOT study.
Future Development
We intend to study Ampion for therapeutic applications outside of osteoarthritis of the knee. We are investigating the possible use of Ampion for pain due to osteoarthritis of the hand and as a potential relief for ocular conditions. If successful, these additional formulations and potential therapeutic indications will supplement the Ampion clinical portfolio and may enable clinical applications in large therapeutic markets where there are significant unmet needs.
6
OPTINA
Optina for Diabetic Macular Edema
Optina is a low-dose formulation of danazol that we are developing to treat diabetic macular edema, or DME. Danazol is a synthetic derivative of modified testosterone ethisterone, and we believe it affects vascular endothelial cell linkage in a biphasic manner. At low doses, danazol decreases vascular permeability by increasing the barrier function of endothelial cells. The lipophilic low-molecular-weight weak androgen has the potential to treat multiple angiopathies. Steroid hormones control a variety of functions through slow genomic and rapid non-genomic mechanisms. Danazol immediately increases intracellular cyclic adenosine monophosphate, or cAMP, through the rapid activation of membrane-associated androgen, steroid binding globulin, and calcium channel receptors. At lower concentrations such as Optina, danazol binds to androgen and steroid binding globulin receptors stimulating the formation of a cortical actin ring. At higher concentrations, activation of the calcium channels shift the balance towards stress fiber formation and increase vascular permeability.
When organized into a cortical ring, filamentous actin, or f-actin, increases the barrier function of endothelial cells by tethering adhesion molecule complexes to the cytoskeleton. In this orientation, increased cortical actin improves tight junctions which strengthen cell-to-cell adhesions. Formation of the cortical actin ring thereby restricts leakage across the cell membrane.
Market Opportunity
Type 1 and Type 2 diabetes mellitus affects 26 million people in the United States. One of the many symptoms of diabetes is the local and systemic inflammation of the microvascular system. Diabetic retinopathy is a complication of diabetes and is characterized by damage to the blood vessels of the retina and can either be proliferative or non-proliferative. Proliferative damage occurs when a reduction in oxygen levels in the retina due to impaired glucose metabolism causes fragile blood vessels to grow in the vitreous humor. Non-proliferative damage occurs when existing vessels experience poor endothelial cell linkage due to increased blood glucose levels and hypertension. Macular edema is the most common form of non-proliferative diabetic retinopathy. In diabetic macular edema, prolonged hyperglycemia compromises endothelial cell linkage leading to vascular permeability. The leakage of fluid, solutes, proteins and immune cells cause the macula to swell and thicken. This leads to damage of the central retinal tissue and can significantly impair sharp central vision. The prevalence of diabetes is 11.3% of the population above the age of 20, with an annual incidence of 1.9 million cases in the United States alone. In this population, the prevalence of diabetic macular edema is estimated at 30% of patients inflicted by the disease for 20 years or more.
Competition
There are no orally administered treatments for DME currently available nor to our knowledge are any being tested in clinical trials. The current standard of care in the United States for the treatment of DME is laser photocoagulation. The first and only approved therapy in the United States is intravitreal ranibizumab-injections. Ranibizumab belongs to a therapeutic class inhibiting vascular endothelial growth factor, or anti-VEGF. It is important to note, there is significant competition from off-label anti-VEGF treatment of DME from bevacizumab. Iluvien (fluocinolone acetonide micro-insert intravitreous implant) is available in six European countries, and is pending approval in the United States while its sponsor reportedly resolves manufacturing issues. Dexamethasone intravitreal implant is available in the United States for macular edema following retinal vein occlusion and noninfectious uveitis and the product's sponsor has submitted applications for U.S. and European approval in the treatment of DME. Aflibercept, another anti-VEGF antibody treatment, is also awaiting U.S. and European approval in the treatment of DME.
Phase II Trial
In 2012, we concluded our Phase II randomized, double-blinded, placebo-controlled, dose-ranging study of Optina in subjects with diabetic macular edema in Canada. The trial established that the dose of Optina should take BMI into account. When stratified for BMI the study demonstrated that 47% of patients who received Optina improved at least one best corrected visual acuity, or BCVA, category and achieved a reduction in central retinal thickness, or CRT, at 12 weeks. The study was stopped early in order to pursue a redesigned trial that would evaluate the safety and efficacy of Optina with drug dosing refined by BMI.
OptimEyes Trial
In 2014 and 2015 we announced the OptimEyes multicenter, placebo-controlled, randomized, dose ranging trial to evaluate the safety and efficacy of oral Optina, which included 355 patients. The trial showed Optina was safe and well tolerated with no drug related
7
adverse events and no differences in side effect rates between placebo and Optina groups. The trial did not meet its primary endpoint for all patients, however we believe we have successfully identified an optimal dose for a BMI subgroup of patients who are refractory to currently available therapies and also utilize RAS inhibitors as a medication. As more than 70% of all DME patients are utilizing RAS inhibitors to control their blood pressure and we believe this combination of drugs shows promise as a painless, safe and efficacious oral treatment for DME, and a rescue medication following anti-VEGF therapy failure. These patients showed a +6.2 letter improvement in visual acuity. We presented these results at the World Ophthalmology Congress in February 2016 and plan to present the results at The Association for Research in Vision and Ophthalmology Conference in May 2016.
Clinical Trials in Support of a §505(b)(2) New Drug Application, or NDA
The FDA has indicated that, for §505(b)(2) NDAs, complete studies of the safety and effectiveness of a candidate product may not be necessary if appropriate bridging studies provide an adequate basis for reliance upon the FDA's findings of safety and effectiveness for a previously approved product.
Future Development
The FDA has previously indicated that a Phase III trial may be necessary following the OptimEyes trial. In late 2015, we announced guidance from the FDA and the outline for a second clinical trial for Optina.
NCE 001
Para-phenoxy-methylphenidate is a novel, small molecule methylphenidate derivative. Its basic mechanism of action is believed to be to increase methylation of the catalytic sub-unit of Protein Phosphatase 2A, or PP2A, with activation of this phosphatase achieving an effect similar to kinase inhibitors. PP2A is known to be largely involved in inflammation, angiogenesis, and cell proliferation, and by decreasing phosphorylation, the intracellular phosphatase inhibits pro-carcinogenic cytokines and chemokines and cell signaling factors. Our pre-clinical research is focused on neuroblastoma, glioblastoma multiforme, renal cell carcinoma, and inflammatory breast cancer.
Ampion Manufacturing Facility
We moved into our new manufacturing facility in the summer of 2014. Since that time we have implemented a quality system, validated the facility for human-use products and produced the product used in the PIVOT study clinical trial. We presented on single use technology in manufacturing at the 24th Annual Aseptic Processing Technology Conference for the International Society for Pharmaceutical Engineers in February of 2015. We are now in the final stages of the FDA required registration batches and have begun to manufacture inventory that could potentially be used commercially. We believe that these steps will shorten our regulatory timelines and significantly reduce our time to commercial market.
Merger/Subsidiary
Aytu BioScience, Inc.
On April 16, 2015, Luoxis Diagnostics, Inc., or Luoxis, and Vyrix Pharmaceuticals, Inc., or Vyrix, each previously a subsidiary of Ampio, entered into an Agreement and Plan of Merger, or the Merger Agreement, by and among Rosewind Corporation, a Colorado corporation and public company, or Rosewind, Luoxis, Vyrix, two major stockholders of Rosewind and two subsidiaries of Rosewind created solely for the purposes of the Merger (as defined below), and which did not survive the Merger.
In the first stage of the transaction, each of Luoxis and Vyrix merged with and into one of Rosewind's merger subsidiaries. Luoxis and Vyrix survived these mergers. The outstanding shares of stock of Luoxis and the outstanding shares of stock of Vyrix were converted into the right to receive shares of common stock in Rosewind. The Luoxis stock and the Vyrix stock were each converted at an exchange factor. The exchange factor for each of them was determined upon the basis of a relative value opinion obtained by Ampio prior to the Merger. The outstanding shares of Rosewind's merger subsidiary that merged with Luoxis were converted into shares of Luoxis as the surviving corporation. The outstanding shares of Rosewind's merger subsidiary that merged with Vyrix were converted into shares of Vyrix as the surviving corporation. After completion of the first stage of the transaction, Luoxis and Vyrix were wholly-owned subsidiaries of Rosewind.
In the second stage of the transaction, which occurred on the same day as the first stage of the transaction, each of Luoxis and Vyrix was merged with and into Rosewind, with Rosewind surviving. The first and second stage mergers are referred to collectively as the Merger. Following the consummation of the Merger, we became the holder of 81.5% of the common stock of Rosewind.
Pursuant to the Merger, Rosewind changed its fiscal year end from August 31 to June 30.
8
On June 1, 2015, the Rosewind shareholders voted to change the state of incorporation from Colorado to Delaware and to change Rosewind's name to Aytu BioScience, Inc., which was effective June 8, 2015.
On June 1, 2015, the Rosewind shareholders voted and approved a reverse stock split that was in effect on June 8, 2015. The reverse stock split was at a ratio of one new share for every 12.174 shares outstanding.
On January, 4, 2016, we distributed a majority of our shares of common stock of Aytu to our shareholders on a pro rata basis. This transaction changed our ownership from 81.5% down to 8.6% of Aytu's outstanding shares on that date.
Government Regulation
FDA Approval Process
In the United States, pharmaceutical products are subject to extensive regulation by the FDA. The Federal Food, Drug, and Cosmetic Act, or the FDC Act, and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, record keeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending NDAs, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties, and criminal prosecution.
Pharmaceutical and biologic product development in the United States typically involves the performance of satisfactory preclinical laboratory and animal studies under the FDA's Good Laboratory Practices, or GLPs, regulation, the development and demonstration of manufacturing processes which conform to FDA mandated current Good Manufacturing Practices, or cGMP, a quality system regulating manufacturing, the submission and acceptance of an IND application which must become effective before human clinical trials may begin in the United States, obtaining the approval of Institutional Review Boards, or IRBs, at each site where we plan to conduct a clinical trial to protect the welfare and rights of human subjects in clinical trials, adequate and well-controlled clinical trials to establish the safety and effectiveness of the drug or biologic for each indication for which FDA approval is sought, and the submission to the FDA for review and approval of a NDA or BLA. Satisfaction of FDA pre-market approval requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity, and novelty of the product or disease. Preclinical tests generally include laboratory evaluation of a product candidate, its chemistry, formulation, stability and toxicity, as well as certain animal studies to assess its potential safety and efficacy. Results of these preclinical tests, together with manufacturing information (in compliance with GLP and cGMP), analytical data and the clinical trial protocol (detailing the objectives of the trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated), must be submitted to the FDA as part of an IND, which must become effective before human clinical trials can begin.
An IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions about the intended conduct of the trials and imposes what is referred to as a clinical hold. Preclinical studies generally take several years to complete, and there is no guarantee that an IND based on those studies will become effective, allowing clinical testing to begin. In addition to FDA review of an IND, each medical site that desires to participate in a proposed clinical trial must have the protocol reviewed and approved by an independent IRB or Ethics Committee, or EC. The IRB considers, among other things, ethical factors, and the selection and safety of human subjects. Clinical trials must be conducted in accordance with the FDA's Good Clinical Practices, or GCP, requirements. The FDA and/or IRB/EC may order the temporary, or permanent, discontinuation of a clinical trial or a specific clinical trial site to be halted at any time, or impose other sanctions for failure to comply with requirements under the appropriate entity jurisdiction.
Clinical Trials to support NDAs and BLAs for marketing approval are typically conducted in three sequential phases, but the phases may overlap. In Phase 1 clinical trials, a product candidate is typically introduced either into healthy human subjects or patients with the medical condition for which the new drug is intended to be used. The main purpose of the trial is to assess a product candidate's safety and the ability of the human body to tolerate the product candidate. Phase 1 clinical trials generally include less than 50 subjects or patients. During Phase II trials, a product candidate is studied in an exploratory trial or trials in a limited number of patients with the disease or medical condition for which it is intended to be used in order to: (i) further identify any possible adverse side effects and safety risks, (ii) assess the preliminary or potential efficacy of the product candidate for specific target diseases or medical conditions, and (iii) assess dosage tolerance and determine the optimal dose for Phase III trial. Phase III trials are generally undertaken to demonstrate clinical efficacy and to further test for safety in an expanded patient population with the goal of evaluating the overall risk-benefit relationship of the product candidate. Phase III trials will generally be designed to reach a specific goal or endpoint, the achievement of which is intended to demonstrate the candidate product's clinical efficacy and adequate information for labeling of the drug or biologic.
After completion of the required clinical testing, a NDA or BLA is prepared and submitted to the FDA. FDA approval of the NDA or BLA is required before marketing of the product may begin in the United States. The NDA must include the results of all preclinical, clinical, and other testing and a compilation of data relating to the product's pharmacology, chemistry, manufacture, and controls. The
9
cost of preparing and submitting a NDA is substantial. Under federal law, the submission of most NDAs is additionally subject to a substantial application user fee, currently exceeding $2.3 million and the manufacturer and/or sponsor under an approved NDA are also subject to annual product and establishment user fees, currently approximately $0.1 million per product and $0.6 million per establishment. These fees are typically increased annually. The FDA will waive the application fee for the first human drug application that a small business or its affiliate submits for review (section 736(d)(1)(E) of the FD&C Act).
The FDA has 60 days from its receipt of a NDA to determine whether the application will be accepted for filing based on the FDA's threshold determination that it is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review. The FDA has agreed to certain performance goals in the review of NDAs. Most such applications for standard review drug products are reviewed within ten months; most applications for priority review drugs are reviewed in six months. The review process for both standard and priority review may be extended by FDA for three additional months to consider certain late-submitted information, or information intended to clarify information already provided in the submission. The FDA may also refer applications for novel drug products, or drug products which present difficult questions of safety or efficacy, to an advisory committee-typically a panel that includes clinicians and other experts-for review, evaluation, and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. Before approving a NDA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. Additionally, the FDA will inspect the facility or the facilities at which the drug is manufactured. The FDA will not approve the product unless compliance with cGMP is satisfactory and the NDA contains data that provide substantial evidence that the drug is safe and effective in the indication studied.
After the FDA evaluates the NDA and the manufacturing facilities, it issues either an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. If, or when, those deficiencies have been addressed to the FDA's satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. As a condition of NDA approval, the FDA may require a risk evaluation and mitigation strategy, or REMS, to help ensure that the benefits of the drug outweigh the potential risks. REMS can include medication guides, communication plans for healthcare professionals, and elements to assure safe use, or ETASU. ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries. The requirement for a REMS can materially affect the potential market and profitability of the drug. Moreover, product approval may require substantial post-approval testing and surveillance to monitor the drug's safety or efficacy. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or problems are identified following initial marketing.
Fast Track Designation
The FDA has developed "Fast Track" policies, which provide the potential for expedited review of a NDA. Fast Track status is potentially provided only for those new and novel therapies that are intended to treat persons with life-threatening and severely debilitating diseases where there is a defined unmet medical need, especially where no satisfactory alternative therapy exists or the new therapy is significantly superior to alternative therapies. During the development of product candidates that qualify for this status, the FDA may expedite consultations and reviews of these experimental therapies. Fast Track status also provides the potential for a product candidate to have a "Priority Review." A Priority Review allows for portions of the NDA to be submitted to the FDA for review prior to the completion of the entire application, which could result in a reduction in the length of time it would otherwise take the FDA to complete its review of the NDA. Fast Track status may be revoked by the FDA at any time if the clinical results of a trial fail to continue to support the assertion that the respective product candidate has the potential to address and unmet medical need. For biologics, priority review is further limited only for therapies intended to treat a serious or life threatening disease.
Orphan Drug Designation
The FDA may grant Orphan Drug status to drugs intended to treat a "rare disease or condition," which is generally a disease or condition that affects fewer than 200,000 individuals in the United States. If and when the FDA grants Orphan Drug status, the generic name and trade name of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Aside from guidance concerning the non-clinical laboratory studies and clinical investigations necessary for approval of the NDA, Orphan Drug status does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. The FDA may grant Orphan Drug status to multiple competing product candidates targeting the same indications. A product that has been designated as an Orphan Drug that subsequently receives the first FDA approval is entitled to Orphan Drug exclusivity. This exclusivity means the FDA may not approve any other applications to market the same drug for the same indication, except in very limited circumstances, for seven years from the date of the initial FDA approval. Orphan Drug approval may also provide certain tax benefits to the company that receives the first FDA approval. Finally, the FDA may fund the development of orphan products through its grants program for clinical studies.
10
Breakthrough Designation
Breakthrough therapy designation is intended to expedite the development and review of drugs for serious or life-threatening conditions. The criteria for breakthrough therapy designation require preliminary clinical evidence that demonstrates the drug may have substantial improvement for at least one clinically significant endpoint compared to available therapy. A breakthrough therapy designation conveys all of the fast track program features, as well as more intensive FDA guidance on an efficient drug development program. The FDA also has an organizational commitment to involve senior management in such guidance.
Accelerated Approval
Under the FDA's accelerated approval regulations, the FDA may approve a drug for a serious or life-threatening illness that provides meaningful therapeutic benefit to patients compared to existing treatments based upon a surrogate endpoint that is reasonably likely to predict clinical benefit. In clinical trials, a surrogate endpoint is a measurement of laboratory tests or clinical signs of a disease or condition that substitutes for a direct measurement of how a patient feels, functions, or survives. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. A drug candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of Phase IV or post-approval clinical trials to confirm the predictability of surrogate endpoints for clinical outcomes. Failure to conduct required post-approval studies, or confirm a clinical benefit during post-marketing studies, will allow the FDA to withdraw the drug from the market on an expedited basis. All promotional materials for drug candidates approved under accelerated regulations are subject to prior review by the FDA.
Foreign Regulatory Approval
Outside of the United States, our ability to market our product candidates will be contingent upon our receiving marketing authorizations from the appropriate foreign regulatory authorities, whether or not FDA approval has been obtained. The foreign regulatory approval process in most industrialized countries generally encompasses risks similar to those we will encounter in the FDA approval process. The requirements governing conduct of clinical trials and marketing authorizations, and the time required to obtain requisite approvals, may vary widely from country to country and differ from that required for FDA approval.
Under European Union regulatory systems, marketing authorizations may be submitted either under a centralized or decentralized procedure. The centralized procedure provides for the grant of a single marketing authorization that is valid for all European Union member states. The decentralized procedure provides for mutual recognition of national approval decisions. Under this procedure, the holder of a national marketing authorization may submit an application to the remaining member states. Within 90 days of receiving the applications and assessment report, each member state must decide whether to recognize approval. The mutual recognition process results in separate national marketing authorizations in the reference member state and each concerned member state. We will seek to choose the appropriate route of European regulatory filing in an attempt to accomplish the most rapid regulatory approvals for our product candidates when ready for review. However, the chosen regulatory strategy may not secure regulatory approvals or approvals of the chosen product indications. In addition, these approvals, if obtained, may take longer than anticipated. We can provide no assurance that any of our product candidates will prove to be safe or effective, will receive required regulatory approvals, or will be successfully commercialized.
The Hatch-Waxman Act
In seeking approval for a drug through a NDA, applicants are required to list with the FDA each patent whose claims cover the applicant's product. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the FDA's Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential competitors in support of approval of an abbreviated new drug application, or ANDA. An ANDA provides for marketing of a drug product that has the same active ingredients in the same strengths and dosage form as the listed drug and has been shown through bioequivalence testing to be therapeutically equivalent to the listed drug. Other than the requirement for bioequivalence testing, ANDA applicants are not required to conduct, or submit results of, pre-clinical or clinical tests to prove the safety or effectiveness of their drug product. Drugs approved in this way are commonly referred to as "generic equivalents" to the listed drug, and can often be substituted by pharmacists under prescriptions written for the original listed drug.
The ANDA applicant is required to certify to the FDA concerning any patents listed for the approved product in the FDA's Orange Book. Specifically, the applicant must certify that: 1) the required patent information has not been filed; 2) the listed patent has expired; 3) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or 4) the listed patent is invalid or will not be infringed by the new product. A certification that the new product will not infringe the already approved product's listed patents, or that such patents are invalid, is called a Paragraph IV certification. If the applicant does not challenge the listed patents, the ANDA application will not be approved until all the listed patents claiming the referenced product have expired.
11
If the ANDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit, or a decision in the infringement case that is favorable to the ANDA applicant.
The ANDA application also will not be approved until any non-patent exclusivity listed in the Orange Book for the referenced product has expired. Federal law provides a period of five years following approval of a drug containing no previously approved active ingredients during which ANDAs for generic versions of those drugs cannot be submitted, unless the submission contains a Paragraph IV challenge to a listed patent-in which case the submission may be made four years following the original product approval. Federal law provides for a period of three years of exclusivity during which FDA cannot grant effective approval of an ANDA based on the approval of a listed drug that contains previously approved active ingredients but is approved in a new dosage form, route of administration or combination, or for a new use; the approval of which was required to be supported by new clinical trials conducted by, or for, the applicant.
Post-Approval Regulation
Even if a product candidate receives regulatory approval, the approval is typically limited to specific clinical indications. Further, even after regulatory approval is obtained, subsequent discovery of previously unknown problems with a product may result in restrictions on its use or even complete withdrawal of the product from the market. Any FDA-approved products manufactured or distributed by us are subject to continuing regulation by the FDA, including record-keeping requirements and reporting of adverse events or experiences. Further, drug manufacturers and their subcontractors are required to register their establishments with the FDA and state agencies, and are subject to periodic inspections by the FDA and state agencies for compliance with cGMP, which impose rigorous procedural and documentation requirements upon us and our contract manufacturers. We cannot be certain that we or our present or future contract manufacturers or suppliers will be able to comply with cGMP regulations and other FDA regulatory requirements. Failure to comply with these requirements may result in, among other things, total or partial suspension of production activities, failure of the FDA to grant approval for marketing, and withdrawal, suspension, or revocation of marketing approvals.
If the FDA approves one or more of our product candidates, we and the contract manufacturers we use for manufacture of clinical supplies and commercial supplies must provide certain updated safety and efficacy information. Product changes, as well as certain changes in the manufacturing process or facilities where the manufacturing occurs or other post-approval changes may necessitate additional FDA review and approval. The labeling, advertising, promotion, marketing and distribution of a drug or biologic product also must be in compliance with FDA and Federal Trade Commission, or FTC, requirements which include, among others, standards and regulations for direct-to-consumer advertising, off-label promotion, industry sponsored scientific and educational activities, and promotional activities involving the Internet. The FDA and FTC have very broad enforcement authority, and failure to abide by these regulations can result in penalties, including the issuance of a warning letter directing us to correct deviations from regulatory standards and enforcement actions that can include seizures, fines, injunctions and criminal prosecution.
Other Regulatory Requirements
We are also subject to regulation by other regional, national, state and local agencies, including the U.S. Department of Justice, the Office of Inspector General of the U.S. Department of Health and Human Services and other regulatory bodies. Our current and future partners are subject to many of the same requirements.
In addition, we are subject to other regulations, including regulations under the Occupational Safety and Health Act, regulations promulgated by the U.S. Drug Enforcement Administration, the Toxic Substance Control Act, the Resource Conservation and Recovery Act, and regulations under other federal, state and local laws.
Violations of any of the foregoing requirements could result in penalties being assessed against us.
Privacy
Most health care providers, including research institutions from whom we or our partners obtain patient information, are subject to privacy and security rules under the Health Insurance Portability and Accountability Act of 1996, or HIPAA, and the recent amendments to HIPAA under the Health Information Technology for Economic and Clinical Health Act, or HITECH. Additionally, strict personal privacy laws in other countries affect pharmaceutical companies' activities in other countries. Such laws include the European Union, or EU, Directive 95/46/EC on the protection of individuals with regard to the processing of personal data, as well as individual EU Member States, implementing laws and additional laws. Although our clinical development efforts are not barred by these privacy regulations, we could face substantial criminal penalties if we knowingly receive individually identifiable health information from a health care provider that has not satisfied HIPAA's or the EU's disclosure standards. Failure by EU clinical trial partners to obey requirements of national laws on private personal data, including laws implementing the EU Data Protection Directive, might result in liability and/or adverse publicity.
12
Information Systems
We believe that our Information Systems, or IS, capabilities are adequate to manage our core business and our internal controls related to IS are operating effectively.
Intellectual Property Summary
Ampion
As of December 31, 2015, the current Ampion patent portfolio consists of 56 issued patents and 84 pending applications worldwide. The portfolio primarily consists of seven families filed in the United States and throughout the world. The first family includes six issued U.S. patents and one issued European Patent Office, or EPO, patent validated in 19 countries with claims relating to methods of treating inflammatory disease and compositions of matter comprising diketopiperazine derivatives, including DA-DKP. This family also includes issued patents in Australia, Canada, China, Hong Kong, Japan and South Africa and one pending application in the United States. The standard 20-year expiration for patents in this family is in 2021.
The second family includes seven issued U.S. patents with claims directed to methods of treating inflammation and T-cell mediated or inflammatory diseases with compositions of matter comprising DA-DKP. This family also includes issued patents in Australia, China, India, New Zealand, Singapore, Hong Kong, Israel, and South Africa and pending applications in the United States, Australia, Canada, China, EPO, Israel, Japan, Korea and Hong Kong. The standard 20-year expiration for patents in this family is in 2024.
The third family includes two issued U.S. patents, a pending U.S. application, an issued Chinese patent and pending applications in Australia, Brazil, Canada, China, Eurasia, EPO, Hong Kong, Indonesia, Israel, Japan, Korea, Mexico, Malaysia, New Zealand, Philippines, Singapore, United States, and South Africa. The claims in this family are directed to the use of DA-DKP for the treatment of degenerative joint diseases. The standard 20-year expiration for patents in this family is in 2032.
The fourth family includes one pending U.S. application and pending applications in Australia, Brazil, Canada, China, Eurasia, EPO, Indonesia, Israel, India, Japan, Korea, Mexico, Malaysia, New Zealand, Philippines, Singapore, and South Africa with claims directed to the use of DA-DKP to mobilize, home, expand and differentiate stem cells in the treatment of subjects. The standard 20-year expiration for patents in this family is in 2034.
The fifth family includes pending applications in the United States, Australia, Brazil, Canada, China, Eurasia, EPO, Indonesia, Israel, India, Japan, Korea, Mexico, Malaysia, New Zealand, Philippines, Singapore and South Africa with claims directed to methods for the manufacture of DA-DKP containing compositions. The standard 20-year expiration for patents in this family is in 2034.
The sixth family includes one pending U.S. application and a Patent Cooperation Treaty international application with claims directed to the use of DA-DKP for the treatment of degenerative joint diseases in a multi-dose treatment regimen. The standard 20-year expiration for patents in this family is in 2035.
The seventh family includes one pending U.S. provisional application with claims directed to the use of DA-DKP in the absence of COX-2 antagonist treatment. The standard 20-year expiration for patents in this family will be in 2036.
Optina
As of December 31, 2015, the Optina patent portfolio currently consists of 134 issued patents and 50 pending applications worldwide. The portfolio consists primarily of three patent families, the first and second of which include claims for the use of low doses of danazol to treat conditions associated with vascular hyperpermeability. These two families include issued patents in the United States, Australia, EPO (validated in 36 countries and Hong Kong), Germany, Japan, Mexico, New Zealand, South Africa, Singapore and Canada with claims relating to methods of treating macular edema or diabetic nephropathy with danazol. These families also include pending applications in Australia, Brazil, China, Canada, Eurasian Patent Organization, EPO, Indonesia, Israel, Japan, Korea, Mexico, Malaysia, New Zealand, Philippines, Singapore, Hong Kong and the United States. The standard 20-year expiration for patents in these families is in 2030. The third family is for the treatment of conditions associated with vascular hyperpermeability with low doses of danazol that correspond to the body fat content of the patient. The standard 20-year expiration for patents in this family is in 2033.
Barriers to Entry – General
We also maintain trade secrets and proprietary know-how that we seek to protect through confidentiality and nondisclosure agreements. We expect to seek U.S. and foreign patent protection for drug and diagnostic products we discover, as well as therapeutic
13
and diagnostic products and processes. We expect also to seek patent protection or rely upon trade secret rights to protect certain other technologies which may be used to discover and characterize drugs and diagnostic products and processes, and which may be used to develop novel therapeutic and diagnostic products and processes. These agreements may not provide meaningful protection or adequate remedies in the event of unauthorized use or disclosure of confidential and proprietary information. If we do not adequately protect our trade secrets and proprietary know-how, our competitive position and business prospects could be materially harmed.
The patent positions of companies such as ours involve complex legal and factual questions and, therefore, their enforceability cannot be predicted with any certainty. Our issued and licensed patents, and those that may be issued to us in the future, may be challenged, invalidated or circumvented, and the rights granted under the patents or licenses may not provide us with meaningful protection or competitive advantages. Our competitors may independently develop similar technologies or duplicate any technology developed by us, which could offset any advantages we might otherwise realize from our intellectual property. Furthermore, even if our product candidates receive regulatory approval, the time required for development, testing, and regulatory review could mean that protection afforded us by our patents may only remain in effect for a short period after commercialization. The expiration of patents or license rights we hold could adversely affect our ability to successfully commercialize our pharmaceutical drugs or diagnostics, thus harming our operating results and financial position.
We will be able to protect our proprietary intellectual property rights from unauthorized use by third parties only to the extent that such rights are covered by valid and enforceable patents or are effectively maintained as trade secrets. If we must litigate to protect our intellectual property from infringement, we may incur substantial costs and our officers may be forced to devote significant time to litigation-related matters. The laws of certain foreign countries do not protect intellectual property rights to the same extent as do the laws of the United States.
Our pending patent applications, or those we may file or license from third parties in the future, may not result in patents being issued. Until a patent is issued, the claims covered by an application for patent may be narrowed or removed entirely, thus depriving us of adequate protection. As a result, we may face unanticipated competition, or conclude that without patent rights the risk of bringing product candidates to market exceeds the returns we are likely to obtain. We are generally aware of the scientific research being conducted in the areas in which we focus our research and development efforts, but patent applications filed by others are maintained in secrecy for at least 18 months and, in some cases in the United States, until the patent is issued. The publication of discoveries in scientific literature often occurs substantially later than the date on which the underlying discoveries were made. As a result, it is possible that patent applications for products similar to our drug or diagnostic candidates may have already been filed by others without our knowledge.
The biotechnology and pharmaceutical industries are characterized by extensive litigation regarding patents and other intellectual property rights, and it is possible that our development of product candidates could be challenged by other pharmaceutical or biotechnology companies. If we become involved in litigation concerning the enforceability, scope and validity of the proprietary rights of others, we may incur significant litigation or licensing expenses, be prevented from further developing or commercializing a product candidate, be required to seek licenses that may not be available from third parties on commercially acceptable terms, if at all, or subject us to compensatory or punitive damage awards. Any of these consequences could materially harm our business.
Competition
The biotechnology and pharmaceutical industries are highly competitive and subject to significant and rapid technological change as researchers learn more about diseases and develop new technologies and treatments. Significant competitive factors in our industry include product efficacy and safety; quality and breadth of an organization's technology; skill of an organization's employees and its ability to recruit and retain key employees; timing and scope of regulatory approvals; government reimbursement rates for, and the average selling price of, products; the availability of raw materials and qualified manufacturing capacity; manufacturing costs; intellectual property and patent rights and their protection; and sales and marketing capabilities.
We cannot assure you that any of our products that we successfully develop will be clinically superior or scientifically preferable to products developed or introduced by our competitors.
Many of our actual and potential competitors have substantially longer operating histories and possess greater name recognition, product portfolios, and significantly greater experience in discovering, developing, manufacturing, and marketing products as well as financial, research, and marketing resources than we do. Among our smaller competitors, many of these companies have established co-development and collaboration relationships with larger pharmaceutical and biotechnology firms, which may make it more difficult for us to attract strategic partners. Our current and potential competitors include major multinational pharmaceutical companies, biotechnology firms, universities and research institutions. Some of these companies and institutions, either alone or together with their collaborators, have substantially greater financial resources and larger research and development staffs than do we. In addition, many of these competitors, either alone or together with their collaborators, have significantly greater experience than us in discovering, developing, manufacturing, and marketing pharmaceutical products and diagnostics. If one of our competitors realizes a significant advance in pharmaceutical drugs or diagnostics that address one or more of the diseases targeted by our product candidates, our products or diagnostics could be rendered uncompetitive or obsolete.
14
Our competitors may also succeed in obtaining FDA or other regulatory approvals for their product candidates more rapidly than we are able to do, which could place us at a significant competitive disadvantage or deny us marketing exclusivity rights. Market acceptance of our product or diagnostic candidates will depend on a number of factors, including: (i) potential advantages over existing or alternative therapies or tests, (ii) the actual or perceived safety of similar classes of products, (iii) the effectiveness of sales, marketing, and distribution capabilities, and (iv) the scope of any approval provided by the FDA or foreign regulatory authorities.
Although we believe our product candidates possess attractive attributes, we cannot assure you that our product candidates will achieve regulatory or market acceptance, or that we will be able to compete effectively in the pharmaceutical drug or diagnostic markets. If our product candidates fail to gain regulatory approvals and acceptance in their intended markets, we may not generate meaningful revenues or achieve profitability.
Research and Development
Our strategy is to minimize fixed overhead by outsourcing much of our research and development activities. Through a sponsored research agreement, our discovery activities are conducted by Trauma Research LLC, or TRLLC, a limited liability company owned by Dr. David Bar-Or. Under the research agreement, TRLLC conducts drug and biomarker discovery and development programs at its research facilities, and we provide funding and some scientific personnel. Intellectual property from discovery programs conducted by TRLLC on our behalf belongs to us, and we are solely responsible for protecting that intellectual property. While we have the right to generally request development work under the research agreement, TRLLC directs such work and is responsible for how the work is performed.
For the years ended December 31, 2015, 2014 and 2013, we recorded $19.0 million, $26.9 million, and $16.6 million, respectively, of research and development expenses. Research and development expenses represented 55.0%, 68.8%, and 68.9% of total operating expenses in the years ended December 31, 2015, 2014 and 2013, respectively. More information regarding our research and development activities can be found in the section entitled "Management's Discussion and Analysis of Financial Condition and Results of Operations" under Item 7 of this Annual Report.
Compliance with Environmental Laws
We believe we are in compliance with current material environmental protection requirements that apply to us or our business. Costs attributable to environmental compliance are not currently material.
Product Liability and Insurance
The development, manufacture and sale of pharmaceutical products involve inherent risks of adverse side effects or reactions that can cause bodily injury or even death. Product candidates we succeed in commercializing could adversely affect consumers even after obtaining regulatory approval and, if so, we could be required to withdraw a product from the market or be subject to administrative or other proceedings. We obtain clinical trial liability coverage for human clinical trials, and will obtain appropriate product liability insurance coverage for products we manufacture and sell for human consumption. The amount, nature and pricing of such insurance coverage will likely vary due to a number of factors such as the product candidate's clinical profile, efficacy and safety record, and other characteristics. We may not be able to obtain sufficient insurance coverage to address our exposure to product recall or liability actions, or the cost of that coverage may be such that we will be limited in the types or amount of coverage we can obtain. Any uninsured loss we suffer could materially and adversely affect our business and financial position.
Employees
As of February 26, 2016, we had 21 full-time employees and utilized the services of a number of consultants on a temporary basis. Overall, we have not experienced any work stoppage and do not anticipate any work stoppage in the foreseeable future. Management believes that relations with our employees are good.
Available Information
Our principal executive offices are located at 373 Inverness Parkway, Suite 200, Englewood, Colorado 80112 USA, and our phone number is (720) 437-6500.
15
We maintain a website on the internet at www.ampiopharma.com . We make available free of charge through our website, by way of a hyperlink to a third-party site that includes filings we make with the SEC website ( www.sec.gov) , our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports electronically filed or furnished pursuant to Section 15(d) of the Exchange Act. The information on our website is not, and shall not be deemed to be, a part of this annual report on Form 10-K or incorporated into any other filings we make with the SEC. In addition, the public may read and copy any materials we file with the SEC at the SEC's Public Reference Room at 100 F Street, N.E., Washington D.C., 20549. Information on the operation of the Public Reference Room may be obtained by calling the SEC at 1-800-SEC-0330. Our Code of Conduct and Ethics and the charters of our Nominating and Governance Committee, Audit Committee, and Compensation Committee of our Board of Directors may be accessed within the Investor Relations section of our website. Amendments and waivers of the Code of Conduct and Ethics will also be disclosed within four business days of issuance on the website. Information found in our website is neither part of this annual report on Form 10-K nor any other report filed with the SEC.
| Item 1A. | Risk Factors |
Risks Related to Our Business
We have incurred significant losses since inception, expect to incur net losses for at least the next several years and may never achieve or sustain profitability.
We have experienced significant net losses since inception. As of December 31, 2015, we had an accumulated deficit of $133.9 million. We expect our annual net losses could continue over the next several years as we advance our development programs and incur significant clinical development costs.
We have not received, and do not currently expect to receive, any revenues from the commercialization of our product candidates in the near term. We may enter into licensing and collaboration arrangements, which may provide us with potential milestone payments and royalties and those arrangements, if obtained, will be our primary source of revenues for the coming years. We cannot be certain that any licensing or collaboration arrangements will be concluded, or that the terms of those arrangements will result in our receiving material revenues. To obtain revenues from product candidates, we must succeed, either alone or with others, in a range of challenging activities, including completing clinical trials of our product candidates, obtaining marketing approval for these product candidates, manufacturing, marketing and selling those products for which we, or our collaborators, may obtain marketing approval, satisfying any post-marketing requirements and obtaining reimbursement for our products from private insurance or government payors. We, and our collaborators, may never succeed in these activities and, even if we do, or one of our collaborators does, we may never generate revenues that are significant enough to achieve profitability.
We will need substantial additional capital to fund our operations. If we are unable to raise capital when needed, we could be forced to delay, reduce or eliminate our product development programs and commercialization efforts.
Developing pharmaceutical products, including conducting pre-clinical studies and clinical trials, is a very time-consuming, expensive and uncertain process that takes years to complete. We expect our expenses to increase in connection with our ongoing activities, particularly as we initiate new clinical trials of, initiate new research and pre-clinical development efforts for and seek marketing approval for, our product candidates. We will require additional capital to fund our operations, including to:
| • | continue to fund clinical trials of Ampion and Optina; |
| • | prepare for and apply for regulatory approval for our product candidates; |
| • | develop additional product candidates; |
| • | conduct additional clinical research and development; |
| • | pursue existing and new claims covered by intellectual property we own or license; and |
| • | sustain our corporate overhead requirements, and hire and retain necessary personnel. |
Until we can generate revenue from collaboration agreements to finance our cash requirements, which we may not accomplish, we expect to finance future cash needs primarily through offerings of our equity securities, including under our "at-the-market" equity program, or debt. Such financing may result in dilution to our stockholders, imposition of debt covenants and repayment obligations, or other restrictions that may adversely affect our business.
We do not know whether additional funding will be available to us on acceptable terms, or at all. If we are unable to secure additional funding when needed, we may have to delay, reduce the scope, or eliminate development of one or more of our product candidates, or substantially curtail or close our operations altogether. Alternatively, we may have to obtain a collaborator for one or more of our product candidates at an earlier stage of development, which could lower the economic value of those product candidates to us.
16
Ampion and Optina are currently undergoing clinical trials that are time-consuming and expensive, the outcomes of which are unpredictable, and for which there is a high risk of failure. If clinical trials of our product candidates fail to satisfactorily demonstrate safety and efficacy to the FDA and other regulators, we, or our collaborators, may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of these product candidates.
Pre-clinical testing and clinical trials are long, expensive and unpredictable processes that can be subject to extensive delays. We cannot guarantee that any clinical studies will be conducted as planned or completed on schedule, if at all. It may take several years to complete the pre-clinical testing and clinical development necessary to commercialize a drug or biologic, and delays or failure can occur at any stage. Interim results of clinical trials do not necessarily predict final results, and success in pre-clinical testing and early clinical trials does not ensure that later clinical trials will be successful. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in advanced clinical trials even after promising results in earlier trials and we cannot be certain that we will not face similar setbacks. The design of a clinical trial can determine whether its results will support approval of a product and flaws in the design of a clinical trial may not become apparent until the clinical trial is well advanced.
Our product development programs are at various stages of development. We continue to work toward completion and analysis of clinical trials for our primary products: Ampion and Optina. An unfavorable outcome in one or more trials for Ampion or Optina would be a major set-back for the development programs for these product candidates and for us. Due to our limited financial resources, an unfavorable outcome in one or more of these trials may require us to delay, reduce the scope of, or eliminate one of these product development programs, which could have a material adverse effect on our business and financial condition and on the value of our common stock.
In connection with clinical testing and trials, we face a number of risks, including:
| • | a product candidate is ineffective, inferior to existing approved medicines, unacceptably toxic, or has unacceptable side effects; |
| • | patients may die or suffer other adverse effects for reasons that may or may not be related to the product candidate being tested; |
| • | the results may not confirm the positive results of earlier testing or trials; and |
| • | the results may not meet the level of statistical significance required by the U.S. Food and Drug Administration, or FDA, or other regulatory agencies to establish the safety and efficacy of our product candidates. |
The results of pre-clinical studies do not necessarily predict clinical success, and larger and later-stage clinical studies may not produce the same results as earlier-stage clinical studies. Frequently, product candidates developed by pharmaceutical companies have shown promising results in early pre-clinical or clinical studies, but have subsequently suffered significant setbacks or failed in later clinical studies. In addition, clinical studies of potential products often reveal that it is not possible or practical to continue development efforts for these product candidates.
If we do not successfully complete pre-clinical and clinical development, we will be unable to market and sell products derived from our product candidates and generate revenues. Even if we do successfully complete clinical trials, those results are not necessarily predictive of results of additional trials that may be needed before an NDA or BLA may be submitted to the FDA. Although there are a large number of drugs and biologics in development in the U.S. and other countries, only a small percentage result in the submission of an NDA or BLA to the FDA, even fewer are approved for commercialization, and only a small number achieve widespread physician and consumer acceptance following regulatory approval. If our clinical studies are substantially delayed or fail to prove the safety and effectiveness of our product candidates in development, we may not receive regulatory approval of any of these product candidates and our business and financial condition will be materially harmed.
Delays, suspensions and terminations in our clinical trials could result in increased costs to us and delay or prevent our ability to generate revenues.
Human clinical trials are very expensive, time-consuming, and difficult to design, implement and complete. We currently expect clinical trials of our product candidates could take from nine to 24 months to complete, but the completion of trials for our product candidates may be delayed for a variety of reasons, including delays in:
| • | demonstrating sufficient safety and efficacy to obtain regulatory approval to commence a clinical trial; |
| • | reaching agreement on acceptable terms with prospective contract research organizations and clinical trial sites; |
| • | manufacturing sufficient quantities of a product candidate; |
| • | obtaining approval of an IND from the FDA; |
| • | obtaining institutional review board approval to conduct a clinical trial at a prospective clinical trial site; |
17
| • | determining dosing and making related adjustments; and |
| • | patient enrollment, which is a function of many factors, including the size of the patient population, the nature of the protocol, the proximity of patients to clinical trial sites, the availability of effective treatments for the relevant disease and the eligibility criteria for the clinical trial. |
The commencement and completion of clinical studies for our product candidates may be delayed, suspended or terminated due to a number of factors, including:
| • | lack of effectiveness of product candidates during clinical studies; |
| • | adverse events, safety issues or side effects relating to the product candidates or their formulation; |
| • | inability to raise additional capital in sufficient amounts to continue clinical trials or development programs, which are very expensive; |
| • | the need to sequence clinical studies as opposed to conducting them concomitantly in order to conserve resources; |
| • | our inability to enter into collaborations relating to the development and commercialization of our product candidates; |
| • | failure by us or our collaborators to conduct clinical trials in accordance with regulatory requirements; |
| • | our inability or the inability of our collaborators to manufacture or obtain from third parties materials sufficient for use in pre-clinical and clinical studies; |
| • | governmental or regulatory delays and changes in regulatory requirements, policy and guidelines, including mandated changes in the scope or design of clinical trials or requests for supplemental information with respect to clinical trial results; |
| • | failure of our collaborators to advance our product candidates through clinical development; |
| • | delays in patient enrollment, variability in the number and types of patients available for clinical studies, and lower than anticipated retention rates for patients in clinical trials; |
| • | difficulty in patient monitoring and data collection due to failure of patients to maintain contact after treatment; |
| • | a regional disturbance where we or our collaborative partners are enrolling patients in our clinical trials, such as a pandemic, terrorist activities or war, or a natural disaster; and |
| • | varying interpretations of data by the FDA and similar foreign regulatory agencies. |
Many of these factors may also ultimately lead to denial of regulatory approval of a current or potential product candidate. If we experience delay, suspensions or terminations in a clinical trial, the commercial prospects for the related product candidate will be harmed, and our ability to generate product revenues will be delayed. For example, in August 2014 we experienced a delay in the STEP Study of Ampion due to a deviation from protocol in temperature excursions. We cannot be certain we will successfully complete the Ampion PIVOT trial or be able to complete future Optina trials within any specific time period, if at all.
In addition, we may encounter delays or product candidate rejections based on new governmental regulations, future legislative or administrative actions, or changes in FDA policy or interpretation during the period of product development. If we obtain required regulatory approvals, such approvals may later be withdrawn. Delays or failures in obtaining regulatory approvals may:
| • | adversely affect the commercialization of any product candidates we develop; |
| • | diminish any competitive advantages that such product candidates may have or attain. |
Furthermore, if we fail to comply with applicable FDA and other regulatory requirements at any stage during this regulatory process, we may encounter or be subject to:
| • | delays in clinical trials or commercialization; |
| • | refusal by the FDA to review pending applications or supplements to approved applications; |
| • | product recalls or seizures; |
| • | suspension of manufacturing; |
| • | withdrawals of previously approved marketing applications; and |
| • | fines, civil penalties, and criminal prosecutions. |
18
If we do not secure collaborations with strategic partners to test, commercialize and manufacture product candidates, we may not be able to successfully develop products and generate meaningful revenues.
An aspect of our current strategy is to selectively enter into collaborations with third parties to conduct clinical testing, as well as to commercialize and manufacture product candidates. Our ability to generate revenues from these arrangements will depend on our collaborators' abilities to successfully perform the functions assigned to them in these arrangements. Collaboration agreements typically call for milestone payments that depend on successful demonstration of efficacy and safety, obtaining regulatory approvals, and clinical trial results. Collaboration revenues are not guaranteed, even when efficacy and safety are demonstrated. The current economic environment may result in potential collaborators electing to reduce their external spending, which may prevent us from developing our product candidates.
Even if we succeed in securing collaborators, the collaborators may fail to develop or effectively commercialize products using our product candidates or technologies. Collaborations involving our product candidates pose a number of risks, including the following:
| • | collaborators may not have sufficient resources or decide not to devote the necessary resources due to internal constraints such as budget limitations, lack of human resources, or a change in strategic focus; |
| • | collaborators may believe our intellectual property or the product candidate infringes on the intellectual property rights of others; |
| • | collaborators may dispute their responsibility to conduct development and commercialization activities pursuant to the applicable collaboration, including the payment of related costs or the division of any revenues; |
| • | collaborators may decide to pursue a competitive product developed outside of the collaboration arrangement; |
| • | collaborators may not be able to obtain, or believe they cannot obtain, the necessary regulatory approvals; |
| • | collaborators may delay the development or commercialization of our product candidates in favor of developing or commercializing another party's product candidate; or |
| • | collaborators may decide to terminate or not to renew the collaboration for these or other reasons. |
Thus, collaboration agreements may not lead to development or commercialization of product candidates in the most efficient manner or at all. For example, our former collaborator that licensed Zertane conducted clinical trials which we believe demonstrated efficacy in treating PE, but the collaborator undertook a merger that we believe altered its strategic focus and thereafter terminated the collaboration agreement. The merger also created a potential conflict with a principal customer of the acquired company, which sells a product to treat PE in certain European markets.
Collaboration agreements are generally terminable without cause on short notice. Once a collaboration agreement is signed, it may not lead to commercialization of a product candidate. We also face competition in seeking out collaborators. If we are unable to secure new collaborations that achieve the collaborator's objectives and meet our expectations, we may be unable to advance our product candidates and may not generate meaningful revenues.
If our product candidates are not approved by the FDA, we will be unable to commercialize them in the United States.
The FDA must approve any new medicine before it can be commercialized, marketed, promoted or sold in the United States. We must provide the FDA with data from pre-clinical and clinical studies that demonstrate that our product candidates are safe and effective for a defined indication before they can be approved for commercial distribution. Clinical testing is expensive, difficult to design and implement, can take many years to complete and is inherently uncertain as to outcome. We will not obtain approval for a product candidate unless and until the FDA approves a NDA for a drug and a BLA for a biologic. The processes by which regulatory approvals are obtained from the FDA to market and sell a new or repositioned product are complex, require a number of years and involve the expenditure of substantial resources. We cannot assure you that any of our product candidates will receive FDA approval in the future, and the time for receipt of any such approval is currently incapable of estimation.
19
If we do not receive marketing approval for Ampion, we may not realize the investment we have made in our manufacturing facility.
In December 2013, we entered into a ten-year lease of a multi-purpose facility containing approximately 19,000 square feet. We have spent approximately $10.4 million dollars to build out this facility in anticipation of receiving approval of our BLA and commencing commercialization of Ampion. If the FDA does not approve our BLA for Ampion, or does not approve of our manufacturing operation, we will not be able to manufacture and commercialize Ampion in our new facility and we will remain obligated to make payments under our lease, which is set to expire in 2024. Any delay or failure to receive BLA approval for Ampion could have a material adverse effect on the carrying value of the manufacturing facility as well as on our results of operations.
We or our collaborators intend to seek FDA approval for most of our product candidates using an expedited process established by the FDA. If we, or our collaborators, are unable to secure clearances to use expedited development pathways from the FDA for certain of our drug product candidates, we, or they, may be required to conduct additional pre-clinical studies or clinical trials beyond those that we, or they, contemplate, which could increase the expense of obtaining, and delay the receipt of, necessary marketing approvals and of any product revenues.
Assuming successful completion of clinical trials, we expect to submit NDAs to the FDA at various times in the future under §505(b)(2) of the Food, Drug and Cosmetic Act, as amended, or the FDCA. NDAs submitted under this section are eligible to receive FDA approval by relying in part on the FDA's findings of safety and efficacy for a previously approved drug. We are currently pursuing in our clinical trials a §505(b)(2) pathway for Optina and may also do so for other product candidates. The FDA's 1999 guidance on §505(b)(2) applications states that new indications for a previously approved drug, a new combination product, a modified active ingredient, or changes in dosage form, strength, formulation, and route of administration of a previously approved product are encompassed within the §505(b)(2) NDA process. Relying on §505(b)(2) is advantageous because we or our collaborators may not be required (i) to perform the full range of safety and efficacy trials that is otherwise required to secure approval of a new drug, and (ii) obtain a "right of reference" from the applicant that obtained approval of the previously approved drug. However, a §505(b)(2) application must support the proposed change of the previously approved drug by including necessary and adequate information, as determined by the FDA, and the FDA may still require us to perform a full range of safety and efficacy trials.
If one of our product candidates achieves clinical trial objectives, we must prepare and submit to the FDA a comprehensive NDA or BLA application. Review of the application may lead the FDA to request more information or require us to perform additional clinical trials, thus adding to product development costs and delaying any marketing approval from the FDA. Additionally, time to review may vary significantly based on the disease to be treated, availability of alternate treatments, severity of the disease, and the risk/benefit profile of the proposed product. Even if one of our products receives FDA marketing approval, we could be required to conduct post-marketing Phase IV studies and surveillance to monitor for adverse effects. If we experience delays in NDA application processing, requests for additional information or further clinical trials, or are required to conduct post-marketing studies or surveillance, our product development costs could increase substantially, and our ability to generate revenues from a product candidate could be postponed, perhaps indefinitely. The resulting negative impact on our operating results and financial condition may cause the value of our common stock to decline, and you may lose all or a part of your investment.
The approval process outside the United States varies among countries and may limit our ability to develop, manufacture and sell our products internationally. Failure to obtain marketing approval in international jurisdictions would prevent our product candidates from being marketed abroad.
In order to market and sell our products in the European Union and many other jurisdictions, we, and our collaborators, must obtain separate marketing approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and can involve additional testing. We may conduct clinical trials for, and seek regulatory approval to market, our product candidates in countries other than the United States. Depending on the results of clinical trials and the process for obtaining regulatory approvals in other countries, we may decide to first seek regulatory approvals of a product candidate in countries other than the U.S., or we may simultaneously seek regulatory approvals in the U.S. and other countries. If we or our collaborators seek marketing approvals for a product candidate outside the U.S., we will be subject to the regulatory requirements of health authorities in each country in which we seek approvals. With respect to marketing authorizations in Europe, we will be required to submit a European marketing authorization application, or MAA, to the European Medicines Agency, or EMA, which conducts a validation and scientific approval process in evaluating a product for safety and efficacy. The approval procedure varies among regions and countries and can involve additional testing, and the time required to obtain approvals may differ from that required to obtain FDA approval. Obtaining regulatory approvals from health authorities in countries outside the U.S. is likely to subject us to all of the risks associated with obtaining FDA approval described above. In addition, marketing approval by the FDA does not ensure approval by the health authorities of any other country, and approval by foreign health authorities does not ensure marketing approval by the FDA.
20
Even if we, or our collaborators, obtain marketing approvals for our product candidates, the terms of approvals and ongoing regulation of our products may limit how we, or they, manufacture and market our products, which could materially impair our ability to generate revenue.
Even if we receive regulatory approval for a product candidate, this approval may carry conditions that limit the market for the product or put the product at a competitive disadvantage relative to alternative therapies. For instance, a regulatory approval may limit the indicated uses for which we can market a product or the patient population that may utilize the product, or may be required to carry a warning in its labeling and on its packaging. Products with boxed warnings are subject to more restrictive advertising regulations than products without such warnings. These restrictions could make it more difficult to market any product candidate effectively.
In addition, manufacturers of approved products and those manufacturers' facilities are required to comply with extensive FDA requirements, including ensuring that quality control and manufacturing procedures conform to cGMPs, which include requirements relating to quality control and quality assurance as well as the corresponding maintenance of records and documentation and reporting requirements. We, our contract manufacturers, our collaborators and their contract manufacturers could be subject to periodic unannounced inspections by the FDA to monitor and ensure compliance with cGMPs.
Accordingly, assuming we, or our collaborators, receive marketing approval for one or more of our product candidates, we, and our collaborators, and our and their contract manufacturers will continue to expend time, money and effort in all areas of regulatory compliance, including manufacturing, production, product surveillance and quality control.
Any of our product candidates for which we, or our collaborators, obtain marketing approval in the future could be subject to post-marketing restrictions or withdrawal from the market and we, and our collaborators, may be subject to substantial penalties if we, or they, fail to comply with regulatory requirements or if we, or they, experience unanticipated problems with our products following approval.
Any of our product candidates for which we, or our collaborators, obtain marketing approval in the future, as well as the manufacturing processes, post-approval studies and measures, labeling, advertising and promotional activities for such products, among other things, will be subject to continual requirements of and review by the FDA and other regulatory authorities. These requirements include submissions of safety and other post-marketing information and reports, registration and listing requirements, requirements relating to manufacturing, quality control, quality assurance and corresponding maintenance of records and documents, requirements regarding the distribution of samples to physicians and recordkeeping. Even if marketing approval of a product candidate is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or to the conditions of approval, including the FDA requirement to implement a Risk Evaluation and Mitigation Strategy (REMS) to ensure that the benefits of a drug or biological product outweigh its risks.
The FDA may also impose requirements for costly post-marketing studies or clinical trials and surveillance to monitor the safety or efficacy of a product. The FDA and other agencies, including the Department of Justice, closely regulate and monitor the post-approval marketing and promotion of products to ensure that they are manufactured, marketed and distributed only for the approved indications and in accordance with the provisions of the approved labeling. The FDA imposes stringent restrictions on manufacturers' communications regarding off-label use and if we, or our collaborators, do not market any of our product candidates for which we, or they, receive marketing approval for only their approved indications, we, or they, may be subject to warnings or enforcement action for off-label marketing. Violation of the FDCA, the Public Health Service Act (PHSA), and other statutes, including the False Claims Act, relating to the promotion and advertising of prescription drugs may lead to investigations or allegations of violations of federal and state health care fraud and abuse laws and state consumer protection laws.
If we do not achieve our projected development and commercialization goals in the timeframes we announce and expect, the commercialization of our product candidates may be delayed, our business will be harmed, and our stock price may decline.
We sometimes estimate for planning purposes the timing of the accomplishment of various scientific, clinical, regulatory and other product development objectives. These milestones may include our expectations regarding the commencement or completion of scientific studies, clinical trials, the submission of regulatory filings, or commercialization objectives. From time to time, we may publicly announce the expected timing of some of these milestones, such as the completion of an ongoing clinical trial, the initiation of other clinical programs, receipt of marketing approval, or a commercial launch of a product. The achievement of many of these milestones may be outside of our control. All of these milestones are based on a variety of assumptions which may cause the timing of achievement of the milestones to vary considerably from our estimates, including:
| • | our available capital resources or capital constraints we experience; |
21
| • | the rate of progress, costs and results of our clinical trials and research and development activities, including the extent of scheduling conflicts with participating clinicians and collaborators, and our ability to identify and enroll patients who meet clinical trial eligibility criteria; |
| • | our receipt of approvals by the FDA and other regulatory agencies and the timing thereof; |
| • | other actions, decisions or rules issued by regulators; |
| • | our ability to access sufficient, reliable and affordable supplies of compounds used in the manufacture of our product candidates; |
| • | the efforts of our collaborators with respect to the commercialization of our products; and |
| • | the securing of, costs related to, and timing issues associated with, product manufacturing as well as sales and marketing activities. |
If we fail to achieve announced milestones in the timeframes we announce and expect, our business and results of operations may be harmed and the price of our stock may decline.
Our success is dependent in large part upon the continued services of our Chief Scientific Officer.
Our success is dependent in large part upon the continued services of our Chief Scientific Officer, Dr. David Bar-Or. We have an employment agreement with Dr. Bar-Or and a research agreement with Trauma Research LLC, an entity owned by Dr. Bar-Or that conducts research and development activities on our behalf. These agreements are terminable on short notice for cause by us or Dr. Bar-Or and may also be terminated without cause under certain circumstances. We do not maintain key-man life insurance on Dr. Bar-Or, although we may elect to obtain such coverage in the future. If we lost the services of Dr. Bar-Or for any reason, our clinical testing and other product development activities may experience significant delays, and our ability to develop and commercialize new product candidates may be diminished.
If we do not obtain the capital necessary to fund our operations, we will be unable to successfully develop, obtain regulatory approval of, and commercialize, pharmaceutical products.
The development of pharmaceutical products is capital-intensive. At December 31, 2015, we had cash and cash equivalents of approximately $27.0 million on a consolidated basis with Aytu and $16.0 million on an Ampio stand-alone basis. Based upon our current plans, it will be necessary to raise additional capital within the next 18 months. In 2014, we obtained a total of approximately $63.4 million in net proceeds from the sale of our common stock in an underwritten public offering, and in 2013, we obtained a total of approximately $29.0 million in net proceeds from the sale of our common stock in a registered direct offering. In February 2016, we entered into a Controlled Equity Offering SM Sales Agreement with a placement agent to implement an "at-the-market" equity program under which we, from time to time, may offer and sell shares of our common stock having an aggregate offering price of up to $25.0 million through the placement agent. We anticipate we will require significant additional financing to continue to fund our operations beyond the next 12 months. Our future capital requirements will depend on, and could increase significantly as a result of, many factors including:
| • | progress in, and the costs of, our pre-clinical studies and clinical trials and other research and development programs; |
| • | the scope, prioritization and number of our research and development programs; |
| • | the achievement of milestones or occurrence of other developments that trigger payments under any collaboration agreements we obtain; |
| • | the extent to which we are obligated to reimburse, or entitled to reimbursement of, clinical trial costs under future collaboration agreements, if any; |
| • | the costs involved in filing, prosecuting, enforcing and defending patent claims and other intellectual property rights; |
| • | the costs of securing manufacturing arrangements for commercial production; and |
| • | the costs of establishing or contracting for sales and marketing capabilities if we obtain regulatory clearances to market our product candidates. |
Until we can generate significant continuing revenues, we expect to satisfy our future cash needs through collaboration arrangements, private or public sales of our securities, debt financings, or by licensing one or more of our product candidates. Dislocations in the financial markets have generally made equity and debt financing more difficult to obtain, and may have a material adverse effect on our ability to meet our fundraising needs. We cannot be certain that additional funding will be available to us on acceptable terms, if at all. If funds are not available, we may be required to delay, reduce the scope of, or eliminate one or more of our research or development programs or our commercialization efforts. Additional funding, if obtained, may significantly dilute existing shareholders if that financing is obtained through issuing equity or instruments convertible into equity.
22
We rely on third parties to conduct our clinical trials and perform data collection and analysis, which may result in costs and delays that prevent us from successfully commercializing product candidates.
Although we design and manage our current pre-clinical studies, we do not have the in-house capability to conduct clinical trials for our product candidates. We rely, and will rely in the future, on medical institutions, clinical investigators, contract research organizations, contract laboratories, and collaborators to perform data collection and analysis and other aspects of our clinical trials. We rely primarily on Trauma Research LLC, a related party, to conduct pre-clinical studies and provide assessments of clinical observations.
Our pre-clinical activities or clinical trials conducted in reliance on third parties may be delayed, suspended, or terminated if:
| • | the third parties do not successfully carry out their contractual duties or fail to meet regulatory obligations or expected deadlines; |
| • | we replace a third party; or |
| • | the quality or accuracy of the data obtained by third parties is compromised due to their failure to adhere to clinical protocols, regulatory requirements, or for other reasons. |
Third party performance failures may increase our development costs, delay our ability to obtain regulatory approval, and delay or prevent the commercialization of our product candidates. While we believe that there are numerous alternative sources to provide these services, in the event that we seek such alternative sources, we may not be able to enter into replacement arrangements without incurring delays or additional costs.
Even if collaborators with which we contract in the future successfully complete clinical trials of our product candidates, those candidates may not be commercialized successfully for other reasons.
Even if we contract with collaborators that successfully complete clinical trials for one or more of our product candidates, those candidates may not be commercialized for other reasons, including:
| • | failure to receive regulatory clearances required to market them as drugs; |
| • | being subject to proprietary rights held by others; |
| • | being difficult or expensive to manufacture on a commercial scale; |
| • | having adverse side effects that make their use less desirable; or |
| • | failing to compete effectively with products or treatments commercialized by competitors. |
Relying on third-party manufacturers may result in delays in our clinical trials and product introductions.
Our core business strategy is to maintain a strong foundation in basic scientific research and combine that foundation with our clinical development capabilities. To date, we have contracted original equipment manufacturers, or OEMs to produce the drug candidate for our Optina clinical trials. Our long-term objective is to build a successful, integrated biopharmaceutical company that provides patients and medical professionals with new and better options for preventing and treating human diseases. However, developing and commercializing new medicines entails significant risks and expenses. We have little experience in the manufacturing of drugs or in designing drug-manufacturing processes. We currently obtain the HSA needed to produce Ampion for our clinical trials from one manufacturer in the United States. Our clinical trials may be delayed if this manufacturer is unable to assure a sufficient quantity of the drug product to meet our study needs. We are currently validating a manufacturing facility in Denver, Colorado where we plan to manufacture Ampion for registration, batching and commercial supply, as well as future clinical supplies. We obtain the active pharmaceutical ingredient, or API, for Optina from an Indian company, which is one of only four suppliers of the API in the world. Our clinical trials and ultimately FDA approval may be delayed if we are unable to obtain a sufficient quantity of the drug product on a timely basis or if we need to establish an alternative source of supply for the API.
Once regulatory approval is obtained, a marketed product and its manufacturer are subject to continual review. The discovery of previously unknown problems with a product or manufacturer may result in restrictions on the product, manufacturer or manufacturing facility, including withdrawal of the product from the market. Any manufacturers with which we contract HSA for Ampion or danazol for Optina supplies are required to operate in accordance with FDA-mandated current good manufacturing practices, or cGMPs. A failure of any of our contract manufacturers to establish and follow cGMPs and to document their adherence to such practices may lead to significant delays in the launch of products based on our product candidates into the market. Failure by
23
third-party manufacturers to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, revocation or suspension of marketing approval for any products granted pre-market approvals, seizures or recalls of products, operating restrictions, and criminal prosecutions.
Our transactions with related parties may not benefit us and may harm us.
We are party to a sponsored research agreement with Trauma Research LLC, a related party controlled by our director and Chief Scientific Officer, Dr. Bar-Or. We rely primarily on Trauma Research LLC to conduct pre-clinical studies and provide assessments of clinical observations.
We believe that we have conducted our related-party transactions on an arm's-length basis and on terms comparable to, or more favorable to us than, similar transactions we would enter into with independent third parties. However, we cannot assure you that all our future transactions with related parties will be beneficial to us.
We intend to enter into agreements with third parties to sell and market any products we develop and for which we obtain regulatory approvals, which may affect the sales of our products and our ability to generate revenues.
We do not currently maintain an organization for the sale, marketing and distribution of pharmaceutical products and may contract with, or license, third parties to market any products we develop that receive regulatory approvals. Outsourcing sales and marketing in this manner may subject us to a variety of risks, including:
| • | our inability to exercise control over sales and marketing activities and personnel; |
| • | failure or inability of contracted sales personnel to obtain access to or persuade adequate numbers of physicians to prescribe our products; |
| • | disputes with third parties concerning sales and marketing expenses, calculation of royalties, and sales and marketing strategies; and |
| • | unforeseen costs and expenses associated with sales and marketing. |
If we are unable to partner with a third party that has adequate sales, marketing, and distribution capabilities, we may have difficulty commercializing our product candidates, which would adversely affect our business, financial condition, and ability to generate product revenues.
We face substantial competition from companies with considerably more resources and experience than we have, which may result in others discovering, developing, receiving approval for, or commercializing products before or more successfully than us.
Our ability to succeed in the future depends on our ability to discover, develop and commercialize pharmaceutical products that offer superior efficacy, convenience, tolerability, and safety when compared to existing treatment methodologies. We intend to do so by identifying product candidates that address new indications using previously approved drugs, use of new combinations of previously approved drugs, or which are based on a modified active ingredient which previously received regulatory approval. Because our strategy is to develop new product candidates primarily for treatment of diseases that affect large patient populations, those candidates are likely to compete with a number of existing medicines or treatments, and a large number of product candidates that are being developed by others.
Many of our potential competitors have substantially greater financial, technical, personnel and marketing resources than we do. In addition, many of these competitors have significantly greater resources devoted to product development and pre-clinical research. Our ability to compete successfully will depend largely on our ability to:
| • | discover and develop product candidates that are superior to other products in the market; |
| • | attract and retain qualified personnel; |
| • | obtain patent and/or other proprietary protection for our product candidates; |
| • | obtain required regulatory approvals; and |
| • | obtain collaboration arrangements to commercialize our product candidates. |
Established pharmaceutical companies devote significant financial resources to discovering, developing or licensing novel compounds that could make our product candidates obsolete. Our competitors may obtain patent protection, receive FDA approval, and commercialize medicines before us. Other companies are engaged in the discovery of compounds that may compete with the product candidates we are developing.
24
Any new product that competes with a currently-approved treatment or medicine must demonstrate compelling advantages in efficacy, convenience, tolerability and/or safety in order to address price competition and be commercially successful. If we are not able to compete effectively against our current and future competitors, our business will not grow and our financial condition and operations will suffer.
Product liability and other lawsuits could divert our resources, result in substantial liabilities and reduce the commercial potential of our product candidates.
The risk that we may be sued on product liability claims is inherent in the development and commercialization of pharmaceutical products. Side effects of, or manufacturing defects in, products that we develop which are commercialized by any collaborators could result in the deterioration of a patient's condition, injury or even death. Once a product is approved for sale and commercialized, the likelihood of product liability lawsuits increases. Claims may be brought by individuals seeking relief for themselves or by individuals or groups seeking to represent a class. These lawsuits may divert our management from pursuing our business strategy and may be costly to defend. In addition, if we are held liable in any of these lawsuits, we may incur substantial liabilities and may be forced to limit or forgo further commercialization of the affected products.
We may be subject to legal or administrative proceedings and litigation other than product liability lawsuits which may be costly to defend and could materially harm our business, financial conditions and operations.
Although we maintain general liability and product liability insurance, this insurance may not fully cover potential liabilities. In addition, inability to obtain or maintain sufficient insurance coverage at an acceptable cost or to otherwise protect against potential product or other legal or administrative liability claims could prevent or inhibit the commercial production and sale of any of our product candidates that receive regulatory approval, which could adversely affect our business. Product liability claims could also harm our reputation, which may adversely affect our collaborators' ability to commercialize our products successfully.
If any of our product candidates are commercialized, this does not assure acceptance by physicians, patients, third-party payors, or the medical community in general. Even if we, or our collaborators, are able to commercialize our product candidates, the products may become subject to unfavorable pricing regulations, third-party payor reimbursement practices or healthcare reform initiatives that could harm our business.
The commercial success of our product candidates will depend substantially, both domestically and abroad, on the extent to which the costs of our product candidates will be paid by health maintenance, managed care, pharmacy benefit and similar healthcare management organizations, or reimbursed by government health administration authorities, private health coverage insurers and other third-party payors. If reimbursement is not available, or is available only to limited levels, we, or our collaborators, may not be able to successfully commercialize our product candidates. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow us, or our collaborators, to establish or maintain pricing sufficient to realize a sufficient return on our or their investments. We cannot be sure that any of our product candidates, if and when approved for marketing, will be accepted by these parties. Even if the medical community accepts a product as safe and efficacious for its indicated use, physicians may choose to restrict the use of the product if we or any collaborator is unable to demonstrate that, based on experience, clinical data, side-effect profiles and other factors, our product is preferable to any existing medicines or treatments. We cannot predict the degree of market acceptance of any product candidate that receives marketing approval, which will depend on a number of factors, including, but not limited to:
| • | the demonstration of the clinical efficacy and safety of the product; |
| • | the approved labeling for the product and any required warnings; |
| • | the advantages and disadvantages of the product compared to alternative treatments; |
| • | our and any collaborator's ability to educate the medical community about the safety and effectiveness of the product; |
| • | the reimbursement policies of government and third-party payors pertaining to the product; and |
| • | the market price of our product relative to competing treatments. |
Government restrictions on pricing and reimbursement, as well as other healthcare payor cost-containment initiatives, may negatively impact our ability to generate revenues if we obtain regulatory approval to market a product.
The continuing efforts of the government, insurance companies, managed care organizations and other payors of health care costs to contain or reduce costs of health care may adversely affect one or more of the following:
| • | our or our collaborators' ability to set a price we believe is fair for our products, if approved; |
| • | our ability to generate revenues and achieve profitability; and |
| • | the availability of capital. |
25
The 2010 enactments of the Patient Protection and Affordable Care Act, or PPACA, and the Health Care and Education Reconciliation Act are expected to significantly impact the provision of, and payment for, health care in the United States. Various provisions of these laws take effect over the next four years, and are designed to expand Medicaid eligibility, subsidize insurance premiums, provide incentives for businesses to provide health care benefits, prohibit denials of coverage due to pre-existing conditions, establish health insurance exchanges, and provide additional support for medical research. Additional legislative proposals to reform healthcare and government insurance programs, along with the trend toward managed healthcare in the United States, could influence the purchase of medicines and reduce demand and prices for our products, if approved. This could harm our or our collaborators' ability to market any products and generate revenues. Cost containment measures that health care payors and providers are instituting and the effect of further health care reform could significantly reduce potential revenues from the sale of any of our product candidates approved in the future, and could cause an increase in our compliance, manufacturing, or other operating expenses. In addition, in certain foreign markets, the pricing of prescription drugs is subject to government control and reimbursement may in some cases be unavailable. We believe that pricing pressures at the federal and state level, as well as internationally, will continue and may increase, which may make it difficult for us to sell our potential products that may be approved in the future at a price acceptable to us or any of our future collaborators.
If we or Trauma Research LLC use hazardous and biological materials in a manner that causes injury or violates applicable law, we may be liable for damages or fines.
The research and development activities conducted at our facility and on our behalf by Trauma Research LLC, a related party controlled by Dr. Bar-Or, involve the controlled use of potentially hazardous substances, including chemical, biological and radioactive materials and produce hazardous waste products. In addition, we are currently validating a manufacturing facility in Denver, Colorado where we plan to manufacture Ampion for registration, batching and commercial supply, as well as future clinical supplies. This manufacturing facility will involve the controlled use of potentially hazardous substances and produce hazardous waste products. Federal, state and local laws and regulations govern the use, manufacture, storage, handling and disposal of hazardous materials. If either we or Trauma Research LLC experience a release of hazardous substances, it is possible that this release could cause personal injury or death, and require decontamination of facilities. Trauma Research LLC has advised us that it believes it is in compliance with laws applicable to the handling of hazardous substances, but such compliance does not assure that a release of hazardous substances will not occur, or assure that such compliance will be maintained in the future. In the event of an accident involving the manufacture of Ampion by us or research being conducted on our behalf by Trauma Research LLC, we could be held liable for damages or face substantial penalties. We do not have any insurance for liabilities arising from the procurement, handling, or discharge of hazardous materials. Compliance with applicable environmental laws and regulations is expensive, and current or future environmental regulations may impair our research, development and production efforts, which could harm our business.
Business interruptions could limit our ability to operate our business.
Our operations are vulnerable to damage or interruption from computer viruses, human error, natural disasters, telecommunications failures, intentional acts of misappropriation, and similar events. We have not established a formal disaster recovery plan, and our back-up operations and our business interruption insurance may not be adequate to compensate us for losses that occur. A significant business interruption could result in losses or damages incurred by us and require us to curtail our operations.
Risks Related to Our Intellectual Property
Our ability to compete may decline if we do not adequately protect our proprietary rights.
Our commercial success depends on obtaining and maintaining proprietary rights to our product candidates and compounds and their uses, as well as successfully defending these rights against third-party challenges. We will only be able to protect our product candidates, proprietary compounds, and their uses from unauthorized use by third parties to the extent that valid and enforceable patents, or effectively protected trade secrets, cover them.
Our ability to obtain patent protection for our product candidates and compounds is uncertain due to a number of factors, including:
| • | we may not have been the first to make the inventions covered by pending patent applications or issued patents; |
| • | we may not have been the first to file patent applications for our product candidates or the compounds we developed or for their uses; |
| • | others may independently develop identical, similar or alternative products or compounds; |
| • | our disclosures in patent applications may not be sufficient to meet the statutory requirements for patentability; |
| • | any or all of our pending patent applications may not result in issued patents; |
26
| • | we may not seek or obtain patent protection in countries that may eventually provide us a significant business opportunity; |
| • | any patents issued to us may not provide a basis for commercially viable products, may not provide any competitive advantages, or may be successfully challenged by third parties; |
| • | our proprietary compounds may not be patentable; |
| • | others may design around our patent claims to produce competitive products which fall outside of the scope of our patents; or |
| • | others may identify prior art which could invalidate our patents. |
Even if we have or obtain patents covering our product candidates or compounds, we may still be barred from making, using and selling our product candidates or technologies because of the patent rights of others. Others have or may have filed, and in the future may file, patent applications covering compounds or products that are similar or identical to ours. There are many issued U.S. and foreign patents relating to chemical compounds and therapeutic products, and some of these relate to compounds we intend to commercialize. Numerous U.S. and foreign issued patents and pending patent applications owned by others exist in the area of metabolic disorders, cancer, inflammatory responses, and the other fields in which we are developing products. These could materially affect our ability to develop our product candidates or sell our products if approved. Because patent applications can take many years to issue, there may be currently pending applications unknown to us that may later result in issued patents that our product candidates or compounds may infringe. These patent applications may have priority over patent applications filed by us.
We periodically conduct searches to identify patents or patent applications that may prevent us from obtaining patent protection for our compounds or that could limit the rights we have claimed in our patents and patent applications. Disputes may arise regarding the source or ownership of our inventions. It is difficult to determine if and how such disputes would be resolved. Others may challenge the validity of our patents. If our patents are found to be invalid, we will lose the ability to exclude others from making, using or selling the compounds or products addressed in those patents. In addition, compounds or products we may license may become important to some aspects of our business. We generally will not control the prosecution, maintenance or enforcement of patents covering licensed compounds or products.
Confidentiality agreements with employees and others may not adequately prevent disclosure of our trade secrets and other proprietary information and may not adequately protect our intellectual property, which could limit our ability to compete.
Because we operate in the highly technical field of drug discovery and development of therapies that can address metabolic disorders, cancer, inflammation and other conditions, we rely in part on trade secret protection in order to protect our proprietary technology and processes. However, trade secrets are difficult to protect. We enter into confidentiality and intellectual property assignment agreements with our employees, consultants, outside scientific collaborators, sponsored researchers, and other advisors. These agreements generally require that the other party keep confidential and not disclose to third parties all confidential information developed by the party or made known to the party by us during the course of the party's relationship with us. These agreements also generally provide that inventions conceived by the party in the course of rendering services to us will be our exclusive property.
However, these agreements may not be honored and may not effectively assign intellectual property rights to us. Enforcing a claim that a party illegally obtained and is using our trade secrets is difficult, expensive and time consuming and the outcome is unpredictable. In addition, courts outside the United States may be less willing to protect trade secrets. The failure to obtain or maintain trade secret protection could adversely affect our competitive position. We have entered into non-compete agreements with certain of our employees, but the enforceability of those agreements is not assured.
A dispute concerning the infringement or misappropriation of our proprietary rights or the proprietary rights of others could be time consuming and costly, and an unfavorable outcome could harm our business.
There is significant litigation in the pharmaceutical industry regarding patent and other intellectual property rights. While we are not currently subject to any pending intellectual property litigation, and are not aware of any such threatened litigation, we may be exposed to future litigation by third parties based on claims that our product candidates, technologies or activities infringe the intellectual property rights of others. In particular, there are many patents relating to repositioned drugs and chemical compounds used to treat metabolic disorders, cancer and inflammation. Some of these may encompass repositioned drugs or compounds that we utilize in our product candidates. If our development activities are found to infringe any such patents, we may have to pay significant damages or seek licenses to such patents. A patentee could prevent us from using the patented drugs or compounds. We may need to resort to litigation to enforce a patent issued to us, to protect our trade secrets, or to determine the scope and validity of third-party proprietary rights. From time to time, we may hire scientific personnel or consultants formerly employed by other companies involved in one or more areas similar to the activities conducted by us. Either we or these individuals may be subject to allegations of trade secret misappropriation or other similar claims as a result of prior affiliations. If we become involved in litigation, it could consume a
27
substantial portion of our managerial and financial resources, regardless of whether we win or lose. We may not be able to afford the costs of litigation. Any legal action against us or our collaborators could lead to:
| • | payment of damages, potentially treble damages, if we are found to have willfully infringed a party's patent rights; |
| • | injunctive or other equitable relief that may effectively block our ability to further develop, commercialize, and sell products; or |
| • | us or our collaborators having to enter into license arrangements that may not be available on commercially acceptable terms, if at all. |
As a result, we could be prevented from commercializing current or future product candidates.
Pharmaceutical patents and patent applications involve highly complex legal and factual questions, which, if determined adversely to us, could negatively impact our patent position.
The patent positions of pharmaceutical companies can be highly uncertain and involve complex legal and factual questions. For example, some of our patents and patent applications cover methods of use of repositioned drugs, while other patents and patent applications cover composition of a particular compound. The interpretation and breadth of claims allowed in some patents covering pharmaceutical compounds may be uncertain and difficult to determine, and are often affected materially by the facts and circumstances that pertain to the patented compound and the related patent claims. The standards of the United States Patent and Trademark Office, or USPTO, are sometimes uncertain and could change in the future. Consequently, the issuance and scope of patents cannot be predicted with certainty. Patents, if issued, may be challenged, invalidated or circumvented. U.S. patents and patent applications may also be subject to interference proceedings, and U.S. patents may be subject to reexamination proceedings in the USPTO. Foreign patents may be subject also to opposition or comparable proceedings in the corresponding foreign patent office, which could result in either loss of the patent or denial of the patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. In addition, such interference, reexamination and opposition proceedings may be costly. Accordingly, rights under any issued patents may not provide us with sufficient protection against competitive products or processes.
In addition, changes in or different interpretations of patent laws in the United States and foreign countries may permit others to use our discoveries or to develop and commercialize our technology and products without providing any compensation to us, or may limit the number of patents or claims we can obtain. The laws of some countries do not protect intellectual property rights to the same extent as U.S. laws and those countries may lack adequate rules and procedures for defending our intellectual property rights. For example, some countries do not grant patent claims directed to methods of treating humans and, in these countries, patent protection may not be available at all to protect our product candidates. In addition, U.S. patent laws may change, which could prevent or limit us from filing patent applications or patent claims to protect our products and/or compounds.
If we fail to obtain and maintain patent protection and trade secret protection of our product candidates, proprietary compounds and their uses, we could lose our competitive advantage and competition we face would increase, reducing any potential revenues and adversely affecting our ability to attain or maintain profitability.
Risks Related to Our Common Stock
The price of our stock has been extremely volatile and may continue to be volatile and fluctuate substantially, which could result in substantial losses for purchasers of our common stock.
The price of our common stock has been extremely volatile and may continue to be so. The stock market in general and the market for pharmaceutical companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies, to a greater extent during the last few years. The following factors, in addition to the other risk factors described in this section, may also have a significant impact on the market price of our common stock:
| • | any actual or perceived adverse developments in clinical trials for Ampion or Optina, such as the delay experienced with the STEP Study of Ampion in August 2014 and the failure of the STRIDE study to reach its primary endpoint; |
| • | any actual or perceived difficulties or delays in obtaining regulatory approval of any of our product candidates in the United States or other countries once clinical trials are completed; |
| • | any finding that our product candidates are not safe or effective, or any inability to demonstrate clinical effectiveness of our product candidates when compared to existing treatments; |
| • | any actual or perceived adverse developments in repurposed drug technologies, including any change in FDA policy or guidance on approval of repurposed drug technologies for new indications; |
28
| • | any announcements of developments with, or comments by, the FDA, the EMA, or other regulatory authorities with respect to product candidates we have under development; |
| • | any announcements concerning our retention or loss of key employees, especially Dr. Bar-Or; |
| • | our success or inability to obtain collaborators to conduct clinical trials, commercialize a product candidate for which regulatory approval is obtained, or market and sell an approved product candidate; |
| • | any actual or perceived adverse developments with respect to our relationship with Trauma Research LLC; |
| • | announcements of patent issuances or denials, product innovations, or introduction of new commercial products by our competitors that will compete with any of our product candidates; |
| • | publicity regarding actual or potential study results or the outcome of regulatory reviews relating to products under development by us, our collaborators, or our competitors; |
| • | economic and other external factors beyond our control; and |
| • | sales of stock by us or by our shareholders. |
In addition, we believe there has been and may continue to be substantial off-market transactions in derivatives of our stock, including short selling activity or related similar activities, which are beyond our control and which may be beyond the full control of the SEC and Financial Institutions Regulatory Authority, or FINRA. While SEC and FINRA rules prohibit some forms of short selling and other activities that may result in stock price manipulation, such activity may nonetheless occur without detection or enforcement. We have held conversations with regulators concerning trading activity in our stock; however, there can be no assurance that should there be any illegal manipulation in the trading of our stock it will be detected, prosecuted or successfully eradicated. Significant short selling or other types of market manipulation could cause our stock trading price to decline, to become more volatile, or both.
The price of our stock may be vulnerable to manipulation.
Our common stock has been the subject of significant short selling efforts by certain market participants. Short sales are transactions in which a market participant sells a security that it does not own. To complete the transaction, the market participant must borrow the security to make delivery to the buyer. The market participant is then obligated to replace the security borrowed by purchasing the security at the market price at the time of required replacement. If the price at the time of replacement is lower than the price at which the security was originally sold by the market participant, then the market participant will realize a gain on the transaction. Thus, it is in the market participant's interest for the market price of the underlying security to decline as much as possible during the period prior to the time of replacement.
Because our unrestricted public float (not subject to lockup restrictions) has been small relative to other issuers, previous short selling efforts have impacted, and may in the future continue to impact, the value of our stock in an extreme and volatile manner to our detriment and the detriment of our shareholders. In addition, market participants with admitted short positions in our stock have published, and may in the future continue to publish, negative information regarding us and our management team on internet sites or blogs that we believe is inaccurate and misleading. We believe that the publication of this negative information has led, and may in the future continue to lead, to significant downward pressure on the price of our stock to our detriment and the further detriment of our shareholders. These and other efforts by certain market participants to manipulate the price of our common stock for their personal financial gain may cause our stockholders to lose a portion of their investment, may make it more difficult for us to raise equity capital when needed without significantly diluting existing stockholders, and may reduce demand from new investors to purchase shares of our stock.
If we cannot continue to satisfy the NYSE MKT listing maintenance requirements and other rules, including the director independence requirements, our securities may be delisted, which could negatively impact the price of our securities.
Although our common stock is listed on the NYSE MKT, we may be unable to continue to satisfy the listing maintenance requirements and rules. If we are unable to satisfy the NYSE MKT criteria for maintaining our listing, our securities could be subject to delisting. To qualify for continued listing on the NYSE MKT, we must remain in compliance.
Under the NYSE MKT rules, shares that are held by "public shareholders" do not include shares held by officers, directors, controlling shareholders and concentrated (10% or greater), affiliated or family holdings.
If the NYSE MKT delists our securities, we could face significant consequences, including:
| • | a limited availability for market quotations for our securities; |
| • | reduced liquidity with respect to our securities; |
29
| • | a determination that our common stock is a "penny stock," which will require brokers trading in our common stock to adhere to more stringent rules and possibly result in reduced trading; |
| • | activity in the secondary trading market for our common stock; |
| • | limited amount of news and analyst coverage; and |
| • | a decreased ability to issue additional securities or obtain additional financing in the future. |
In addition, we would no longer be subject to the NYSE MKT rules, including rules requiring us to have a certain number of independent directors and to meet other corporate governance standards.
Concentration of our ownership limits the ability of our shareholders to influence corporate matters.
As of December 31, 2015, holders of more than 5% of our common stock and our directors, executive officers and their affiliates beneficially owned approximately 27.0% of our outstanding common stock. These shareholders may control effectively the outcome of actions taken by us that require shareholder approval.
Anti-takeover provisions in our charter and bylaws and in Delaware law could prevent or delay a change in control of Ampio.
Provisions of our certificate of incorporation and bylaws may discourage, delay or prevent a merger or acquisition that shareholders may consider favorable, including transactions in which shareholders might otherwise receive a premium for their shares. These provisions include:
| • | requiring supermajority shareholder voting to effect certain amendments to our certificate of incorporation and bylaws; |
| • | restricting the ability of shareholders to call special meetings of shareholders; |
| • | prohibiting shareholder action by written consent except in certain circumstances; and |
| • | establishing advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted on by shareholders at shareholder meetings. |
Increased costs associated with corporate governance compliance may significantly impact our results of operations.
As a public company, we incur significant legal, accounting, and other expenses due to our compliance with regulations and disclosure obligations applicable to us, including compliance with the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, as well as rules implemented by the SEC, and the NYSE MKT. The SEC and other regulators have continued to adopt new rules and regulations and make additional changes to existing regulations that require our compliance. In July 2010, the Dodd-Frank Wall Street Reform and Consumer Protection Act, or the Dodd-Frank Act, was enacted. There are significant corporate governance and executive compensation related provisions in the Dodd-Frank Act that have required the SEC to adopt additional rules and regulations in these areas. Stockholder activism, the current political environment, and the current high level of government intervention and regulatory reform may lead to substantial new regulations and disclosure obligations, which may lead to additional compliance costs and impact, in ways we cannot currently anticipate, the manner in which we operate our business. Our management and other personnel devote a substantial amount of time to these compliance programs and monitoring of public company reporting obligations, and as a result of the new corporate governance and executive compensation related rules, regulations, and guidelines prompted by the Dodd-Frank Act, and further regulations and disclosure obligations expected in the future, we will likely need to devote additional time and costs to comply with such compliance programs and rules. These rules and regulations will cause us to incur significant legal and financial compliance costs and will make some activities more time-consuming and costly.
The Sarbanes-Oxley Act requires that we maintain effective disclosure controls and procedures and internal control over financial reporting. We are continuing to develop and refine our disclosure controls and other procedures that are designed to ensure that information required to be disclosed by us in the reports that we file with the SEC is recorded, processed, summarized, and reported within the time periods specified in SEC rules and forms, and that information required to be disclosed in reports under the Exchange Act is accumulated and communicated to our principal executive and financial officers. Our current controls and any new controls that we develop may become inadequate, and weaknesses in our internal control over financial reporting may be discovered in the future. Any failure to develop or maintain effective controls could adversely affect the results of periodic management evaluations and annual independent registered public accounting firm attestation reports regarding the effectiveness of our internal control over financial reporting, which we may be required to include in our periodic reports we file with the SEC under Section 404 of the Sarbanes-Oxley Act, and could harm our operating results, cause us to fail to meet our reporting obligations, or result in a restatement of our prior period financial statements. In the event that we are not able to demonstrate compliance with the Sarbanes-Oxley Act, that our internal control over financial reporting is perceived as inadequate, or that we are unable to produce timely or accurate financial statements, investors may lose confidence in our operating results, and the price of our common stock could decline.
30
We are required to comply with certain of the SEC rules that implement Section 404 of the Sarbanes-Oxley Act, which requires management to certify financial and other information in our quarterly and annual reports and provide an annual management report on the effectiveness of our internal control over financial reporting. This assessment needs to include the disclosure of any material weaknesses in our internal control over financial reporting identified by our management or our independent registered public accounting firm. During the evaluation and testing process, if we identify one or more material weaknesses in our internal control over financial reporting or if we are unable to complete our evaluation, testing, and any required remediation in a timely fashion, we will be unable to assert that our internal control over financial reporting is effective.
These developments could make it more difficult for us to retain qualified members of our Board of Directors, or qualified executive officers. We are presently evaluating and monitoring regulatory developments and cannot estimate the timing or magnitude of additional costs we may incur as a result. To the extent these costs are significant, our general and administrative expenses are likely to increase.
If securities analysts do not publish research or reports about our business or if they downgrade our stock after instituting coverage, the price of our common stock could decline.
The research and reports that industry or financial analysts publish about us or our business may vary widely and may not predict accurate results, but will likely have an effect on the trading price of our common stock. If an industry analyst decides not to cover us, or if an industry analyst institutes coverage and later decides to cease covering us, we could lose visibility in the market, which in turn could cause our stock price to decline. If an industry analyst who covers our stock decides to downgrade that stock, our stock price would likely decline rapidly in response.
We have no plans to pay cash dividends on our common stock.
We have no plans to pay cash dividends on our common stock. We generally intend to invest future earnings, if any, to fund our growth. Any payment of future dividends will be at the discretion of our Board of Directors and will depend on, among other things, our earnings, financial condition, capital requirements, level of indebtedness, statutory and contractual restrictions applying to the payment of dividends and other considerations our Board of Directors deem relevant. Any future credit facilities or preferred stock financing we obtain may further limit our ability to pay cash dividends on our common stock.
| Item 1B. | Unresolved Staff Comments |
None.
| Item 2. | Properties |
We maintain our headquarters in leased space in Englewood, Colorado, for monthly rental payments of approximately $27,000. The lease expires in September 2024. We anticipate that the lease can be renewed on terms similar to those now in effect.
| Item 3. | Legal Proceedings |
As previously disclosed, on May 8, 2015 and May 14, 2015, purported stockholders of the Company brought two putative class action lawsuits in the United States District Court in the Central District of California, Napoli v. Ampio Pharmaceuticals, Inc., et al., Case No. 2:15-cv-03474-TJH and Stein v. Ampio Pharmaceuticals, Inc., et al., Case No. 2:15-cv-03640-TJH (the "Securities Class Actions"), alleging that Ampio and certain of its current and former officers violated federal securities laws by misrepresenting and/or omitting information regarding the STEP study. The cases were consolidated, and on February 8, 2016, plaintiffs filed a consolidated amended complaint alleging claims under Sections 10(b) and 20(a) and Rule 10b-5 under the Exchange Act and Sections 11 and 15 under the Securities Act of 1933 on behalf of a putative class of purchasers of common stock from January 13, 2014 through August 21, 2014, including purchasers in the Company's offering on February 28, 2014. The lawsuits seek unspecified damages, pre-judgment and post-judgment interest, and attorneys' fees and costs.
On August 6, 2015 and September 25, 2015, purported stockholders of the Company brought derivative actions in the United States District Court in the Central District of California, Oglina v. Macaluso et al ., Case No. 2:15-cv-05970-TJH-PJW (" Oglina action") and the Colorado state court in Denver, Loyd v. Giles et al ., Case No. 2015CV33429 (" Loyd action"), alleging primarily that the directors and officers of Ampio breached their fiduciary duties because of their alleged misstatements and/or omissions regarding the STEP study. Pursuant to the parties' stipulation, the United States District Court in the Central District of California has stayed the proceedings in the Oglina action at the present time in accordance with the terms of the parties' stipulation. Pursuant to the parties' stipulation, the Colorado state court in Denver has stayed the Loyd action at the present time in accordance with the terms of the parties' stipulation.
31
The Company believes these claims are without merit and intends to defend these lawsuits vigorously. We currently believe the likelihood of a loss contingency related to these matters is remote and, therefore, no provision for a loss contingency is required.
| Item 4. | Mine Safety Disclosures. |
Not applicable.
32
PART II
| Item 5. | Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Market Data
On June 17, 2013, our common stock began trading on the NYSE MKT under the ticker symbol "AMPE". It was previously quoted on the NASDAQ Capital Market under the same ticker symbol "AMPE". The following table sets forth the high and low last reported sale price information for our common stock for each quarter for the past two fiscal years.
Fiscal Year ended December 31, 2015 | High | Low | ||||||
First Quarter | $ | 7.60 | $ | 3.68 | ||||
Second Quarter | $ | 7.38 | $ | 1.79 | ||||
Third Quarter | $ | 3.12 | $ | 2.00 | ||||
Fourth Quarter | $ | 3.28 | $ | 2.51 | ||||
Fiscal Year ended December 31, 2014 | High | Low | ||||||
First Quarter | $ | 9.73 | $ | 5.70 | ||||
Second Quarter | $ | 8.35 | $ | 5.44 | ||||
Third Quarter | $ | 8.59 | $ | 3.33 | ||||
Fourth Quarter | $ | 4.16 | $ | 3.25 | ||||
As of February 1, 2016, there were approximately 8,400 holders of record of our common stock.
We have never paid cash dividends and intend to employ all available funds in the development of our business. We have no plans to pay cash dividends in the near future. If we issue in the future any preferred stock or obtain financing from a bank, the terms of those financings may contain restrictions on our ability to pay dividends for so long as the preferred stock or bank financing is outstanding.
33
Performance Graph
We have presented below the cumulative return to our stockholders during the period from December 31, 2010 through December 31, 2015 in comparison to the cumulative return NASDAQ Biotechnology Index and the Russell 2000 Index. The comparisons are based on historical data and are not indicative of, nor intended to forecast, the future performance of our common stock.
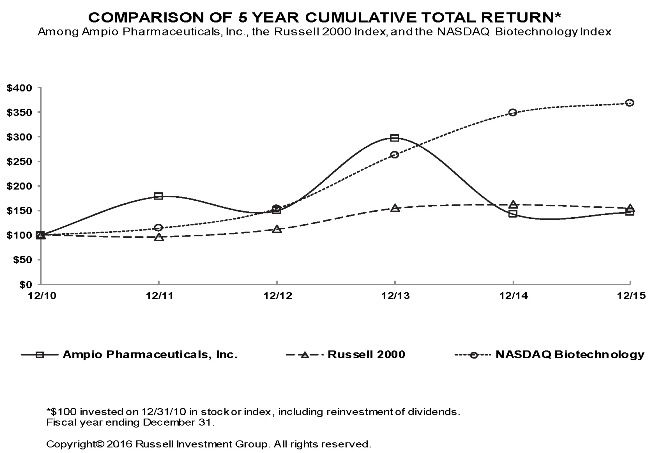
| 12/10 | 12/11 | 12/12 | 12/13 | 12/14 | 12/15 | |||||||||||||||||||
Ampio Pharmaceuticals, Inc. | 100.00 | 177.92 | 149.58 | 297.08 | 142.92 | 145.83 | ||||||||||||||||||
Russell 2000 | 100.00 | 95.82 | 111.49 | 154.78 | 162.35 | 155.18 | ||||||||||||||||||
NASDAQ Biotechnology | 100.00 | 113.92 | 153.97 | 263.29 | 348.49 | 369.06 | ||||||||||||||||||
The information under "Performance Graph" is not deemed to be "soliciting material" or "filed" with the Securities and Exchange Commission or subject to Regulation 14A or 14C, or to the liabilities of Section 18 of the Exchange Act and is not to be incorporated by reference in any filing of Ampio Pharmaceuticals, Inc. under the Securities Act of 1933, as amended, or the Exchange Act, whether made before or after the date of this Annual Report on Form 10-K and irrespective of any general incorporation language in those filings.
Unregistered Sales of Equity Securities and Use of Proceeds
Information regarding unregistered sales of equity securities and use of proceeds is incorporated by reference to Item 15 of Part IV, Notes to Consolidated Financial Statements – Note 9 – Common Stock of this annual report on Form 10-K.
34
Equity Compensation Plan Information
In March 2010, our shareholders approved the adoption of a stock and option award plan, or the "2010 Plan", under which 2,500,000 shares were reserved for future issuance under restricted stock awards, options, and other equity awards. The 2010 Plan permits grants of equity awards to employees, directors and consultants. In August 2010, the number of shares issuable under the 2010 Plan was increased to 4,500,000 shares by consent of our majority shareholders. At the annual shareholders' meeting, held in December 2011, the number of shares issuable under the 2010 Plan was increased to 5,700,000. At the annual shareholders' meeting held in December 2012, the number of shares issuable under the 2010 Plan was further increased to 8,200,000 and in December 2013, total shares issuable was increased to 11,700,000. The following table displays equity compensation plan information as of December 31, 2015.
Plan Category | Number of Securities to be Issued upon Exercise of Outstanding Options (a) | Weighted-Average Exercise Price of Outstanding Options (b) | Number of Securities Remaining Available for Issuance under Equity Compensation Plans (Excluding Securities Reflected in Column (a)) (c) | |||||||||
Equity compensation plans approved by security holders | 7,315,832 | $ | 3.71 | 2,989,773 | ||||||||
Equity compensation plans not approved by security holders | - | - | - | |||||||||
|
|
|
|
|
| |||||||
Total | 7,315,832 | $ | 3.71 | 2,989,773 | ||||||||
|
|
|
|
|
| |||||||
| Item 6. | Selected Financial Data |
Our selected consolidated financial data shown below should be read together with Item 7- "Management's Discussion and Analysis of Financial Condition and Results of Operations" and our consolidated financial statements and respective notes included in Item 8 "Financial Statements and Supplementary Data" referencing Item 15 of Part IV. The data shown below is not necessarily indicative of results to be expected for any future period. See Note 16 – Subsequent Events to financial statements for unaudited pro forma financial information giving effect to our dividend of a majority of our Aytu shares to our shareholders.
35
| Years Ended December 31, | ||||||||||||||||||||
| 2015 | 2014 | 2013 | 2012 | 2011 | ||||||||||||||||
Selected Statements of Operations Data: | ||||||||||||||||||||
Product and service revenue | $ | 1,076,749 | $ | - | $ | - | $ | - | $ | - | ||||||||||
License revenue | 85,713 | 76,787 | 50,000 | 50,000 | 18,750 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Total revenue | 1,162,462 | 76,787 | 50,000 | 50,000 | 18,750 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Cost of sales | 369,309 | - | - | - | - | |||||||||||||||
Research and development | 18,960,545 | 26,922,988 | 16,596,477 | 6,044,337 | 5,686,784 | |||||||||||||||
General and administrative | 15,134,727 | 12,224,834 | 7,477,396 | 5,826,419 | 5,466,107 | |||||||||||||||
Other expense (income) | 411,798 | (22,263 | ) | 504,553 | (227,711 | ) | 7,142,593 | |||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Net loss, before income tax | (33,713,917 | ) | (39,048,772 | ) | (24,528,426 | ) | (11,593,045 | ) | (18,276,734 | ) | ||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Foreign tax expense | - | - | - | - | 82,500 | |||||||||||||||
Net loss applicable to non-controlling interests | 1,703,675 | 923,357 | 519,868 | - | - | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Net loss applicable to Ampio | $ | (32,010,242 | ) | $ | (38,125,415 | ) | $ | (24,008,558 | ) | $ | (11,593,045 | ) | $ | (18,359,234 | ) | |||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Per share data: | ||||||||||||||||||||
Weighted average number of Ampio common shares outstanding | 51,992,048 | 50,226,555 | 38,294,259 | 33,983,590 | 26,013,838 | |||||||||||||||
Basic and diluted Ampio net loss per common share | $ | (0.62 | ) | $ | (0.76 | ) | $ | (0.63 | ) | $ | (0.34 | ) | $ | (0.71 | ) | |||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Selected Balance Sheets Data: | ||||||||||||||||||||
Cash and cash equivalents | $ | 26,957,938 | $ | 50,320,656 | $ | 26,309,449 | $ | 17,682,517 | $ | 11,362,325 | ||||||||||
Total current assets | $ | 29,228,422 | $ | 51,259,157 | $ | 26,441,435 | $ | 17,847,407 | $ | 11,405,445 | ||||||||||
Total assets | $ | 50,418,593 | $ | 70,268,410 | $ | 36,018,752 | $ | 25,847,165 | $ | 19,482,599 | ||||||||||
Total current liabilities | $ | 5,515,113 | $ | 3,679,983 | $ | 2,472,632 | $ | 1,635,893 | $ | 1,291,533 | ||||||||||
Total long term liabilities | $ | 6,976,492 | $ | 1,129,909 | $ | 331,250 | $ | 381,250 | $ | 431,250 | ||||||||||
Working capital | $ | 23,713,309 | $ | 47,579,174 | $ | 23,968,803 | $ | 16,211,514 | $ | 10,113,912 | ||||||||||
Total stockholders' equity | $ | 37,926,988 | $ | 65,458,518 | $ | 33,214,870 | $ | 23,830,022 | $ | 17,759,816 | ||||||||||
| Item 7. | Management's Discussion and Analysis of Financial Condition and Results of Operations |
You should read the following discussion and analysis of our financial condition and results of operations together with our financial statements and the related notes appearing elsewhere in this report. Some of the information contained in this discussion and analysis, including information with respect to our plans and strategy for our business and related financing, includes forward-looking statements that involve risks and uncertainties. You should read the "Risk Factors" section of this Form 10-K for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
36
Overview
We are a biopharmaceutical company focused primarily on developing compounds that decrease inflammation by (i) inhibiting specific pro-inflammatory compounds by affecting specific pathways at the protein expression and at the transcription level; (ii) activating specific phosphatase or depleting available phosphate needed for the inflammation process; and (iii) decreasing vascular permeability. Through Aytu, until our distribution of Aytu shares in January 2016, we were also focused on acquiring, developing and commercializing products focused primarily on the urological disorders market, specifically sexual dysfunction, urological cancer, urinary tract infections and male infertility.
AMPION
Ampion for Osteoarthritis and Other Inflammatory Conditions
Ampion is a sub 5000 molecular weight fraction of commercial human serum albumin, or HSA. The primary constituent ingredient is aspartyl-alanyl diketopiperazine, or DA-DKP an endogenous immunomodulatory molecule derived from the N-terminus of HSA. Based on Ampio's published in-vitro findings, DA-DKP appears to play a significant role in the homeostasis of inflammation. DA-DKP is believed to reduce inflammation by suppressing pro-inflammatory cytokine production in T-cells. Ampion also contains other known small molecules that confer anti-inflammatory effects to complement the activity of DA-DKP and derive in-vitro and in-vivo effects. We believe the non-steroidal, low molecular weight, anti-inflammatory biologic has the potential to be used in a wide variety of acute and chronic inflammatory conditions as well as immune-mediated diseases. We are currently developing Ampion as an intra-articular injection to treat pain due to osteoarthritis of the knee.
Ampion is manufactured as the low molecular weight filtration product of commercial human serum albumin containing DA-DKP, N-acetyltryptophan, caprylate, and other small molecules either contained in HSA or added to HSA during the processing and production of commercial HSA products. DA-DKP, the primary constituent ingredient contained in Ampion, is a locally generated molecule formed as a physiological result of the cleavage and cyclization of the N-terminal aspartic acid and alanine residues of human albumin. The molecule was originally discovered in the blood and cerebrospinal fluid of patients several days after suffering severe closed head injuries. A high concentration of DA-DKP has also been detected in biofilms found on endotracheal tubes recovered from intubated patients and on implanted orthopedic plates and screws. Together these findings suggest a mechanism by which DA-DKP contributes to the ability to reduce the body's inflammatory response following insult or injury.
DA-DKP is believed to reduce inflammation through the activation of Ras-related protein 1, or Rap1. Rap1 interrupts the kinase cascade by regulating the amount of rapidly accelerated fibrosarcoma, or Raf, kinases available for interaction with Ras, inhibiting antigen-specific Ras activation. This decrease disrupts the mitogen-activation protein kinase, or MAPK, cascade and results in decreased immunoinflammatory cytokine gene transcription. The clinical results which are detailed below also suggest an effect other than anti-inflammatory properties are at work and imply more prolonged healing-like effects.
We have published several scientific papers on Ampion. Most recently in June 2015 we announced three peer-reviewed publications, "The Low Molecular Weight Fraction of Commercial Human Serum Albumin (LMWF5A-Ampion) Induces Morphologic and Transcriptional Changes of Bone Marrow-Derived Mesenchymal Stem Cells" and "Anti-Inflammatory Activity in the Low Molecular Weight Fraction of Commercial Human Serum Albumin (LMWF5A)" and "Inflammatory pathways in knee osteoarthritis: potential targets for treatment".
AIK Study Trial Results
In 2011 and 2012 we conducted our Phase I Ampion™ trial in Australia. The AIK study established that Ampion™ was safe for human use and showed efficacy treating patients with pain due to OA of the knee. The trial was conducted in Australia because the biologics legislation governing the Australian Therapeutic Goods Administration, or TGA, allowed us to move Ampion™ directly into human clinical trials as the TGA recognized that HSA has an already established safety profile in humans by virtue of its longstanding commercial use. The AIK trial was conducted in patients diagnosed with moderately-severe to severe osteoarthritis of the knee.
SPRING Pivotal Trial Results
In the second half of 2013 we announced results of our first pivotal trial, the SPRING study, of Ampion™ for the treatment of pain due to osteoarthritis of the knee. The results of this study establish the safety and efficacy of Ampion for reduction of pain due to OA at 12 weeks after a single intra-articular, or IA, injection in the knee. The SPRING study was a U.S. multicenter, randomized, double-blind, vehicle controlled trial. Three hundred twenty-nine patients were randomized to receive one of two doses (4 mL or 10 mL) of Ampion™ or corresponding saline control via intra-articular injection. Both doses of Ampion™, 4 mL and 10 mL, showed a statistically significant reduction in pain compared to control, and there were no significant differences between the efficacy of the two Ampion™ doses. As such, the lowest required dose, 4 mL, was selected as the optimal dose. Patients who received Ampion™ experienced, on average, greater than a 40% reduction in pain from baseline at 12 weeks. Patients who received Ampion also showed a significant improvement in function and quality of life (quality of life was assessed using the Patient Global Assessment, PGA) compared to patients who received saline control at 12 weeks. Furthermore, the trial included severely diseased patients (defined as
37
Kellgren-Lawrence IV) and those patients who received Ampion had a significantly greater reduction in pain than those patients who received saline control. Ampion™ was well tolerated with minimal adverse events, or AEs, reported equally across Ampion™ and saline groups in the study. There were no drug-related serious adverse events, or SAEs.
STEP Trial
In early 2014 we announced the STEP study clinical trial of Ampion for the treatment of pain due to osteoarthritis of the knee. The STEP study was a randomized, vehicle controlled, double-blind study in which 538 patients with osteoarthritis knee pain were randomized to receive either a 4 mL single injection of Ampion or saline control. A deviation of temperature protocols occurred during the drug distribution process of the STEP Study, which interfered with efficacy analysis. There were minimal adverse events reported and there were no drug-related SAEs in the STEP study.
STRUT Trial
In mid-2014 we announced the beginning of a Phase I multiple injection study, the STRUT study, at a single site for patients with pain due to mostly severe or very severe osteoarthritis of the knee. Patients showed a 65% improvement in pain and a 74% improvement in function from baseline at one-month post-injection. No drug-related SAEs were reported. Following these results, we initiated the randomized, double-blind, vehicle controlled (Phase II) portion of the multiple injection STRUT study.
In 2015, we announced the results from the Phase II STRUT study, which showed that patients who received Ampion demonstrated a significant improvement in pain when compared to patients who received saline control. Patients who received Ampion demonstrated, on average, a 64% reduction in pain at 20 weeks compared to baseline. The safety profile of Ampion in this trial was highly favorable, with no treatment-related SAEs. The results of this study establish the safety and efficacy of Ampion for reduction of pain at 20 weeks after a series of three IA injections administered two weeks apart in the knee of patients with OA.
STRIDE Trial
In late 2014 we enrolled 329 patients in the vehicle controlled, multiple injection, multi-center STRIDE study. Enrollment in this study differed from previous trials in both disease severity and patient Body Mass Index, or BMI. In the STRIDE study 68% of patients had severe osteoarthritis (Kellgren-Lawrence IV), compared to 23% in the SPRING study. Patients in this study were also significantly heavier and had a larger BMI than in any previous trials. In mid- 2015 we announced that, although patients showed a marked reduction in pain from baseline to 20 weeks when treated with Ampion, the study failed to reach its primary endpoint.
PIVOT Trial
Together with the U.S. Food and Drug Administration, or the FDA, we agreed on a Special Protocol Assessment, an agreement on the trial design and size to form the primary basis of efficacy for a BLA filing, for the confirmatory pivotal trial the PIVOT study. The PIVOT trial is a randomized, vehicle controlled, double-blind study, planned to have approximately 484 patients with osteoarthritis knee pain who are randomized to receive either a 4mL single injection of Ampion or saline control. The primary objective of this study is to evaluate the efficacy of 4 mL Ampion versus 4 mL placebo intra-articular injection in improving knee pain, when administered to patients suffering from OA of the knee. The study started in October 2015 and in January 2016 we announced enrollment for this confirmatory study was approximately 80% complete. We expect the study to be completed by mid-2016.
Clinical Development Pathway
Upon conclusion of the AIK trial, pre-clinical and clinical data were presented to the blood products division of the Center for Biologics Evaluation and Research, or CBER, of the FDA for guidance toward an Ampion novel biologic BLA filing. The FDA provides novel biologics twelve years of market exclusivity against would-be "biosimilar" competitors. The FDA granted an active Investigational New Drug, or IND, for Ampion for the treatment of pain due to osteoarthritis of the knee in March 2013.
We met with the FDA in late 2013 and the FDA confirmed the SPRING study is the first of two pivotal clinical trials required to demonstrate efficacy in a BLA. In collaboration with the FDA we confirmed the trial design of the PIVOT study and the FDA granted us a Special Protocol Assessment, or SPA, an agreement on the design and size of the clinical trial intended to form the primary basis of an efficacy claim in the BLA, for the PIVOT study.
Future Development
We intend to study Ampion for therapeutic applications outside of osteoarthritis of the knee. We are investigating the possible use of Ampion for pain due to osteoarthritis of the hand and as a potential relief for ocular conditions. If successful, these additional formulations and potential therapeutic indications will supplement the Ampion clinical portfolio, and may enable clinical applications in large therapeutic markets where there are significant unmet needs.
38
OPTINA
Optina for Diabetic Macular Edema
Optina is a low-dose formulation of danazol that we are developing to treat diabetic macular edema, or DME. Danazol is a synthetic derivative of modified testosterone ethisterone, and we believe it affects vascular endothelial cell linkage in a biphasic manner. At low doses, danazol decreases vascular permeability by increasing the barrier function of endothelial cells. The lipophilic low-molecular-weight weak androgen has the potential to treat multiple angiopathies. Steroid hormones control a variety of functions through slow genomic and rapid non-genomic mechanisms. Danazol immediately increases intracellular cyclic adenosine monophosphate, or cAMP through the rapid activation of membrane-associated androgen, steroid binding globulin, and calcium channel receptors. At lower concentrations such as Optina, danazol binds to androgen and steroid binding globulin receptors stimulating the formation of a cortical actin ring. At higher concentrations, activation of the calcium channels shift the balance towards stress fiber formation and increase vascular permeability.
When organized into a cortical ring, filamentous actin, or f-actin increases the barrier function of endothelial cells by tethering adhesion molecule complexes to the cytoskeleton. In this orientation, increased cortical actin improves tight junctions which strengthen cell-to-cell adhesions. Formation of the cortical actin ring thereby restricts leakage across the cell membrane.
Phase II Trial
In 2012, we concluded our Phase II randomized, double-blinded, placebo-controlled, dose-ranging study of Optina in subjects with diabetic macular edema in Canada. The trial established that the dose of Optina should take BMI into account. When stratified for BMI the study demonstrated that 47% of patients who received Optina improved at least one best corrected visual acuity, or BCVA, category and achieved a reduction in central retinal thickness, or CRT, at 12 weeks. The study was stopped early in order to pursue a redesigned trial that would evaluate the safety and efficacy of Optina with drug dosing refined by BMI.
OptimEyes Trial
In 2014 and 2015 we announced the OptimEyes multicenter, placebo-controlled, randomized, dose ranging trial to evaluate the safety and efficacy of oral Optina, which included 355 patients. The trial showed Optina was safe and well tolerated with no drug related adverse events and no differences in side effect rates between placebo and Optina groups. The trial did not meet its primary endpoint for all patients, however we believe we have successfully identified an optimal dose for a BMI subgroup of patients who are refractory to currently available therapies and also utilize RAS inhibitors as a medication. As more than 70% of all DME patients are utilizing RAS inhibitors to control their blood pressure and we believe this combination of drugs shows promise as a painless, safe and efficacious oral treatment for DME, and a rescue medication following anti-VEGF therapy failure. These patients showed a +6.2 letter improvement in visual acuity. We presented these results at the World Ophthalmology Congress in February 2016 and plan to present the results at The Association for Research in Vision and Ophthalmology Conference in May 2016.
Aytu Products:
PROSTASCINT
In May 2015, Aytu acquired ProstaScint from Jazz Pharmaceuticals. ProstaScint is the only commercially available diagnostic imaging agent approved by the FDA that specifically targets prostate cancer cells that have spread to tissue outside of the prostate gland. ProstaScint was approved by the FDA in October 1996 and was initially commercialized by Cytogen which was acquired by EUSA Pharma. Jazz Pharmaceuticals acquired EUSA Pharma and its product portfolio including ProstaScint in 2012.
PRIMSOL
In October 2015 Aytu acquired Primsol (trimethoprim hydrochloride) from FSC Laboratories, Inc. Primsol is the only FDA-approved trimethoprim-only oral solution and is standard therapy for urinary tract infections. Primsol is a sulfa-free, pleasant tasting liquid that is appropriate for patients that are sulfa allergic and individuals that have difficulty swallowing pills. Primsol was approved by the FDA in 2000 and was originally marketed by Ascent Pediatrics. FSC Laboratories acquired Primsol from Taro Pharmaceutical.
REDOXSYS
RedoxSYS is a novel, diagnostic platform comprised of a first-in-class, point-of-care device and disposable testing strips that together measure the presence of oxidative stress and antioxidant reserves. Aytu believes this device could also be used as a key indicator in male reproductive health as a stand-alone application. Currently, the device is being studied in over 60 research collaborations that continued through 2016.
39
MIOXSYS
Prior to the Aytu spin-off, Aytu intended to leverage its RedoxSYS research tool to develop a clinical application – known as MiOXSYS – to assess oxidative stress levels in infertile males. Proof of concept studies in male infertility have been conducted with a leading center in the United States and determined that oxidation-reduction potential effectively measures oxidative stress levels in semen and seminal fluid. Semen analysis studies are routinely conducted to assess causes of infertility, so they expect clinicians and oxidative stress researchers to readily integrate MiOXSYS into routine use. Additional studies are now underway that will determine the MiOXSYS system's performance in semen analysis as it relates to infertility.
ZERTANE
Zertane is an oral drug subject to an open Investigational New Drug Application for a Phase III clinical trial for the treatment of premature ejaculation. The FDA has accepted Aytu's Investigational New Drug application and a Phase III clinical study may now begin in the United States.
We owned these assets during the years ended December 31, 2015, 2014 and 2013, respectively, but they were subsequently spun-off on January 4, 2016 and since we no longer own a majority of Aytu's stock, the products will not be consolidated into Ampio in fiscal 2016.
Recent Financing Activities
In January 2013, we formed a subsidiary, Luoxis Diagnostics, Inc., or Luoxis, to focus on the development and commercialization of our Oxidation Reduction Potential, or ORP, technology platform. Luoxis was funded through a private placement which had a final closing in May 2013 with $4.7 million in gross proceeds. Net proceeds were $4.0 million after placement agent and legal fees. The placement agent also received 465,250 warrants to purchase Luoxis common stock valued at $313,064 in connection with the closing. Prior to the private placement, Ampio incurred all of the costs associated with the development of the ORP platform. As a result of the private placement, we owned 80.9% of Luoxis.
In September 2013, we closed on the sale of 4,600,319 shares of common stock at $5.50 per share, for a total of $25.3 million of gross proceeds and $25.0 million net proceeds after offering costs. The sale of the common stock was made pursuant to the Form S-3 Shelf Registration.
In December 2013, we filed an additional shelf registration statement on Form S-3 with the Securities and Exchange Commission to register our common stock and warrants in an aggregate amount of up to $100.0 million for offering from time to time in the future, as well as 1.5 million shares of common stock available for sale by selling shareholders. The shelf registration was declared effective in January 2014 by the Securities and Exchange Commission. As a result of the subsequent equity raises, approximately $86.3 million remains available under the Form S-3 filed in December 2013.
In March 2014, we completed an underwritten public offering for the sale of 9,775,000 shares of common stock at a price of $7.00 per share. Gross proceeds to us were $68.4 million with net proceeds of $63.4 million after underwriter fees and cash offering expenses.
In February 2016, we entered into a Controlled Equity Offering SM Sales Agreement with a placement agent to implement an "at-the-market" equity program under which we, from time to time, may offer and sell shares of our common stock having an aggregate offering price of up to $25 million through the placement agent.
Known Trends or Future Events; Outlook
We have not generated any significant revenues and have therefore incurred significant net losses totaling $133.9 million since our inception in December 2008. The assets we purchased from BioSciences in April 2009 generated minimal revenues prior to their acquisition. We expect to generate operating losses for the foreseeable future, but intend to try to limit the extent of these losses by entering into co-development or collaboration agreements with one or more strategic partners. Although we have raised capital in the past and with net proceeds of $63.4 million and $29.0 million through the sale of common stock in 2014 and 2013, respectively, we cannot assure you that we will be able to secure such additional financing, if needed, or that it will be adequate to execute our business strategy. Even if we obtain additional financing, it may be costly and may require us to agree to covenants or other provisions that will favor new investors over existing shareholders.
Our primary focus is advancing the clinical development of our core assets: Ampion and Optina. In December 2013, we entered into a ten-year lease of a multi-purpose facility containing approximately 19,000 square feet. This facility includes an FDA compliant clean room to manufacture Ampion and our corporate offices.
On January 4, 2016 we distributed to our shareholders the majority of our shares in Aytu, and since we no longer own a majority of Aytu's stock, our focus will be solely on Ampio products Ampion and Optina in fiscal 2016.
40
Significant Accounting Policies and Estimates
Our consolidated financial statements have been prepared in accordance with accounting policies generally accepted in the United States of America. The preparation of the consolidated financial statements requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the consolidated financial statements and the reported amounts of expenses during the reporting period. On an on-going basis, management evaluates its estimates and judgments, including those related to recoverability and useful lives of long-lived assets, fair value of our derivative instruments, allowances and contingencies. Management bases its estimates and judgments on historical experience and on various other factors that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. The methods, estimates, and judgments used by us in applying these most critical accounting policies have a significant impact on the results we report in our consolidated financial statements.
Below is a discussion of the policies and estimates that we believe involve a high degree of judgment and complexity.
Principals of Consolidation
These consolidated financial statements include the accounts of Ampio and its majority-owned subsidiary. All material intercompany transactions and balances have been eliminated.
Cash and Cash Equivalents
We consider all highly liquid instruments purchased with an original maturity of three months or less to be cash equivalents. Cash equivalents consist primarily of money market fund investments. Our investment policy is to preserve principal and maintain liquidity. We periodically monitor our positions with, and the credit quality of, the financial institutions with which we invest. Periodically, throughout the year, we have maintained balances in excess of federally insured limits.
Accrued Compensation
The accrued compensation consists of a 2015 employee bonus accrual. As of the filing date of this report, a majority of the bonus had not been paid. The majority of the bonus is contingent upon our completion of a successful Ampion trial. The Compensation Committee will re-evaluate the payment of the bonus during the first half of 2016.
Revenue Recognition/Deferred Revenue
License Agreements and Royalties
Payments received upon signing of license agreements are for the right to use the license and are deferred and amortized over the lesser of the license term or patent life of the licensed drug. Milestone payments relate to obtaining regulatory approval in the territories, cumulative sales targets, and other projected milestones and are recognized at the time the milestone requirements are achieved. Royalties will be recognized as revenue when earned.
Product & Service Sales
We recognize revenue from product and service sales when there is persuasive evidence that an arrangement exists, delivery has occurred or service has been rendered, the price is fixed or determinable and collectability is reasonably assured.
Estimated Sales Returns and Allowances
We record estimated reductions in revenue for potential returns of products by customers. As a result, management must make estimates of potential future product returns and other allowances related to current period product revenue. In making such estimates, management analyzes historical returns, current economic trends and changes in customer demand and acceptance of our products. If management were to make different judgments or utilize different estimates, material differences in the amount of our reported revenue could result.
Fixed Assets
Fixed assets are recorded at cost and after being placed in service, are depreciated using the straight-line method over estimated useful lives.
In-Process Research and Development
In-process research and development, or IPRD, relates to the Zertane product and clinical trial data acquired in connection with the 2011 business combination of BioSciences. The $7,500,000 recorded was based on an independent, third party appraisal of the fair value of the assets acquired. IPRD is considered an indefinite-lived intangible asset and its fair value will be assessed annually and
41
written down if impaired. Once the Zertane product obtains regulatory approval and commercial production begins, IPRD will be reclassified to an intangible that will be amortized over its estimated useful life. If Aytu decided to abandon the Zertane product, the IPRD would be expensed.
Patents
Costs of establishing patents, consisting of legal and filing fees paid to third parties, are expensed as incurred. The fair value of the Zertane patents, determined by an independent, third party appraisal to be $500,000, acquired in connection with the 2011 acquisition of BioSciences is being amortized over the remaining U.S. patent life since our acquisition of approximately 11 years. The fair value of the Luoxis patents was $380,000 when they were acquired in connection with the 2013 formation of Luoxis and is being amortized over the remaining U.S. patent life since our acquisition of approximately 15 years.
Business Combinations
The Company accounts for its business acquisitions under the acquisition method of accounting as indicated in the Financial Accounting Standards Board's, or FASB, Accounting Standards Codification, or ASC, 805, "Business Combinations", which requires the acquiring entity in a business combination to recognize the fair value of all assets acquired, liabilities assumed, and any non-controlling interest in the acquiree; and establishes the acquisition date as the fair value measurement point. Accordingly, we recognize assets acquired and liabilities assumed in business combinations, including contingent assets and liabilities and non-controlling interest in the acquiree, based on the fair value estimates as of the date of acquisition. In accordance with ASC 805, we recognize and measure goodwill as of the acquisition date, as the excess of the fair value of the consideration paid over the fair value of the identified net assets acquired.
Goodwill
The ProstaScint and Primsol purchase price allocations were based upon an analysis of the fair value of the assets and liabilities acquired. The final purchase price may be adjusted up to one year from the date of the acquisition. Identifying the fair value of the tangible and intangible assets and liabilities acquired required the use of estimates by management, and were based upon currently available data, as noted below.
We allocated the excess of purchase price over the identifiable intangible and net tangible assets to goodwill. Such goodwill is not deductible for tax purposes and represents the value placed on entering new markets and expanding market share.
We test our goodwill for impairment annually, or whenever events or changes in circumstances indicate an impairment may have occurred, by comparing the carrying value to its implied fair value. Impairment may result from, among other things, deterioration in the performance of the acquired business, adverse market conditions, adverse changes in applicable laws or regulations and a variety of other circumstances. If we determine that an impairment has occurred, we are required to record a write-down of the carrying value and charge the impairment as an operating expense in the period the determination is made. In evaluating the recoverability of the carrying value of goodwill, we must make assumptions regarding estimated future cash flows and other factors to determine the fair value of the acquired assets. Changes in strategy or market conditions could significantly impact those judgments in the future and require an adjustment to the recorded balances. The goodwill was recorded as part of the acquisition of ProstaScint that occurred in May 2015 and the acquisition of Primsol that occurred in October 2015. There was no impairment of goodwill for the year ended December 31, 2015.
Use of Estimates
The preparation of consolidated financial statements in accordance with Generally Accepted Accounting Principles in the United States of America, or GAAP, requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, disclosures of contingent assets and liabilities as of the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting periods. Significant items subject to such estimates and assumptions include the fair value of warrant derivative liability, hybrid debt instruments, valuation allowances, stock-based compensation, useful lives of fixed assets, estimates of contingent consideration in business combinations and assumptions in evaluating impairment of indefinite lived assets. Actual results could differ from these estimates.
Derivatives
We accounted for hybrid financial instruments (debentures with embedded derivative features – conversion options, down-round protection and mandatory conversion provisions) and related warrants by recording the fair value of each hybrid instrument in its entirety and recording the fair value of the warrant derivative liability. The fair value of the hybrid financial instruments and related warrants was calculated using a binomial-lattice-based valuation model. We recorded a derivative expense at the inception of each instrument reflecting the difference between the fair value and cash received. Changes in the fair value in subsequent periods were
42
recorded as unrealized gain or loss on fair value of debt instruments for the hybrid financial instruments and to derivative income or expense for the warrants. Accounting for hybrid financial instruments and derivatives is discussed more fully in Note 4 – Derivative Financial Instruments Related to Ampio. The fair value of warrants issued in connection with the common stock offerings was valued using a Black-Scholes option pricing model. The fair value of the warrants issued in connection with the convertible debt were valued using the Monte Carlo valuation methodology.
Income Taxes
Deferred taxes are provided on an asset and liability method whereby deferred tax assets are recognized for deductible temporary differences and operating loss and tax credit carry forwards and deferred tax liabilities are recognized for taxable temporary differences. Temporary differences are the differences between the reported amounts of assets and liabilities and their tax bases. Deferred tax assets are reduced by a valuation allowance when, in the opinion of management, it is more likely than not that some portion or all of the deferred tax assets will not be realized. Deferred tax assets and liabilities are adjusted for the effects of changes in tax laws and rates on the date of enactment.
Net Loss per Common Share
Basic net loss per share includes no dilution and is computed by dividing the net loss available to common stockholders by the weighted-average number of shares outstanding during the period. Diluted net loss per share reflects the potential of securities that could share in the net loss of Ampio. Basic and diluted loss per share was the same in 2015, 2014 and 2013. Although there were common stock equivalents of 7,814,908, 7,084,577 and 5,662,748 shares outstanding at December 31, 2015, 2014 and 2013, respectively, consisting of stock options and warrants; they were not included in the calculation of the diluted net loss per share because they would have been anti-dilutive.
Stock-Based Compensation
We account for share based payments by recognizing compensation expense based upon the estimated fair value of the awards on the date of grant. We determine the estimated grant fair value using the Black-Scholes option pricing model and recognize compensation costs ratably over the vesting period using the graded method.
Research and Development
Research and development costs are expensed as incurred with expense recorded in the respective periods.
Fair Value of Financial Instruments
The carrying amounts of financial instruments, including cash and cash equivalents, accounts receivable, accounts payable and accrued liabilities and convertible notes are carried at cost which approximates fair value due to the short maturity of these instruments. Hybrid financial instruments such as convertible debentures and related warrants were recorded at estimated fair value based on a binomial-lattice-based valuation model , see Note – 4 Derivative Financial Instruments Related to Ampio for additional information . The fair value of warrants issued in connection with the Aytu convertible notes was valued using a Monte Carlo option pricing model. The accounting for financial instruments and the Aytu derivatives is discussed more fully in Note 5 – Fair Value Considerations.
Impairment of Long-Lived Assets
We routinely perform an annual evaluation of the recoverability of the carrying value of our long-lived assets to determine if facts and circumstances indicate that the carrying value of assets or intangible assets may be impaired and if any adjustment is warranted. Based on our evaluation as of December 31, 2015, no impairment existed for long-lived assets.
43
Recent Accounting Pronouncements
Refer to our discussion of recently adopted accounting pronouncements and other recent accounting pronouncements in Note 1 – Basis of Presentation, Merger and Business Combination to the accompanying audited financial statements included elsewhere in the Annual Report on Form 10-K.
Results of Operations-Year Ended December 31, 2015, 2014 and 2013
Results of operations for the years ended December 31, 2015, 2014 and 2013 reflected losses applicable to Ampio of $32.0 million, $38.1 million and $24.0 million, respectively. These losses include non-cash charges related to stock-based compensation, depreciation, amortization and accretion expense, amortization of prepaid research and development-related party, amortization of debt issuance costs, common stock issued for services, derivative expense, and loss on disposal of fixed assets totaling $7.8 million, $8.6 million and $4.2 million in 2015, 2014 and 2013 respectively. We expect that non-cash expenses will decrease in fiscal 2016 compared to 2015 as we will no longer be consolidating Aytu into Ampio. We also expect to see depreciation and expense to increase in 2016 compared to 2015 as additional assets start to depreciate in 2016 from our build out of the manufacturing facility.
Revenue
The $1,077,000 product and service revenue recognized in 2015 represents sales from our Aytu segment which includes the ProstaScint product, the Primsol product and the RedoxSYS System. We did not generate any product and service revenue in 2014 or 2013.
The $86,000, $77,000 and $50,000 license revenue recognized in 2015, 2014 and 2013 respectively, represents the amortization of the upfront payments received from our license agreements. The initial payment of $500,000 from the license agreement of Zertane with a Korean pharmaceutical company was deferred and is being recognized over ten years. The initial payment of $250,000 from the license agreement of Zertane with a Canadian-based supplier was deferred and is being recognized over seven years.
As is customary in the pharmaceutical industry, our gross product sales are subject to a variety of deductions in arriving at reported net product sales. Provisions for these deductions are recorded concurrently with the recognition of gross product sales revenue and includes discounts, chargebacks, distributor fees, processing fees, as well as allowances for returns and Medicaid rebates. Provision balances relating to estimated amounts payable to direct customers are netted against accounts receivable and balances relating to indirect customers are included in accounts payable and accrued liabilities.
Since all of the revenues during the last three years were generated at the Aytu subsidiary which was spun-out on January 4, 2016, we do not expect to have any revenue in fiscal 2016.
Expenses
Research and Development
Research and development costs consist of clinical trials and sponsored research, labor, stock-based compensation, consultants and sponsored research – related party. These costs relate solely to research and development without an allocation of general and administrative expenses and are summarized as follows:
| Year Ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
Clinical trials and sponsored research | $ | 11,372,000 | $ | 19,071,000 | $ | 12,078,000 | ||||||
Labor | 3,294,000 | 2,227,000 | 1,862,000 | |||||||||
Stock-based compensation | 2,428,000 | 4,641,000 | 1,997,000 | |||||||||
Consultants and other | 1,531,000 | 680,000 | 613,000 | |||||||||
Sponsored Research - related party | 336,000 | 304,000 | 46,000 | |||||||||
|
|
|
|
|
| |||||||
| $ | 18,961,000 | $ | 26,923,000 | $ | 16,596,000 | |||||||
|
|
|
|
|
| |||||||
Comparison of Years Ended December 31, 2015 and 2014
Research and development expenses decreased $7,962,000, or 30%, in 2015 over 2014. This was due primarily to a decrease in clinical trials due to the completion of prior trials for Optina and Ampion. The increase in labor and consultants and other is due to additional costs related to preparing our facility to become operational and the additional professional staffing. Research and development expense in 2016 is expected to decrease with where it was in 2015, but this could materially change based upon the outcome of the current Ampion Phase III trial, which we expect to finalize in the first half of 2016. We expect research and development labor and stock-based compensation to increase slightly in 2016 compared to 2015.
44
Comparison of Years Ended December 31, 2014 and 2013
Research and development expenses increased $10,327,000, or 62%, in 2014 over 2013. This was due primarily to costs associated with the production of study drugs, clinical trials of Ampion and Optina and the Luoxis development of its ORP platform. Stock-based compensation increased due to stock options granted in Ampio, Luoxis and Vyrix as well as the continuing vesting of stock option awards granted in previous years.
Selling, General and Administrative
Selling, general and administrative expenses consist of personnel costs for employees in executive, business development and operational functions and director fees; stock-based compensation; patents and intellectual property; professional fees include legal, auditing and accounting; occupancy, travel and other includes rent, governmental and regulatory compliance, insurance, investor/public relations and professional subscriptions. These costs are summarized as follows:
| Year Ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
Occupancy, travel and other | $ | 3,962,000 | $ | 2,481,000 | $ | 1,721,000 | ||||||
Labor | 3,560,000 | 2,326,000 | 1,538,000 | |||||||||
Stock-based compensation | 3,351,000 | 3,242,000 | 1,539,000 | |||||||||
Professional fees | 2,316,000 | 1,689,000 | 735,000 | |||||||||
Patent costs | 1,675,000 | 2,241,000 | 1,738,000 | |||||||||
Directors fees | 271,000 | 246,000 | 206,000 | |||||||||
|
|
|
|
|
| |||||||
| $ | 15,135,000 | $ | 12,225,000 | $ | 7,477,000 | |||||||
|
|
|
|
|
| |||||||
Comparison of Years Ended December 31, 2015 and 2014
General and administrative costs increased $2,910,000, or 24%, in 2015 over 2014. Occupancy, travel and other increased primarily due to insurance premiums, regulatory and compliance fees and travel expenses. The increase in labor costs is primarily a result of increased staffing due to commercialization efforts for our ProstaScint and Primsol products. The increase in professional fees is associated with the additional expenses to meet the requirements of the public company formation of Aytu. During 2016, we would expect that general and administrative costs will decrease as compared to 2015 as we have completed the spin-off of the Aytu subsidiary and explore options on the best ways to complete the regulatory process for Ampion and Optina.
Comparison of Years Ended December 31, 2014 and 2013
General and administrative costs increased $4,748,000 or 64%, in 2014 over 2013. The increase in labor costs and stock-based compensation primarily relates to increased professional staffing, bonuses earned and stock options granted in Ampio, Luoxis and Vyrix as well as the continuing vesting of stock option awards granted in previous years. The increase in professional fees is related to our legal defense costs for a case that we were found to have no liability and had to pay no settlement fee. Also, the legal costs associated with Vyrix trying to complete an initial public offering during 2014 were expensed. The increase in occupancy, travel and other in 2014 was due to moving into our new facility.
Derivative Expense
We recorded $78,000 in non-cash derivative expense in 2015 in connection with warrants associated with Aytu's convertible notes. See Note 5 – Fair Value Considerations. We incurred $517,000 in non-cash derivative expense in 2013 in connection with our hybrid financial instruments consisting of debentures and related warrants. This expense relates to the fair value at inception and subsequent changes in fair value of the debentures issued in 2011 and 2010 stemming from the embedded derivative features (conversion options, down-round protection and mandatory conversion provisions) and the changes in fair value of warrants issued in conjunction with the debentures. The debentures were redeemed in 2011 and in 2013; all of the warrants were exercised prior to their expiration date of December 31, 2013. Since all of these warrants expired prior to 2014, there was no expense or income related to these warrants in 2014 or 2015. Since the 2015 derivative expense relates to Aytu warrants, we do not expect to see this expense in 2016 as the Aytu subsidiary was spun-off in January 2016.
45
Net Cash Used in Operating Activities
During 2015, our operating activities used $26.4 million in cash. The use of cash was $7.3 million lower than the net loss due primarily to non-cash charges for stock-based compensation, depreciation, amortization and accretion. Cash provided in operating activities also included a $1,142,000 increase in accrued compensation and a $162,000 increase in interest payable which were offset by a $1,332,000 increase in prepaid expenses and a $426,000 decrease in accounts payable.
During 2014, our operating activities used $30.7 million in cash. The use of cash was $8.3 million lower than the net loss due primarily to non-cash charges for stock-based compensation, depreciation and amortization. Cash provided in operating activities also included a $1,020,000 increase in accounts payable and $721,000 increase in deferred rent which were offset by increased prepaid research and development related party of $1,340,000 and prepaid expense of $541,000.
During 2013, our operating activities used approximately $19.1 million in cash. The use of cash was $5.4 million lower than the net loss due primarily to non-cash charges for stock-based compensation, depreciation and amortization and derivative expense. Net cash provided in operating activities also included a $522,000 increase in accrued compensation and $699,000 increase in accounts payable.
Net Cash Used in Investing Activities
During 2015, $1,540,000 million of cash was used to acquire the ProstaScint and Primsol businesses. Purchases of fixed assets decreased in 2015 which reflects the near completion of our manufacturing facility.
During 2014, cash was used to acquire fixed assets which consists of the purchase of machinery and build out related to the in process manufacturing facility/clean room.
During 2013, cash was used to acquire ORP patents on behalf of Luoxis. Fixed assets reflect purchases of machinery related to the in process manufacturing facility/clean room, a new server, a laboratory research instrument and a Luoxis ORP manufacturing device.
Net Cash from Financing Activities
Net cash provided by financing activities in 2015 was $4.9 million which was primarily related to the Aytu convertible promissory notes which reflects gross proceeds of $5.2 million offset by the cash portion of the debt issuance costs related to the convertible promissory notes of $298,000.
Net cash provided by financing activities in 2014 was $63.4 million which reflects net proceeds from our completed underwritten public offering and of stock option exercises.
Net cash provided by financing activities in 2013 was $29.4 million which reflects net proceeds from the registered direct placement of $25.0 million, Luoxis' private financing of $4.0 million and $0.4 million from the exercise of stock options and warrants.
Contractual Obligations and Commitments
The following table summarizes the commitments and contingencies including Aytu (see Note 16 – Subsequent Events) as of December 31, 2015 which are described below:
| Total | 2016 | 2017 | 2018 | 2019 | 2020 | Thereafter | ||||||||||||||||||||||
Ampion supply agreement | $ | 5,100,000 | $ | - | $ | 2,550,000 | $ | 2,550,000 | $ | - | $ | - | $ | - | ||||||||||||||
Clinical research and trial obligations | 4,275,000 | 4,275,000 | - | - | - | - | - | |||||||||||||||||||||
Facility leases | 3,309,000 | 436,000 | 450,000 | 418,000 | 326,000 | 335,000 | 1,344,000 | |||||||||||||||||||||
Sponsored research agreement with related party | 1,406,000 | 395,000 | 395,000 | 395,000 | 151,000 | 70,000 | - | |||||||||||||||||||||
Primsol business | 1,250,000 | 1,250,000 | - | - | - | - | - | |||||||||||||||||||||
Manufacturing agreement | 1,000,000 | 1,000,000 | - | - | - | - | - | |||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||
| $ | 16,340,000 | $ | 7,356,000 | $ | 3,395,000 | $ | 3,363,000 | $ | 477,000 | $ | 405,000 | $ | 1,344,000 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||
Ampion Supply Agreement
In connection with the manufacturing facility/clean room, in October 2013, we entered into a human serum albumin ingredient and purchase sale agreement with a remaining commitment of $5,100,000. Per an amendment to the original agreement, we are not committed to purchases in 2016 and have extended the agreement to 2018.
46
Clinical Research and Trial Obligations
In connection with current and recent clinical trials, as of December 31, 2015, we have a remaining commitment of $4,263,000 on contracts related to the Ampion study trial expense and $12,000 remaining contract commitment related to the Optina study trial expense.
Facility Leases
In December 2013, we entered into a 125-month non-cancellable operating lease for new office space and the manufacturing facility effective May 1, 2014. The new lease has initial base rent of $23,000 per month, with the total base rent over the term of the lease of approximately $3.3 million and includes rent abatements and leasehold incentives. We recognize rental expense of the facility on a straight-line basis over the term of the lease. Differences between the straight-line net expenses on rent payments are classified as liabilities between current deferred rent and long-term deferred rent.
In June 2015, Aytu entered into a 37-month operating lease for a space in Raleigh, North Carolina. This lease has initial base rent of $2,900 a month, with total base rent over the term of the lease of approximately $112,000. In September 2015, Aytu entered into a 37- month operating lease in Englewood, Colorado. This lease has an initial base rent of $8,500 a month with a total base rent over the term of the lease of approximately $318,000. Aytu recognizes rental expense of the facilities on a straight-line basis over the term of the lease. Differences between the straight-line net expenses on rent payments are classified as liabilities between current deferred rent and long-term deferred rent.
Sponsored Research Agreement with Related Party
We entered into a Sponsored Research Agreement with TRLLC, a related party, in September 2009. Under the terms of the Sponsored Research Agreement, we are to provide personnel and pay for leased equipment. The Sponsored Research Agreement may be terminated without cause by either party on 180 days' notice. As further noted in Note 11 – Related Party Transactions, in March 2014, the Sponsored Research Agreement was extended through March 2019, including a "no termination" period through March 2017. In a subsequent Addendum, the parties also agreed to increase the equivalent value of the personnel provided by us from $264,000 to $325,000 per year.
Aytu entered into a Sponsored Research Agreement with TRLLC, a related party, in June 2013. Under the terms of the Sponsored Research Agreement, TRLLC agreed to work collaboratively in advancing the RedoxSYS System diagnostic platform through research and development efforts. The Sponsored Research Agreement may be terminated without cause by either party on 30 days' notice.
Primsol Business
In October 2015, Aytu entered into an agreement with FSC Laboratories, Inc. for the purchase of Primsol ( see Note 1 – Business Combination-Primsol).
Manufacturing Agreement
In October 2015, Aytu entered into a Master Services Agreement with Biovest International, Inc., or Biovest. The agreement provides that Aytu may engage Biovest from time to time to provide services in accordance with mutually agreed upon project addendums and purchase orders. Aytu expects to use the agreement from time to time for manufacturing services, including without limitation, the manufacturing, processing, quality control testing, release or storage of its products for the ProstaScint product. Aytu is obligated to pay Biovest $1.0 million for time and materials as they develop a plan to reproduce the manufacturing process.
Liquidity and Capital Resources
We have not generated significant revenue as our primary activities are focused on research and development, advancing our primary product candidates, and raising capital. As of December 31, 2015, we had cash and cash equivalents totaling $27.0 million available to fund our operations and $4.3 million in accounts payable and accrued compensation. Since our Aytu subsidiary was spun-off January 4, 2016, we believe that it is appropriate to review Ampio's liquidity and capital resources on a stand-alone basis going into 2016 as Aytu will no longer be consolidated into Ampio. Ampio, on a stand-alone basis, had cash and cash equivalents of $16.0 million at December 31, 2015. Based upon our current plans, we believe our capital resources at December 31, 2015 will be sufficient to fund our currently planned operations through 2016 and into early 2017. This projection is based on a number of assumptions that may prove to be wrong, and we could exhaust our available cash and cash equivalents earlier than presently anticipated. We will be required to seek additional capital within the next 18 months to expand our clinical and commercial development activities for Ampion and Optina based on the positive results of our ongoing clinical trials, if we face challenges or delays in connection with our clinical trials, or to maintain minimum cash balances that we deem reasonable and prudent. We intend to evaluate the capital markets from time to time to determine whether to raise additional capital in the form of equity, convertible debt or otherwise, depending on market conditions relative to our need for funds at such time, and we will seek to raise additional capital within the next 18 months when we conclude that such capital is available on terms that we consider to be in the best interests of us and our stockholders.
47
We have prepared a budget for 2016 which reflects cash requirements for fixed, on-going expenses such as payroll, legal and accounting, patents and overhead at an average cash burn rate of approximately $800,000 per month. Additional funds are planned for regulatory approvals, clinical trials, outsourced research and development and commercialization consulting. We have no planned additional funding to be provided to Aytu during 2016. Accordingly, it will be necessary to raise additional capital and/or enter into licensing or collaboration agreements. At this time, we expect to satisfy our future cash needs through our Controlled Equity Offering Sales Agreement that we entered into in February 2016, private or public sales of our securities or debt financings. We cannot be certain that financing will be available to us on acceptable terms, or at all. Over the last three years, volatility in the financial markets has adversely affected the market capitalizations of many pharmaceutical companies and generally made equity and debt financing more difficult to obtain. This volatility, coupled with other factors, may limit our access to additional financing.
If we cannot raise adequate additional capital in the future when we require it, we will be required to delay, reduce the scope of, or eliminate one or more of our research or development programs or our future commercialization efforts. We also may be required to relinquish greater or all rights to product candidates at an earlier stage of development or on less favorable terms than we would otherwise choose. This may lead to impairment or other charges, which could materially affect our balance sheet and operating results.
The $1.1 million in product and service revenue recognized in 2015 represents sales from our Aytu segment which includes the ProstaScint and Primsol products and the RedoxSYS system. We did not generate any product and service revenue in 2014 or 2013. We do not expect to generate any revenue in 2016 as our Aytu subsidiary was spun-off in January 2016.
Off Balance Sheet Arrangements
We do not have off-balance sheet arrangements, financings, or other relationships with unconsolidated entities or other persons, also known as "variable interest entities."
Impact of Inflation
In general, we believe that our operating expenses can be negatively impacted by increases in the cost of clinical trials due to inflation and rising health care costs.
| Item 7A. | Quantitative and Qualitative Disclosures about Market Risks |
Our business is not currently subject to material market risk related to financial instruments, equity or commodities.
| Item 8. | Financial Statements and Supplementary Data |
The Financial Statements and Supplementary Data required by this item are located in Item 15 of Part IV, "Index to Financial Statements" at page F-1 of this annual report on Form 10-K and are incorporated herein by reference.
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
None
| Item 9A. | Controls and Procedures |
Evaluation of Disclosure Controls and Procedures
We maintain "disclosure controls and procedures," as such term is defined in Rules 13a-15(e) and 15d-15(e) of the Securities Exchange Act of 1934 (the "Exchange Act"), that are designed to ensure that information required to be disclosed by us in reports that we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in SEC rules and forms, and that such information is accumulated and communicated to our management, including our chief executive officer and chief financial officer, as appropriate, to allow timely decisions regarding required disclosure.
As of the end of the period covered by this report, we carried out an evaluation, under the supervision and with the participation of senior management, including the chief executive officer and the chief financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures pursuant to Exchange Act Rules 13a-15(b) and 15d-15(b). Based upon this evaluation, the chief executive officer and the chief financial officer concluded that our disclosure controls and procedures as of the end of the period covered by this report were effective at the reasonable assurance level.
48
Management's Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as such term is defined in Rules 13a-15(f) under the Exchange Act). Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2015. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control-Integrated Framework (2013) . Our management has concluded that, as of December 31, 2015, our internal control over financial reporting is effective based on these criteria.
EKS&H LLLP, the independent registered public accounting firm that audited our consolidated financial statements included in this Annual Report on Form 10-K, has issued an attestation report on our internal control over financial reporting, which is included herein at F-2.
As permitted by the SEC, management's evaluation of internal control over financial reporting did not include an evaluation of the internal controls of the ProstaScint business which we acquired in May 2015 and the Primsol business which we acquired in October 2015, which are included in our December 31, 2015 consolidated financial statements. The acquired assets related to these acquisitions constitute $3.7 million or 7.4% of total assets as of December 31, 2015 and contributed net revenue of $1.0 million for the period from the acquisition dates through December 31, 2015. Aytu acquired ProstaScint and Primsol. After January 4, 2016, Aytu will not be consolidated with Ampio.
Changes in Internal Control over Financial Reporting
There were no changes in our internal controls over financial reporting, known to the chief executive officer or the chief financial officer that occurred during the period covered by this report that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
| Item 9B. | Other Information |
None.
49
PART III
| Item 10. | Directors and Executive Officers, and Corporate Governance |
The following table sets forth the names, ages and positions of our executive officers and directors as of February 1, 2016.
Name | Age | Position With Ampio | Principal Occupation and Areas of Relevant Experience For Directors | Director Since | ||||
| Michael Macaluso | 64 | Chief Executive Officer and Chairman of the Board | Mr. Macaluso founded Life Sciences and has been a member of the board of directors of Life Sciences, our predecessor, since its inception. Mr. Macaluso has also been a member of our Board of Directors since the merger with Chay Enterprises in March 2010 and our Chief Executive Officer since January 9, 2012. Mr. Macaluso was appointed president of Isolagen, Inc. (AMEX: ILE) and served in that position from June 2001 to August 2001, when he was appointed chief executive officer. In June 2003, Mr. Macaluso was re-appointed as president of Isolagen and served as both chief executive officer and president until September 2004. Mr. Macaluso also served on the board of directors of Isolagen from June 2001 until April 2005. From October 1998 until June 2001, Mr. Macaluso was the owner of Page International Communications, a manufacturing business. Mr. Macaluso was a founder and principal of International Printing and Publishing, a position Mr. Macaluso held from 1989 until 1997, when he sold that business to a private equity firm.
Mr. Macaluso's experience in executive management and marketing within the pharmaceutical industry, monetizing company opportunities, and corporate finance led to the conclusion of our Board of Directors that he should serve as a director of our company in light of our business and structure. | March 2010 | ||||
| David Bar-Or, MD. | 67 | Chief Scientific Officer and Director | Dr. Bar-Or has served as our chief scientific officer since March 2010. Dr. Bar-Or also served as our chairman of the Board from March 2010 until May 2010. From April 2009 until March 2010, he served as chairman of the board and chief scientific officer of Life Sciences. Dr. Bar-Or is currently the director of Trauma Research at Swedish Medical Center, Englewood, Colorado, St. Anthony's Hospital, Lakewood, Colorado and The Medical Center of Plano, Plano, Texas. Dr. Bar-Or is the founder of Ampio Pharmaceuticals Inc. Dr. Bar-Or is principally responsible for all patented and proprietary technologies acquired by us from BioSciences in April 2009 and for all patents issued and applied for since then, having been issued over 291patents and having filed or co-filed almost 242 patent applications. Dr. Bar-Or has authored or co-authored over 125 peer-reviewed journal articles and several book chapters. Dr. Bar-Or is a reviewer for over 20 peer reviewed scientific and clinical journals. Is the recipient of the Gustav Levi Award from the Mount Sinai Hospital, New York, New York, the Kornfeld Award for an outstanding MD Thesis, the Outstanding Resident Research Award from the Denver General Hospital, and the Outstanding Clinician Award for the Denver General Medical Emergency Resident Program. Dr. Bar-Or received his medical degree from The Hebrew University, Hadassah Medical School, Jerusalem, Israel, following which he completed a biochemistry fellowship at Hadassah Hospital under Professor Alisa Gutman and undertook post-graduate Residency training at Denver Health | March 2010 | ||||
50
Name | Age | Position With Ampio | Principal Occupation and Areas of Relevant Experience For Directors | Director Since | ||||
Medical Center, specializing in emergency medicine, a discipline in which he is board certified. He completed the first research fellowship in Emergency Medicine at Denver Health Medical Center under the direction of Prof Peter Rosen. Dr. Bar-Or practiced Emergency Medicine for 20 years at Swedish Medical Center, Englewood, Colorado and 5 years as the Emergency Department Medical Director.
Among other experience, qualifications, attributes and skills, Dr. Bar-Or's medical training, extensive involvement and inventions in researching and developing our product candidates, and leadership role in his hospital affiliations led to the conclusion of our Board of Directors that he should serve as a director of our company in light of our business and structure. | ||||||||
| Philip H. Coelho (1)(2)(3) | 72 | Director | Mr. Coelho has served as a member of our Board of Directors since April 2010. Mr. Coelho is the Chief Technology Officer and Co-Founder of SynGen Inc., a firm inventing and commercializing products that provide advanced cell separation and purification tools and accessories to aid regenerative medicine workflows. Prior to founding SynGen Inc. in October 2009, Mr. Coelho was the President and CEO of PHC Medical, Inc., a consulting firm, from August 2008 through October 2009. From August 2007 through May 2008, Mr. Coelho served as the Chief Technology Architect of ThermoGenesis Corp., a medical products company he founded in 1986 that focused on the regenerative medicine market. From 1989 through July 2007, he was Chairman and Chief Executive Officer of ThermoGenesis Corp. Mr. Coelho served as Vice President of Research & Development of ThermoGenesis from 1986 through 1989. Mr. Coelho has been in the senior management of high technology consumer electronic or medical device companies for over 30 years. He was President of Castleton Inc. from 1982 to 1986, and President of ESS Inc. from 1971 to 1982. Mr. Coelho also serves as a member of the board of directors of Nasdaq-listed company, Catalyst Pharmaceuticals Partners, Inc. (CPRX) (since October 2002), and served as a member of the Board of Directors of NASDAQ-listed Mediware Information Systems, Inc. (MEDW) (from December 2001 until July 2006, and commencing again in May 2008 until it was sold in December 2012). Mr. Coelho received a B.S. degree in thermodynamic and mechanical engineering from the University of California, Davis and has been awarded more than 35 U.S. patents in the areas of cell cryopreservation, cryogenic robotics, cell selection, blood protein harvesting and surgical homeostasis.
Mr. Coelho's long tenure as a chief executive officer of a public medical device company, as director of a public pharmaceutical company, prior and current public company board experience, and knowledge of corporate finance and governance as an executive and director, as well as his demonstrated success in developing patented technologies, led to the conclusion of our Board of Directors that he should serve as a director of our company in light of our business and structure. | April 2010 | ||||
51
Name | Age | Position With Ampio | Principal Occupation and Areas of Relevant Experience For Directors | Director Since | ||||
| Richard B. Giles (1)(2)(3) | 66 | Director | Mr. Giles, CPA, has served as a member of our Board of Directors since August 2010. Mr. Giles is the Chief Financial Officer of Ludvik Electric Co., an electrical contractor headquartered in Lakewood, Colorado, a position he has held since 1985. Ludvik Electric is a private electrical contractor with 2015 revenues of $70 million that has completed electrical contracting projects throughout the United States, South Africa and Germany totaling more than $1.8 billion. As CFO and Treasurer of Ludvik Electric, Mr. Giles oversees accounting, risk management, financial planning and analysis, financial reporting, regulatory compliance, and tax-related accounting functions. He serves also as the trustee of Ludvik Electric Co.'s 401(k) plan. Prior to joining Ludvik Electric, Mr. Giles was for three years an audit partner with Higgins Meritt & Company, then a Denver, Colorado CPA firm, and during the preceding nine years he was an audit manager and a member of the audit staff of Price Waterhouse, one of the legacy firms which now comprises PricewaterhouseCoopers. While with Price Waterhouse, Mr. Giles participated in a number of public company audits, including one for a leading computer manufacturer. Mr. Giles received a B.S. degree in accounting from the University of Northern Colorado. He is a member of the American Institute of Certified Public Accountants, Colorado Society of Certified Public Accountants, Construction Financial Management Association and Financial Executives International.
Mr. Giles' experience in executive financial management, accounting and financial reporting, and corporate accounting and controls led to the conclusion of our Board of Directors that he should serve as a director of our company in light of our business and structure. | August 2010 | ||||
David R. Stevens, Ph.D. (1)(2)(3) | 66 | Director | Dr. Stevens has served as a member of our Board of Directors since June 2011. Dr. Stevens is currently a board member of Cetya, Inc., a privately held development stage pharmaceutical company and of Micro-Imaging Solutions, LLC, a private medical device company. He has served on the boards of several other public and private life science companies, including Cedus, Inc., (2006-2014), Poniard Pharmaceuticals, Inc. (2006-2012), Aqua Bounty Technologies, Inc. (2002-2012), and Smart Drug Systems, Inc. (1999-2006), and was an advisor to Bay City Capital from 1999-2006. Dr. Stevens was previously President and CEO of Deprenyl Animal Health, Inc., a public veterinary pharmaceutical company, from 1990 to 1998, and Vice President, Research and Development, of Agrion Corp., a private biotechnology company, from 1986 to 1988. He began his career in pharmaceutical research and development at the former Upjohn Company, where he contributed to the preclinical evaluation of Xanax and Halcion. Dr. Stevens received B.S. and D.V.M. degrees from Washington State University, and a Ph.D. in Comparative Pathology from the University of California, Davis. He is a Diplomate of the American College of Veterinary Pathologists, and serves as a consulting experimental pathologist to Premier Laboratory, LLC. | June 2011 | ||||
52
Name | Age | Position With Ampio | Principal Occupation and Areas of Relevant Experience For Directors | Director Since | ||||
| Dr. Stevens has worked in the pharmaceutical and biotechnology industries since 1978. Dr. Stevens' experience in executive management in the pharmaceutical industry, and knowledge of the medical device industry led to the conclusion of our Board of Directors that he should serve as a director of our company in light of our business and structure. | ||||||||
| Dr. Vaughan L. Clift | 54 | Chief Regulatory Affairs Officer | Dr. Clift has been employed by us since March 2010 and was employed by Life Sciences from May 2009 until March 2010. From 2005 to 2009, Dr. Clift was the chief executive officer of Detectachem LLC, a Houston, Texas-based manufacturer of a hand-held explosive and narcotics detection device. Dr. Clift was the Vice President of Operations, including all FDA regulatory matters, for Isolagen from 2002 until 2005. From January 2001 to May 2002, Dr. Clift researched home oxygen therapy systems while developing an oxygen system for NASA. From July 1997 to January 2001, he was Chief Scientist of DBCD, Inc., a medical device company that manufactures a range of blood diagnostic products for the human and veterinary markets. From May 1992 to June 1997, Dr. Clift was Chief Scientist for the Science Payload Development, Engineering and Operations project at Lockheed Martin's Human Spaceflight Division. Dr. Clift has received a number of international and federal awards and was nominated as one of NASA's top ten inventors in 1995. | |||||
| Gregory A. Gould | 49 | Chief Financial Officer, Treasurer and Secretary | Mr. Gould has been employed by us since June 2014. Prior to joining us, he provided financial and operational consulting services to the biotech industry through his consulting company, Gould LLC from April 2012 until June 2014. Mr. Gould was Chief Financial Officer, Treasurer and Secretary of SeraCare Life Sciences, Inc. from November 2006 until the company was sold to Linden Capital Partners in April 2012. During the period from July 2011 until April 2012 Mr. Gould also served as the Interim President and Chief Executive Officer of SeraCare Life Sciences. Mr. Gould has held several other executive positions at publicly traded life sciences companies including the Chief Financial Officer role at Atrix Laboratories, Inc., an emerging specialty pharmaceutical company focused on advanced drug delivery. During Mr. Gould's tenure at Atrix he was instrumental in the negotiation and sale of the company to QLT, Inc. for over $855 million. He also played a critical role in the management of several licensing agreements including the global licensing agreement with Sanofi-Synthelabo of the Eligard ® products. Mr. Gould was the Chief Financial Officer at Colorado MedTech, Inc., a publicly traded medical device design and manufacturing company where he negotiated the transaction to sell the company to KRG Capital Partners. Mr. Gould began his career as an auditor with Arthur Andersen, LLP. He currently serves on the board of directors of CytoDyn, Inc., a publicly traded drug development company pursuing ant-viral agents for the treatment of HIV. Mr. Gould graduated from the University of Colorado with a BS in Business Administration and is a Certified Public Accountant. | |||||
53
| (1) | Member of our Audit Committee |
| (2) | Member of our Compensation Committee |
| (3) | Member of our Nominating and Governance Committee |
Family Relationships
There are no family relationships between any of our directors. Raphael Bar-Or, a non-executive officer, is the son of David Bar-Or, our chief scientific officer and a director. Rick Giles, a former non-executive employee, is the son of Richard B. Giles, one of our directors. For our subsidiary Aytu that was spun-off in January 2016, Jarrett Disbrow, is the Chief Operating Officer of Aytu, and the brother of Joshua Disbrow, the Chief Executive Officer of Aytu.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires our executive officers, directors and persons who beneficially own greater than 10% of our Common Stock to file certain reports, Forms 3, 4 and 5, with the SEC with respect to ownership and changes in ownership of our Common Stock. To our knowledge, no shareholder beneficially owns more than 10% of our Common Stock. Based solely on our review of the copies of such forms received by us, or written representations from certain reporting persons, we believe during the period from January 1, 2015 to December 31, 2015, all filing requirements applicable to its officers, directors and 10% beneficial owners were complied with.
Code of Business Conduct and Ethics
We have adopted a code of business conduct and ethics that is applicable to all of our employees, officers and directors. The code is available on our web site, www.ampiopharma.com , under the "Investor Relations" tab. We intend to disclose future amendments to, or waivers from, certain provisions of our code of ethics, if any, on the above website within four business days following the date of such amendment or waiver.
Meetings
During the year ended December 31, 2015, there were held (i) eleven meetings of the Board of Directors, (ii) eight meetings of the Audit Committee, (iii) eight meetings of the Compensation Committee, and (iv) one meeting of the Nominating and Governance Committee. No incumbent director attended fewer than seventy-five percent (75%) of the aggregate of (1) the total number of meetings of the Board, and (2) the total number of meetings held by all committees of the Board during the period that such director served.
Annual Meeting Attendance, Executive Sessions and Shareholder Communications
Since 2011, our policy has been that directors attend the annual meeting of stockholders. We previously did not have a policy concerning director attendance at annual meetings. Commencing in 2011, our policy has been that our non-employee directors are also required to meet in separate sessions without management on a regularly scheduled basis four times a year. Generally, these meetings are expected to take place in conjunction with regularly scheduled meetings of the Board throughout the year in conjunction with committee meetings.
54
We have not implemented a formal policy or procedure by which our shareholders can communicate directly with our Board of Directors. Nevertheless, every effort has been made to ensure that the views of shareholders are heard by the Board of Directors or individual directors, as applicable, and that appropriate responses are provided to shareholders in a timely manner. We believe that we are responsive to shareholder communications, and therefore have not considered it necessary to adopt a formal process for shareholder communications with our Board. During the upcoming year, our Board will continue to monitor whether it would be appropriate to adopt such a policy. Communications will be distributed to the Board, or to any individual director or directors as appropriate, depending on the facts and circumstances outlined in the communications. Items that are unrelated to the duties and responsibilities of the Board may be excluded, such as:
| • | junk mail and mass mailings |
| • | resumes and other forms of job inquiries |
| • | surveys; and |
| • | solicitations or advertisements. |
In addition, any material that is unduly hostile, threatening, or illegal in nature may be excluded, provided that any communication that is excluded will be made available to any outside director upon request.
Involvement in Certain Legal Proceedings
No director, executive officer, promoter or control person of our Company has, during the last ten years: (i) been convicted in or is currently subject to a pending a criminal proceeding (excluding traffic violations and other minor offenses); (ii) been a party to a civil proceeding of a judicial or administrative body of competent jurisdiction and as a result of such proceeding was or is subject to a judgment, decree or final order enjoining future violations of, or prohibiting or mandating activities subject to any Federal or state securities or banking or commodities laws including, without limitation, in any way limiting involvement in any business activity, or finding any violation with respect to such law, nor (iii) any bankruptcy petition been filed by or against the business of which such person was an executive officer or a general partner, whether at the time of the bankruptcy or for the two years prior thereto.
We are not engaged in, nor are we aware of any pending or threatened, litigation in which any of our directors, executive officers, affiliates or owner of more than 5% of our common stock is a party adverse to us or has a material interest adverse to us.
Leadership Structure of the Board
The Board of Directors does not currently have a policy on whether the same person should serve as both the chief executive officer and chairman of the board or, if the roles are separate, whether the chairman should be selected from the non-employee directors or should be an employee. The Board believes that it should have the flexibility to make these determinations at any given point in time in the way that it believes best to provide appropriate leadership for us at that time. Our current chairman, Michael Macaluso, was appointed our chief executive officer effective January 2012. Mr. Macaluso has served as a member of our Board since March 2010, and has been a member of the Board of Directors of Life Sciences from December 2009.
Risk Oversight
The Board oversees risk management directly and through its committees associated with their respective subject matter areas. Generally, the Board oversees risks that may affect our business as a whole, including operational matters. The Audit Committee is responsible for oversight of our accounting and financial reporting processes and also discusses with management our financial statements, internal controls and other accounting and related matters. The Compensation Committee oversees certain risks related to compensation programs and the Nominating and Governance Committee oversees certain corporate governance risks. As part of their roles in overseeing risk management, these committees periodically report to the Board regarding briefings provided by management and advisors as well as the committees' own analysis and conclusions regarding certain risks faced by us. Management is responsible for implementing the risk management strategy and developing policies, controls, processes and procedures to identify and manage risks.
Board Committees
Our Board of Directors has an Audit Committee, a Compensation Committee and a Nominating and Governance Committee, each of which has the composition and the responsibilities described below. The Audit Committee, Compensation Committee and Nominating and Governance Committee all operate under charters approved by our Board of Directors, which charters are available on our website.
55
Audit Committee. Our Audit Committee oversees our corporate accounting and financial reporting process and assists the Board of Directors in monitoring our financial systems and our legal and regulatory compliance. Our Audit Committee is responsible for, among other things:
| • | selecting and hiring our independent auditors; |
| • | appointing, compensating and overseeing the work of our independent auditors; |
| • | approving engagements of the independent auditors to render any audit or permissible non-audit services; |
| • | reviewing the qualifications and independence of the independent auditors; |
| • | monitoring the rotation of partners of the independent auditors on our engagement team as required by law; |
| • | reviewing our financial statements and reviewing our critical accounting policies and estimates; |
| • | reviewing the adequacy and effectiveness of our internal controls over financial reporting; and |
| • | reviewing and discussing with management and the independent auditors the results of our annual audit, our quarterly financial statements and our publicly filed reports. |
The members of our Audit Committee are Messrs. Giles, Coelho and Stevens. Mr. Giles is our Audit Committee chairman and was appointed to our Audit Committee in August 2010. Our Board of Directors has determined that each member of the Audit Committee meets the financial literacy requirements of the national securities exchanges and the SEC, and Mr. Giles qualifies as our Audit Committee financial expert as defined under SEC rules and regulations. Our Board of Directors has concluded that the composition of our Audit Committee meets the requirements for independence under the current requirements of the NYSE MKT and SEC rules and regulations. We believe that the functioning of our Audit Committee complies with the applicable requirements of SEC rules and regulations, and applicable requirements of the NYSE MKT.
Compensation Committee. Our Compensation Committee oversees our corporate compensation policies, plans and programs. The Compensation Committee is responsible for, among other things:
| • | reviewing and recommending policies, plans and programs relating to compensation and benefits of our directors, officers and employees; |
| • | reviewing and recommending compensation and the corporate goals and objectives relevant to compensation of our chief executive officer; |
| • | reviewing and approving compensation and corporate goals and objectives relevant to compensation for executive officers other than our chief executive officer; |
| • | evaluating the performance of our executive officers in light of established goals and objectives; |
| • | developing in consultation with our Board of Directors and periodically reviewing a succession plan for our chief executive officer; and |
| • | administering our equity compensations plans for our employees and directors. |
The members of our Compensation Committee are Messrs. Coelho, Giles and Stevens. Mr. Coelho is the chairman of our Compensation Committee. Each member of our Compensation Committee is a non-employee director, as defined in Rule 16b-3 promulgated under the Exchange Act, is an outside director, as defined pursuant to Section 162(m) of the Internal Revenue Code of 1986, as amended, or the Code, and satisfies the independence requirements of the NYSE MKT. We believe that the composition of our Compensation Committee meets the requirements for independence under, and the functioning of our Compensation Committee complies with, any applicable requirements of the NYSE MKT and SEC rules and regulations.
Our Compensation Committee and our Board of Directors have not yet established a succession plan for our chief executive officer.
In fulfilling its responsibilities, the Committee is permitted under the Compensation Committee charter to delegate any or all of its responsibilities to a subcommittee comprised of members of the Compensation Committee or the Board, except that the Committee may not delegate its responsibilities for any matters that involve compensation of any officer or any matters where it has determined such compensation is intended to comply with Section 162(m) of the Code or is intended to be exempt from Section 16(b) under the Exchange Act pursuant to Rule 16b-3 by virtue of being approved by a committee of independent or nonemployee directors.
56
Nominating and Governance Committee . Our Nominating and Governance Committee oversees and assists our Board of Directors in reviewing and recommending corporate governance policies and nominees for election to our Board of Directors. The Nominating and Governance Committee is responsible for, among other things:
| • | evaluating and making recommendations regarding the organization and governance of the Board of Directors and its committees; |
| • | assessing the performance of members of the Board of Directors and making recommendations regarding committee and chair assignments; |
| • | recommending desired qualifications for Board of Directors membership and conducting searches for potential members of the Board of Directors; and |
| • | reviewing and making recommendations with regard to our corporate governance guidelines. |
The members of our Nominating and Governance Committee are currently Messrs. Giles, Stevens and Coelho. Mr. Coelho is the chairman of our Nominating and Governance Committee. Our Board of Directors has determined that each member of our Nominating and Governance Committee is independent within the meaning of the independent director guidelines of the NYSE MKT.
Our Board of Directors may from time to time establish other committees.
Non-Employee Director Compensation
Our Compensation Committee established the following fees for payment to members of our Board of Directors or committees, as the case may be for the fiscal year ended December 31, 2015:
Committee or Committees | Cash Compensation | Common Stock | ||||||||
Board Annual Retainer: | ||||||||||
Chairman | $ | 20,000 | ||||||||
Each non-employee director | 10,000 | |||||||||
Board Meeting Fees: | ||||||||||
Each meeting attended in-person | $ | 1,500 | ||||||||
Each meeting attended telephonically or via web | 1,000 | |||||||||
Committee Annual Retainer: | ||||||||||
Chairman of each committee | Audit; Compensation; Nominating and Governance | $ | 20,000 | |||||||
Each non-chair member | Audit | 12,000 | ||||||||
Each non-chair member | Compensation; Nominating and Governance | 10,000 | ||||||||
Committee Chairman Meeting Fees: | ||||||||||
Each meeting attended in-person | Audit; Compensation; Nominating and Governance | $ | 2,500 | |||||||
Each meeting attended telephonically or via web | Audit; Compensation; Nominating and Governance | 1,500 | ||||||||
Committee Member Meeting Fees: | ||||||||||
Each meeting attended in-person | Audit; Compensation; Nominating and Governance | $ | 1,500 | |||||||
Each meeting attended telephonically or via web | Audit; Compensation; Nominating and Governance | 1,000 | ||||||||
Annual Stock Award: | $ | 10,000 | ||||||||
In December 2015, the Compensation Committee amended the Non-Employee Director Compensation for fiscal 2016 by increasing the annual stock award to $20,000 and granting each Director options to purchase 30,000 shares of our stock on the date of our annual shareholder meeting of stockholders, vesting monthly over the succeeding twelve months.
Director Compensation for 2015
The table below summarizes the compensation paid by us to non-employee directors for the year ended December 31, 2015. Our employee directors do not receive additional compensation for their services as a member of our Board of Directors.
57
| Name | Fees Earned or Paid in Cash | Stock Option Awards (1)(2) | Stock Awards (3) | All Other (4) | Total | |||||||||||||||
Philip H. Coelho | $ | 102,000 | $ | 79,133 | $ | 10,000 | $ | 108,081 | $ | 299,214 | ||||||||||
Richard B. Giles | $ | 94,500 | $ | 79,133 | $ | 10,000 | $ | 108,081 | $ | 291,714 | ||||||||||
David Stevens, PhD | $ | 74,583 | $ | 79,133 | $ | 10,000 | $ | 108,081 | $ | 271,797 | ||||||||||
| (1) | In December 2015, Messrs. Coelho, Giles and Dr. Stevens were granted options to purchase 30,000 shares of common stock. These options have an exercise price of $3.33 per share, which was the closing price of our common stock on the date of grant (December 12, 2015). These options fully vest on December 12, 2016 and have a term of 10 years from the grant date. The amounts in this column reflect the grant date fair values of the stock awards based on the last reported sale price of the common stock at the dates of grant. Please see Item 15 of Part IV, "Notes to Consolidated Financial Statements – Note 10 – Equity Instruments." |
| (2) | At December 31, 2015, Messrs. Coelho, Giles and Dr. Stevens held options to acquire 595,554, 680,000 and 255,000 shares of common stock, respectively. |
| (3) | Annual stock award. In January 2015, each of Messrs. Coelho, Giles and Dr. Stevens was awarded 2,666 shares of common stock pursuant to the 2010 Plan, at a price of $3.75 per share equivalent to $10,000, which was the closing price of our common stock on the date of grant (January 2, 2015). |
| (4) | During the year ended December 31, 2015, Messrs. Coelho, Giles and Dr. Stevens each received options to acquire 125,000 shares of Aytu common stock with an exercise price of $1.51 which were fully vested at the grant date and have a 10-year term with a corresponding fair value of $108,081. Additionally, each director received a cash payment of $350 for option shares equal to the difference between the consideration payable per share of common stock pursuant to the Luoxis Rosewind Merger and the exercise price of the option. |
| Item 11. | Executive Compensation |
Executive Compensation
Compensation Discussion and Analysis
Overview. The following Compensation Discussion and Analysis describes the material elements of compensation for our executives identified in the Summary Compensation Table, or the Named Executive Officers. The Compensation Committee of the Board of Directors assists the Board of Directors in discharging the Board's responsibilities regarding compensation of our executives, including the Named Executive Officers. In particular, the Compensation Committee makes recommendations to the Board of Directors regarding the corporate goals and objectives relevant to executive compensation, evaluates executives' performance in light of such goals and objectives, and recommends the executives' compensation levels to the Board of Directors based on such evaluations. The Compensation Committee's recommendations relating to compensation matters are subject to approval by the Board.
Compensation Philosophy and Objectives . Our executive compensation program is designed to retain our executive officers and to motivate them to increase stockholder value on both an annual and longer term basis. These objectives are to be accomplished primarily by positioning us to maximize our product development efforts and to transform, over time, those efforts into collaboration revenues and income. To that end, compensation packages include significant incentive forms of stock-based compensation to ensure that each executive officer's interest is aligned with the interests of our stockholders.
Named Executive Officers
For our most recently completed fiscal year (the year ended December 31, 2015), our Named Executive Officers were: (i) Michael Macaluso, our Chief Executive Officer, who has served as our Chief Executive Officer since January 2012, (ii) Gregory A. Gould, our Chief Financial Officer, who has served as our Chief Financial officer since June 2014, (iii) David Bar-Or, M.D., our current Chief Scientific Officer, who has served as our Chief Scientific Officer since March 2010, (iv) Vaughan Clift, our current Chief Regulatory Affairs Officer, who has served as our Chief Regulatory Affairs Officer since March 2010, and (v) Joshua Disbrow, our former Chief Operating Officer, who served as our Chief Operating Officer from December 2012 until April 2015 and has been Aytu's Chief Executive Officer from April 2015 until present. We had no other executive officers serving during the year ended December 31, 2015.
58
Executive Compensation Components
Our compensation program for our Named Executive Officers consists of three components: (i) a base salary, (ii) discretionary bonuses based on performance, and (iii) equity compensation. Each of these components is reflected in the Summary Compensation Table below.
Salaries . The initial cash salaries paid to Messrs. Macaluso, Gould, Disbrow and Drs. Bar-Or and Clift were established at the time they became officers. Each of these persons has an employment agreement with us, a copy of which is an exhibit to, or incorporated by reference herein. Since the respective dates of their becoming Named Executive Officers, any increases in the salaries of our Named Executive Officers have been made at the discretion of the Compensation Committee. Mr. Macaluso and Dr. Bar-Or receive no additional compensation for serving on our Board of Directors.
Cash Incentive Compensation . Cash incentive or bonus compensation is discretionary under our employment agreements with Drs. Bar-Or and Clift and Messrs. Macaluso, Gould and Disbrow. However, each employment agreement contains performance objectives tailored to the individual officer's duties, and our performance. All cash incentive compensation grants are intended to be paid in accordance with Section 162(m) of the Code. For 2015, we awarded a cash bonus to Mr. Macaluso of $5,000, to Dr. Bar-Or of $5,000, to Dr. Clift of $5,000, Mr. Disbrow of $122,500 and to Mr. Gould of $98,750 which were awarded on a discretionary basis by the Compensation Committee based on the Compensation Committee's assessment of 2015 performance.
Equity Compensation . In 2015, we granted stock options to certain of our officers, directors and consultants for their services, all of which were granted pursuant to written agreements under the 2010 Plan. Included in such stock options were 470,000 performance-based options granted to two executive officers which are based upon the outcome of the ongoing Ampion trial. All future grants are expected to be made under the 2010 Plan. The vesting period for option grants vary.
Perquisites . We offer health benefits for all of our employees. None of our Named Executive Officers receives any further perquisites.
Why Each Element of Compensation is Paid; How the Amount of Each Element is Determined . The Compensation Committee intends to pay each of these elements in order to ensure that a desirable overall mix is established between base compensation and incentive compensation, cash and non-cash compensation, and annual and long-term compensation. The Compensation Committee also intends to evaluate on a periodic basis the overall competitiveness of our executive compensation packages as compared to packages offered in the marketplace for which we compete with executive talent. Overall, our Compensation Committee believes that our executive compensation packages are currently appropriately balanced and structured to retain and motivate our Named Executive Officers, while necessarily taking into account our presently limited financial resources.
How Each Compensation Element Fits into Overall Compensation Objectives and Affects Decisions Regarding Other Elements. In establishing compensation packages for executive officers, numerous factors are considered, including the particular executive's experience, expertise and performance, our operational and financial performance, and compensation packages available in the marketplace for similar positions. In arriving at amounts for each component of compensation, our Compensation Committee strives to strike an appropriate balance between base compensation and incentive compensation. The Compensation Committee also endeavors to properly allocate between cash and non-cash compensation and between annual and long-term compensation.
Risk Assessment . Our Compensation Committee has reviewed our compensation program and believes that the program, including our cash incentive compensation and equity incentive compensation, does not encourage our Named Executive Officers to engage in any unnecessary or excessive risk-taking. As a result, the Compensation Committee has to date not implemented a provision for recovery by us of cash or incentive compensation bonuses paid to our Named Executive Officers.
Role of Compensation Consultants in Executive Compensation Decisions . The Compensation Committee has the authority to retain the services of third-party executive compensation specialists in connection with the establishment of our compensation policies. The Compensation Committee did not use a compensation consultant in connection with setting 2014 executive compensation, and relied upon the professional and market experience of the Committee members in determining 2014 executive compensation. The Compensation Committee may engage a compensation consultant in the future if it deems such services to be appropriate and cost-justified.
Role of Executives in Executive Compensation Decisions. The Compensation Committee seeks input and specific recommendations from our Chief Executive Officer when discussing the performance of, and compensation levels for, executives other than himself. The Chief Executive Officer provides recommendations to the Compensation Committee regarding each executive officer's level of individual achievement other than himself. However, he is not a member of the Compensation Committee and does not vote. The Compensation Committee also works with our Chief Executive Officer and our Chief Financial Officer to evaluate the financial, accounting, tax and retention implications of our various compensation programs. Neither our Chief Executive Officer nor any of our other executives participates in deliberations relating to his or her own compensation.
Tax and Accounting Implications
Deductibility of Executive Compensation . Section 162(m) of the Code limits the tax deduction to $1 million for compensation paid to certain executives of public companies. However, performance-based compensation that has been approved by stockholders is not
59
subject to the $1 million limit under Section 162(m) if, among other requirements, the compensation is payable only upon attainment of pre-established, objective performance goals, and the Board of Directors committee that establishes such goals consists only of "outside directors." All members of the Compensation Committee qualify as outside directors. Additionally, stock options will qualify for the performance-based exception where, among other requirements, the exercise price of the option is not less than the fair market value of the stock on the date of the grant, and the plan includes a per-executive limitation on the number of shares for which options may be granted during a specified period.
Compensation Committee Interlocks and Insider Participation
None of the members of our Compensation Committee is an officer or employee of our Company. None of our executive officers currently serves, or in the past year has served, as a member of the Board of Directors or Compensation Committee of any entity that has one or more executive officers serving on our Board of Directors or Compensation Committee.
Compensation Committee Report
The Compensation Committee of the Board of Directors has reviewed and discussed the Compensation Discussion and Analysis required by Item 402(b) of Regulation S-K with management and, based on such review and discussions, the Compensation Committee recommended to the Board of Directors that the Compensation Discussion and Analysis be included in this Annual Report on Form 10-K and in the Company's Proxy Statement.
| Submitted by the Compensation Committee of the Board of Directors |
| Philip H. Coelho |
Richard B. Giles David R. Stevens, Ph.D. |
60
The following table sets forth all cash compensation earned, as well as certain other compensation paid or accrued in 2015, 2014 and 2013, to each of the following named executive officers.
Summary Compensation of Named Executive Officers
Name and Principal Position (a) | Year (b) | Salary ($) (c) | Bonus ($) (d) | Stock Award ($) (e) | Option Award ($)(1) (f) | Non-Equity Incentive Plan Compensation ($) (g) | Change in Pension Value and Nonqualified Deferred Compensation Earnings ($) (h) | All Other Compensation ($) (i) | Total ($) (j) | |||||||||||||||||||||||||||
Current Named | ||||||||||||||||||||||||||||||||||||
Michael Macaluso | ||||||||||||||||||||||||||||||||||||
Chief Executive | 2015 | 300,000 | 5,000 | - | - | - | - | 108,433 | (2) (4) | 413,433 | ||||||||||||||||||||||||||
effective January | 2014 | 300,000 | 155,000 | - | 1,095,433 | - | - | - | 1,550,433 | |||||||||||||||||||||||||||
| 2013 | 221,250 | (3) | 155,000 | - | - | - | - | - | 376,250 | |||||||||||||||||||||||||||
David Bar-Or, M.D. | ||||||||||||||||||||||||||||||||||||
Chief Scientic Officer and | 2015 | 300,000 | 5,000 | - | - | - | - | 224,617 | (2) (4) | 529,617 | ||||||||||||||||||||||||||
Former Chairman | 2014 | 300,000 | 5,000 | - | 1,538,943 | - | - | - | 1,843,943 | |||||||||||||||||||||||||||
| 2013 | 300,000 | 155,000 | - | 469,352 | - | - | - | 924,352 | ||||||||||||||||||||||||||||
Gregory A. Gould | ||||||||||||||||||||||||||||||||||||
Chief Financial | 2015 | 250,000 | 98,750 | (14) | - | 212,162 | - | - | 232,801 | (4) | 793,713 | |||||||||||||||||||||||||
since June 2014 | 2014 | 138,450 | (5) | 5,000 | - | 1,435,243 | - | - | 21,620 | (6) | 1,600,313 | |||||||||||||||||||||||||
Vaughan Clift, M.D. | ||||||||||||||||||||||||||||||||||||
Chief Regulatory | 2015 | 250,000 | 5,000 | - | - | - | - | - | 255,000 | |||||||||||||||||||||||||||
Officer | 2014 | 250,000 | 5,000 | - | 872,067 | - | - | - | 1,127,067 | |||||||||||||||||||||||||||
| 2013 | 250,000 | 130,000 | - | 265,966 | - | - | - | 645,966 | ||||||||||||||||||||||||||||
Mark D. McGregor | ||||||||||||||||||||||||||||||||||||
Chief Financial | 2015 | - | - | - | 125,901 | (7) | - | - | - | 125,901 | ||||||||||||||||||||||||||
since April 2011 | 2014 | 103,125 | (8) | 29,000 | - | - | - | - | 75,000 | (9) | 207,125 | |||||||||||||||||||||||||
| 2013 | 152,216 | 20,000 | - | 212,694 | (9) | - | - | - | 384,910 | |||||||||||||||||||||||||||
Joshua R. Disbrow | ||||||||||||||||||||||||||||||||||||
Former Chief | 2015 | 255,587 | (13) | 122,500 | (15) | 691,948 | (10) | - | - | 558,722 | (4) | 1,628,757 | ||||||||||||||||||||||||
and Chief Executive Officer | 2014 | 245,000 | 180,000 | (11) | - | - | - | - | 425,000 | |||||||||||||||||||||||||||
of Aytu BioScience, | 2013 | 228,958 | (12) | 127,500 | - | - | - | - | - | 356,458 | ||||||||||||||||||||||||||
| (1) | Option awards are reported at fair value at the date of grant. See Item 15 of Part IV, "Notes to Consolidated Financial Statements – Note 10 – Equity Instruments." |
| (2) | Compensation includes a cash payment per option share equal to the difference between the consideration payable per share of common stock pursuant to the Luoxis Rosewind Merger and the exercise price of the option (total payment was $27,000) and the fair value of Aytu options granted in November 2015. |
| (3) | Mr. Macaluso's salary was increased from $195,000 to $300,000 annually effective October 2013. |
| (4) | Compensation includes the fair value of Aytu options granted in November 2015. |
| (5) | Mr. Gould was appointed to Chief Financial Officer effective June 2014. |
| (6) | Compensation related to Mr. Gould's expense to move his family to Colorado. |
| (7) | Mr. McGregor's options were modified in May 2015 which extended the expiration date an additional year to August 15, 2016. |
| (8) | Mr. McGregor resigned as Chief Financial Officer effective June 2014. |
| (9) | Mr. McGregor's retirement severance and modified options which accelerated the vesting of 96,181 options and extended the exercise period from 90 days after termination to August 15, 2015 for 275,000 options. All of the $130,000 of expense related to this modification was recognized in 2014. |
| (10) | Mr. Disbrow's options were modified in April 2015 which accelerated the vesting and extended the exercise period from ninety days after termination to April 15, 2020. |
| (11) | In 2014, Mr. Disbrow received a bonus of $175,000 related to his superior performance as Chief Executive Officer of Luoxis. |
| (12) | Effective June 2013, Mr. Disbrow began receiving a $35,000 annual salary from Luoxis. |
| (13) | Mr. Disbrow resigned as Chief Operating Officer effective April 2015 and took the position of Chief Executive Officer at Aytu. |
| (14) | Mr. Gould received $25,000 of this bonus which related to his performance for Aytu. |
| (15) | Mr. Disbrow received a bonus of $122,500 related to his superior performance as Chief Executive Officer of Aytu. |
Our executive officers are reimbursed by us for any out-of-pocket expenses incurred in connection with activities conducted on our behalf.
61
Grants of Plan-Based Awards - Ampio
The following table sets forth certain information regarding grants of plan-based awards to the Named Executive Officers as of December 31, 2015:
| Name | Grant Date | All Other Option Awards: Number of Securities Underlying Options (#) | Exercise Price of Option Awards ($/Share) | Grant Date Fair Value of Option Awards | ||||||||||||
Current Named Exective Officers | ||||||||||||||||
Gould, Gregory | 7/30/2015 | 100,000 | $ | 2.60 | $ | 212,162 | ||||||||||
Clift, Vaughan | 7/31/2015 | 170,000 | $ | 2.68 | - | |||||||||||
Bar-Or, David M.D. | 8/3/2015 | 300,000 | $ | 2.60 | - | |||||||||||
In April 2015, we modified options held by our former Chief Operating Officer which accelerated vesting of 27,790 options and extended the exercise period from 90 days after termination to April 15, 2020 for 400,000 options. All of the $692,000 expense related to this modification was recognized in the period ended June 30, 2015.
In July 2015, each of Drs. Bar-Or and Clift was granted options to purchase 300,000 and 170,000 shares of common stock, respectively. These options have an exercise price of $2.60 and $2.68 per share, respectively, which was the closing price of our common stock on the date of grant (August 3, 2015 and July 31, 2015). These options vest on the date that we meet all endpoints in connection with the Ampion clinical trial as determined in the sole discretion of the Compensation Committee. Also, in July 2015, Mr. Gould was granted options to purchase 100,000 shares of common stock. These options have an exercise price of $2.60 per share which was the closing price of our common stock on the date of grant July 30, 2015. These options vest annually over three years beginning on the date of grant.
Grants of Plan-Based Awards - Aytu
The following table sets forth certain information regarding grants of plan-based awards to the Named Executive Officers as of December 31, 2015:
| Name | Grant Date | All Other Option Awards: Number of Securities Underlying Options (#) | Exercise Price of Option Awards ($/Share) | Grant Date Fair Value of Option Awards | ||||||||||||
Current Named Exective Officers | ||||||||||||||||
Macaluso, Michael | 11/11/2015 | 125,000 | $ | 1.51 | $ | 108,081 | ||||||||||
Bar-Or, David M.D. | 11/11/2015 | 250,000 | $ | 1.51 | $ | 216,163 | ||||||||||
Gould, Gregory | 11/11/2015 | 250,000 | $ | 1.51 | $ | 232,801 | ||||||||||
Disbrow, Joshua | 11/11/2015 | 600,000 | $ | 1.51 | $ | 558,722 | ||||||||||
62
Outstanding Equity Awards
The following table provides a summary of equity awards outstanding for each of the Named Executive Officers as of December 31, 2015:
| Option Awards | Stock Awards | |||||||||||||||||||||||||||||||||||||||
Name (a) | Number of Securities Underlying Unexercised Options Exercisable (#) (b) | Number of Securities Underlying Unexercised Options Unexercisable (#) (c) | Equity Incentive Plan Awards: Number of Securities Underlying Unexercised Unearned Options (#) (d) | Option Exercise Price ($) (e) | Option Expiration Date (f) | Number of Shares or Units of Stock That Have Not Vested (#) (g) | Market Value of Shares or Units of Stock That Have Not Vested ($) (h) | Equity Incentive Plan Awards: Number of Unearned Shares, Units or Other Rights That Have Not Vested (#) (i) | Equity Incentive Plan Awards: Market or Payout Value of Unearned Shares, Units or Other Rights That Have Not Vested ($) (j) | |||||||||||||||||||||||||||||||
Current Named Exective Officers | ||||||||||||||||||||||||||||||||||||||||
Michael Macaluso | (1) | 133,333 | 266,667 | - | 3.46 | 12/20/2024 | - | - | - | - | ||||||||||||||||||||||||||||||
Michael Macaluso | 250,000 | - | - | 2.76 | 5/7/2022 | - | - | - | - | |||||||||||||||||||||||||||||||
Michael Macaluso | 220,000 | - | - | 1.03 | 8/12/2020 | - | - | - | - | |||||||||||||||||||||||||||||||
Michael Macaluso | 180,000 | - | - | 1.70 | 8/27/2020 | - | - | - | - | |||||||||||||||||||||||||||||||
David Bar-Or, M.D. | (2) | - | 300,000 | - | 2.60 | 8/3/2025 | - | - | - | - | ||||||||||||||||||||||||||||||
David Bar-Or, M.D. | 300,000 | - | - | 6.48 | 8/11/2024 | - | - | - | - | |||||||||||||||||||||||||||||||
David Bar-Or, M.D. | 300,000 | - | - | 6.15 | 7/15/2023 | - | - | - | - | |||||||||||||||||||||||||||||||
David Bar-Or, M.D. | 200,000 | - | - | 2.76 | 5/7/2022 | - | - | - | - | |||||||||||||||||||||||||||||||
David Bar-Or, M.D. | 400,000 | - | - | 1.03 | 8/12/2020 | - | - | - | - | |||||||||||||||||||||||||||||||
Gregory A. Gould | (4) | 200,000 | 100,000 | - | 7.14 | 7/30/2025 | - | - | - | - | ||||||||||||||||||||||||||||||
Gregory A. Gould | (3) | 33,333 | 66,667 | - | 2.60 | 6/10/2024 | - | - | - | - | ||||||||||||||||||||||||||||||
Vaughan Clift, M.D. | (2) | - | 170,000 | - | 2.68 | 8/3/2025 | - | - | - | - | ||||||||||||||||||||||||||||||
Vaughan Clift, M.D. | 170,000 | - | - | 6.48 | 8/11/2024 | - | - | - | - | |||||||||||||||||||||||||||||||
Vaughan Clift, M.D. | 170,000 | - | - | 6.15 | 7/15/2023 | - | - | - | - | |||||||||||||||||||||||||||||||
Vaughan Clift, M.D. | 150,000 | - | - | 2.76 | 5/7/2022 | - | - | - | - | |||||||||||||||||||||||||||||||
Vaughan Clift, M.D. | 365,000 | - | - | 1.03 | 8/12/2020 | - | - | - | - | |||||||||||||||||||||||||||||||
Mark D. McGregor | 75,000 | - | - | 2.76 | 8/15/2016 | - | - | - | - | |||||||||||||||||||||||||||||||
Mark D. McGregor | 100,000 | - | - | 2.50 | 8/15/2016 | - | - | - | - | |||||||||||||||||||||||||||||||
Mark D. McGregor | 100,000 | - | - | 8.62 | 8/15/2016 | - | - | - | - | |||||||||||||||||||||||||||||||
Joshua R. Disbrow | 400,000 | - | - | 3.53 | 4/15/2020 | - | - | - | - | |||||||||||||||||||||||||||||||
| (1) | Unexercisable options vest annually and become fully vested January 1, 2017. |
| (2) | Unexercisable options vest upon the outcome of the ongoing Ampion trial (performance-based). |
| (3) | Unexercisable options vest annually and become fully vested July 30, 2017. |
| (4) | Unexercisable options vest annually and become fully vested June 10, 2016. |
On November 10, 2015, Aytu granted exercisable options at an exercise price of $1.51 to Mr. Macaluso and Dr. David Bar-Or. Mr. Macaluso received 125,000 options and Dr. David Bar-Or received 250,000. Also, on November 10, 2015, Aytu granted options that vest over three years at an exercise price of $1.51 to Mr. Gould and Mr. Disbrow. Mr. Gould received 250,000 options and Mr. Disbrow received 600,000 which vest annually and become fully vested on November 11, 2018.
Employment Agreements
We entered into an employment agreement with Mr. Michael Macaluso, our Chief Executive Officer, effective January 9, 2012 which provided for an annual salary of $195,000, with an initial term ending January 9 2015. On October 1, 2013, Ampio increased Mr. Macaluso's annual salary from $195,000 to $300,000. On December 20, 2014, Ampio extended the Employment Agreement of Mr. Macaluso for three additional years, expiring January 9, 2017. In connection with this Amendment, Mr. Macaluso was awarded 400,000 options to purchase our common stock at an exercise price of $3.46 vesting annually over three years beginning on January 1, 2015.
In August 2010, we entered into employment agreements with Dr. David Bar-Or, our Chief Scientific Officer, and Dr. Vaughan Clift, our Chief Regulatory Affairs Officer. The employment agreement with Dr. Bar-Or supersedes his prior agreement with Life Sciences. Dr. Clift's employment agreement was amended on October 1, 2010 and May 26, 2011. The terms of the employment agreements with Dr. Bar-Or and Dr. Clift are substantially identical except as noted below. Each agreement had an initial term ending July 31,
63
2013. The agreements provide for annual salaries of $300,000 for Dr. Bar-Or and $250,000 for Dr. Clift. On July 15, 2013, we extended the Employment Agreements of Dr. David Bar-Or and Dr. Vaughan Clift for one additional year, expiring July 31, 2014. In connection with these Amendments, Dr. Bar-Or and Dr. Clift were awarded 300,000 and 170,000 options, respectively, to purchase our common stock at an exercise price of $6.15 with 50% vesting upon grant and 50% after one year. On August 11, 2014, we extended the Employment Agreements of Dr. David Bar-Or and Dr. Vaughan Clift for one additional year, expiring July 31, 2015. In connection with these Amendments, Dr. Bar-Or and Dr. Clift were awarded 300,000 and 170,000 options, respectively, to purchase our common stock at an exercise price of $6.48 with 50% vesting upon grant and 50% after one year. On August 3 and July 31, 2015, we extended the Employment Agreements of Dr. Bar-Or and Dr. Clift, respectively, for one additional year, expiring July 31, 2016. In connection with these Amendments, Dr. Bar-Or and Dr. Clift were awarded 300,000 and 170,000 options, respectively, to purchase our common stock at exercise prices of $2.60 and $2.68, respectively, with such options vesting on the date that we meet all endpoints in connection with the Ampion clinical trial as determined in the sole discretion of our Compensation Committee.
We entered into an employment agreement with Mr. Joshua Disbrow, our former Chief Operating Officer, effective December 15, 2012. This agreement had an initial term ending December 15, 2015 and provided for an annual salary of $210,000. Mr. Disbrow also received an annual salary of $35,000 from Luoxis effective June 16, 2013. He terminated his position at Ampio Pharmaceuticals, Inc. in April 2015 and became the Chief Executive Officer of Aytu BioScience, Inc. Aytu entered into an employment agreement with Joshua Disbrow in connection with his employment as Aytu's Chief Executive Officer. The agreement is for a term of 24 months beginning on April 16, 2015, subject to termination by Aytu with or without Cause or as a result of officer's disability, or by Mr. Disbrow with or without Good Reason (as discussed below). Mr. Disbrow is entitled to receive $250,000 in annual salary, plus a discretionary performance bonus with a target of 125% of his base salary and 600,000 stock options with 50% vesting upon grant and the remainder vesting on the following two anniversaries of the grant date. Mr. Disbrow is also eligible to participate in the benefit plans maintained by Aytu from time to time, subject to the terms and conditions of such plans.
We entered into an employment agreement with Mr. Gregory Gould, our Chief Financial Officer, on June 10, 2014, which provided for an annual salary of $250,000, with an initial term ending June 10, 2017. In connection with this employment agreement, Mr. Gould was awarded 300,000 options to purchase common stock at an exercise price of $7.14 vesting annually over two years beginning on June 10, 2014.
Each officer is eligible to receive a discretionary annual bonus each year that will be determined by the Compensation Committee of the Board of Directors based on individual achievement and Company performance objectives established by the Compensation Committee. Included in those objectives, as applicable for the responsible officer, are (i) obtaining successful phase II and phase III clinical trial results, (ii) preparation and compliance with a fiscal budget, (iii) the launch of clinical trials for additional products approved by the Board of Directors, (iv) the sale of intellectual property not selected for clinical trials by us at prices, and times, approved by the Board of Directors and (v) making significant scientific discoveries acceptable to the Board of Directors. The targeted amount of Dr. Bar-Or and Dr. Clift and Messrs. Macaluso and Gould annual bonus is 50% of the applicable base salary, although the actual bonus may be higher or lower. Mr. Disbrow's target bonus is 125% of his base salary.
The employment agreements for Dr. Bar-Or and Dr. Clift provided for an initial grant of stock options in the amount of 700,000 (subsequently reduced to 400,000) and 365,000 options, respectively. Each option is exercisable for a period of ten years at an exercise price per share equal to the quoted closing price of our common stock on August 11, 2010. Mr. Disbrow was granted 450,000 (subsequently reduced to 400,000) stock options which upon his departure from Ampio, we modified by accelerating vesting of 27,790 options and extending the exercise period from 90 days after termination to April 15, 2020 for 400,000 options. All of the $692,000 expense related to this modification was recognized in the period ended June 30, 2015.
Potential Payments upon Termination or Change in Control
If the employment of Dr. Bar-Or, Dr. Clift, Mr. Disbrow or Mr. Gould is terminated at our election at any time, for reasons other than death, disability, cause (as defined in the agreement) or a voluntary resignation, or if an officer terminates his employment for good reason, the officer in question shall be entitled to receive a lump sum severance payment equal to two times his base salary and of the continued payment of premiums for continuation of the officer's health and welfare benefits pursuant to COBRA or otherwise, for a period of two years from the date of termination, subject to earlier discontinuation if the officer is eligible for comparable coverage from a subsequent employer. Mr. Macaluso is not entitled to any such termination payments pursuant to the terms of his employment agreement. All severance payments, less applicable withholding, are subject to the officer's execution and delivery of a general release of us and our subsidiaries and affiliates and each of their officers, directors, employees, agents, successors and assigns in a form acceptable to us, and a reaffirmation of the officer's continuing obligation under the propriety information and inventions agreement (or an agreement without that title, but which pertains to the officer's obligations generally, without limitation, to maintain and keep confidential all of our proprietary and confidential information, and to assign all inventions made by the officer to us, which inventions are made or conceived during the officer's employment). If the employment is terminated for cause, no severance shall be payable by us.
"Good Reason" means:
| • | a material reduction in the officer's overall responsibilities or authority or scope of duties; |
64
| • | a material reduction of the officer's compensation; or |
| • | relocation of the officer to a facility or location not within 40 miles of the state capitol building in Denver, Colorado. |
"Cause" means:
| • | willful malfeasance or willful misconduct in connection with employment; |
| • | conviction of, or entry of a plea of guilty or nolo contendere to, any crime other than a traffic violation or misdemeanor; |
| • | willful and deliberate violation of a company policy; |
| • | unintended but material breach of any written policy applicable to all employees which is not cured within 30 business days; |
| • | unauthorized use or disclosure of any proprietary information or trade secrets of the company; |
| • | willful and deliberate breach of the employment agreement; |
| • | any other material breach of the employment agreement which is not cured within 30 business days; or |
| • | gross negligence in the performance of duties. |
"Change in Control" means the occurrence of any of the following events:
| • | The acquisition by an individual, entity, or group, other than us or any of our subsidiaries, of beneficial ownership of 50% or more of the combined voting power or economic interests of our then outstanding voting securities entitled to vote generally in the election of directors (excluding any issuance of securities by us in a transaction or series of transactions made principally for bona fide equity financing purposes); |
| • | The acquisition of us by another entity by means of any transaction or series of related transactions to which we are a party (including, without limitation, any stock acquisition, reorganization, merger or consolidation but excluding any issuance of securities by us in a transaction or series of related transactions made principally for bona fide equity financing purposes) other than a transaction or series of related transactions in which the holders of our voting securities outstanding immediately prior to such transaction or series of related transactions retain, immediately after such transaction or series of related transactions, as a result of our shares held by such holders prior to such transaction or series of related transactions, at least a majority of the total voting power represented by our outstanding voting securities or such other surviving or resulting entity (or if we are or such other surviving or resulting entity is a wholly-owned subsidiary immediately following such acquisition, its parent); or |
| • | The sale or other disposition of all or substantially all of our assets in one transaction or series of related transactions. |
In the event of a Change of Control, all outstanding stock options, restricted stock and other stock-based grants held by Mr. Macaluso, Dr. Bar-Or, Mr. Gould and Dr. Clift become fully vested and exercisable, and all such stock options remain exercisable from the date of the Change in Control until the expiration of the term of such stock options.
Notwithstanding the foregoing, a Change in Control shall not be deemed to have occurred by virtue of the consummation of any transaction or series of integrated transactions immediately following which the record holders of our common stock immediately prior to such transaction or series of transactions continue to have substantially the same proportionate ownership in an entity which owns all or substantially all of our assets immediately following such transaction or series of transactions.
The employment agreements do not provide for the payment of a "gross-up" payment under Section 280G of the Code. The following table provides estimates of the potential severance and other post-termination benefits that each of Dr. Bar-Or, Dr. Clift, Mr. Macaluso, Mr. Disbrow, and Mr. Gould would have been entitled to receive assuming their respective employment was terminated as of December 31, 2015 for the reason set forth in each of the columns.
65
Recipient and Benefit | Cause; Without good reason; | Without Cause; Good reason | Death; Disability | Change in Control | ||||||||||||
Michael Macaluso | ||||||||||||||||
Stock Options (3) | $ | - | $ | - | $ | - | $ | - | ||||||||
|
|
|
|
|
|
|
| |||||||||
Total | $ | - | $ | - | $ | - | $ | - | ||||||||
|
|
|
|
|
|
|
| |||||||||
David Bar-Or, M.D. | ||||||||||||||||
Salary | $ | - | $ | 600,000 | $ | - | $ | - | ||||||||
Stock Options (2) | $ | - | $ | - | $ | - | $ | 147,000 | ||||||||
Value of health benefits provided after | ||||||||||||||||
termination (1) | $ | - | $ | 45,264 | $ | - | $ | - | ||||||||
|
|
|
|
|
|
|
| |||||||||
Total | $ | - | $ | 645,264 | $ | - | $ | 147,000 | ||||||||
|
|
|
|
|
|
|
| |||||||||
Gregory Gould | ||||||||||||||||
Salary | $ | - | $ | 500,000 | $ | - | $ | - | ||||||||
Stock Options (3) | $ | - | $ | - | $ | - | $ | - | ||||||||
Value of health benefits provided after | ||||||||||||||||
termination (1) | $ | - | $ | 61,704 | $ | - | $ | - | ||||||||
|
|
|
|
|
|
|
| |||||||||
Total | $ | - | $ | 561,704 | $ | - | $ | - | ||||||||
|
|
|
|
|
|
|
| |||||||||
Vaughan Clift, M.D. | ||||||||||||||||
Salary | $ | - | $ | 500,000 | $ | - | $ | - | ||||||||
Stock Options (2) | $ | - | $ | - | $ | - | $ | 69,700 | ||||||||
Value of health benefits provided after | ||||||||||||||||
termination (1) | $ | - | $ | 61,704 | $ | - | $ | - | ||||||||
|
|
|
|
|
|
|
| |||||||||
Total | $ | - | $ | 561,704 | $ | - | $ | 69,700 | ||||||||
|
|
|
|
|
|
|
| |||||||||
Joshua Disbrow | ||||||||||||||||
Salary | $ | - | $ | 500,000 | $ | - | $ | - | ||||||||
Stock Options (3) | $ | - | $ | - | $ | - | $ | - | ||||||||
Value of health benefits provided after | ||||||||||||||||
termination (1) | $ | - | $ | 56,510 | $ | - | $ | - | ||||||||
|
|
|
|
|
|
|
| |||||||||
Total | $ | - | $ | 556,510 | $ | - | $ | - | ||||||||
|
|
|
|
|
|
|
| |||||||||
| (1) | The value of such benefits is determined based on the estimated cost of providing health benefits to the Named Executive Officer for a period of two years. |
| (2) | Amounts represent the intrinsic value (that is, the value based upon our stock price on December 31, 2015 of $3.09 per share), minus the exercise price of the equity awards that would have become exercisable as of December 31, 2015. |
| (3) | The unvested options of these officers have a value higher than the stock price on December 31, 2015 of $3.09 per share, therefore there is no intrinsic value. |
66
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters. |
The following table sets forth information regarding beneficial ownership of our common stock as of December 31, 2015 by:
| • | each person or group of affiliated persons known by us to be the beneficial owner of more than 5% of our common stock; |
| • | each of our named executive officers; |
| • | each of our directors; and |
| • | all executive officers and directors as a group. |
We have determined beneficial ownership in accordance with SEC rules. The information does not necessarily indicate beneficial ownership for any other purpose. Under these rules, the number of shares of common stock deemed outstanding includes shares issuable upon exercise of options and warrants held by the respective person or group which may be exercised or converted within 60 days after December 31, 2015. For purposes of calculating each person's or group's percentage ownership, stock options, debentures convertible, and warrants exercisable within 60 days after December 31, 2015 are included for that person or group but not the stock options, debentures, or warrants of any other person or group.
Applicable percentage ownership is based on 51,998,306 shares of common stock outstanding at December 31, 2015.
Unless otherwise indicated and subject to any applicable community property laws, to our knowledge, each stockholder named in the following table possesses sole voting and investment power over the shares listed. Unless otherwise noted below, the address of each stockholder listed on the table is c/o Ampio Pharmaceuticals, Inc., 373 Inverness Parkway, Suite 200, Englewood, Colorado 80112.
| Name of Beneficial Owner | Number of Shares Beneficially Owned | Percentage of Shares Beneficially Owned | ||||||
ACT Capital Management, LLLP (1) | 3,692,500 | 7.10 | % | |||||
Knoll Capital Management (2) | 3,388,322 | 6.52 | % | |||||
Michael Macaluso (3) | 2,703,418 | 5.10 | % | |||||
Vaughan Clift (4) | 1,377,200 | 2.60 | % | |||||
David Bar-Or (5) | 1,200,000 | 2.30 | % | |||||
Richard B. Giles (6) | 891,480 | 1.70 | % | |||||
Philip H. Coelho (7) | 584,413 | 1.10 | % | |||||
Joshua R. Disbrow (8) | 400,000 | 0.80 | % | |||||
David R. Stevens (9) | 233,921 | 0.40 | % | |||||
Gregory A. Gould (10) | 233,333 | 0.40 | % | |||||
All executive officers and directors (eight persons) | 7,623,765 | 13.40 | % | |||||
| (1) | Based solely on a Schedule 13G filed on January 28, 2016 by ACT Capital Management, LLLP reporting beneficial ownership as of December 31, 2015. |
| (2) | Based solely on a Schedule 13G filed on February 17, 2015 by Knoll Capital Management, LP reporting beneficial ownership as of February 17, 2015. |
| (3) | Includes an aggregate of 916,666 shares of common stock issuable to Mr. Macaluso by virtue of (i) exercise of currently exercisable stock options, (ii) exercise of warrants, and (iii) his service as a non-management director and currently as an officer. |
| (4) | Includes (i) 855,000 shares of common stock Dr. Clift has the right to acquire on exercise of currently exercisable stock options, and (ii) 522,200 shares of common stock owned of record by Kristin Clift, Dr. Clift's spouse after selling 52,800 shares in the underwritten offering in July 2012. |
| (5) | Includes 1,200,000 shares of common stock which Dr. Bar-Or has the right to acquire through the exercise of stock options. Excludes 982,783 shares of common stock owned of record by Raphael Bar-Or, Dr. Bar-Or's son, as to which Dr. Bar-Or disclaims beneficial ownership. |
67
| (6) | Includes 650,000 shares of common stock issuable to Mr. Giles by virtue of (i) exercise of currently exercisable stock options, and (ii) exercise of warrants. |
| (7) | Includes 565,554 shares of common stock issuable to Mr. Coelho on exercise of currently exercisable stock options. |
| (8) | Includes 400,000 shares of common stock issuable to Mr. Disbrow on exercise of currently exercisable stock options. |
| (9) | Includes 225,000 shares of common stock issuable to Dr. Stevens on exercise of currently exercisable stock options. |
| (10) | Includes 233,333 shares of common stock issuable to Mr. Gould on exercise of currently exercisable stock options. |
| Item 13. | Certain Relationships, Related Transactions, and Director Independence |
Related Party Transactions
In addition to the director and executive compensation arrangements discussed above in Item 11 "Executive Compensation", we or Life Sciences have been a party to the following transactions since January 2013 in which the amount involved exceeded or will exceed $120,000, and in which any director, executive officer or holder of more than 5% of any class of our voting stock, or any member of the immediate family of or entities affiliated with any of them, had or will have a material interest.
We entered into a sponsored research agreement with TRLLC, an entity controlled by our director and Chief Scientific Officer, Dr. Bar-Or, on September 1, 2009, which has been amended five times with the last amendment occurring in March of 2014. Under the amended terms of the research agreement, we will provide personnel with an equivalent value of $325,000 per year. With the most recent amendment, we also agreed to pay a sum of $725,000 which is being amortized over the contractual term of 60.5 months and is divided between current and long-term on the balance sheet. In return, TRLLC will assign any intellectual property rights it develops on our behalf under the research agreement and undertake additional activities to support our commercial activities and business plan. This agreement is set to expire on March 31, 2019 and cannot be terminated prior to March 31, 2017.
In June 2013, our former subsidiary Luoxis also entered into an agreement with TRLLC. The agreement, which was amended in September 2013, provides for Luoxis to pay $8,000 per month to TRLLC in consideration for services related to research and development of Luoxis' Oxidation Reduction Potential platform. Starting in March of 2014, Luoxis also agreed to pay a sum of $615,000 which is being amortized over the contractual term of 60.5 months and is divided between current and long-term on the balance sheet. This agreement has the same termination and expiration as the agreement between us and TRLLC.
Immediately prior to the Merger on March 2, 2010, Chay accepted subscriptions for an aggregate of 1,325,000 shares of common stock from six officers and employees of Life Sciences, for a purchase price of $150,000. The purchase price was advanced to the six officers and employees by Chay at the time the subscriptions were accepted. These shares were issued immediately before the closing of the Merger but after the shareholders of Chay had approved the merger. The advances are non-interest bearing and due on demand and are classified as a reduction to stockholders' equity. During 2012 and 2011, advances of $37,000 and $23,000 were repaid to us, respectively. As of December 31, 2015, $91,000 of advances to stockholders remained outstanding.
The convertible promissory notes include $275,000 invested by relatives of senior management of Aytu (see Note 8- Convertible Promissory Notes Pertaining to the Company's Subsidiary Aytu).
In July 2015, we entered into an agreement with Aytu whereby Aytu agreed to pay us $30,000 per month for shared overhead which includes costs related to the shared facility, corporate staff, and other miscellaneous overhead expenses. These agreements will be in effect until they are terminated in writing by both parties. On January 20, 2016, Aytu entered into subscription agreements with Joshua R. Disbrow, Aytu's Chief Executive Officer, and Jarrett T. Disbrow, Aytu's Chief Operating Officer, pursuant to which each officer agreed to purchase 153,846 shares of Aytu's common stock (for an aggregate of 307,692 shares) at a price of $0.65 per share. These sales triggered Aytu's obligation to notify noteholders who held convertible notes issued by Aytu in July and August 2015 of their resulting conversion option under the notes.
Policies and Procedures for Related Party Transactions
We have adopted a formal written policy that our executive officers, directors, nominees for election as directors, beneficial owners of more than 5% of any class of our common stock and any member of the immediate family of any of the foregoing persons, are not permitted to enter into a related party transaction with us without the prior consent of our Audit Committee, subject to the pre-approval exceptions described below. If advance approval is not feasible then the related party transaction will be considered at the Audit Committee's next regularly scheduled meeting. In approving or rejecting any such proposal, our Audit Committee is to consider the relevant facts and circumstances available and deemed relevant to our Audit Committee, including, but not limited to, whether the transaction is on terms no less favorable than terms generally available to an unaffiliated third party under the same or similar circumstances and the extent of the related party's interest in the transaction. Our Board of Directors has delegated to the chair of our Audit Committee the authority to pre-approve or ratify any request for us to enter into a transaction with a related party, in which the amount involved is less than $120,000 and where the chair is not the related party. Our Audit Committee will also review certain types
68
of related party transactions that it has deemed pre-approved even if the aggregate amount involved will exceed $120,000 including, employment of executive officers, director compensation, certain transactions with other organizations, transactions where all stockholders receive proportional benefits, transactions involving competitive bids, regulated transactions and certain banking-related services.
Director Independence
Our common stock is listed on the NYSE MKT. The listing rules of the NYSE MKT require that a majority of the members of the board of directors be independent. The rules of the NYSE MKT require that, subject to specified exceptions, each member of our Audit, Compensation and Nominating and Governance Committees be independent. Audit Committee members must also satisfy the independence criteria set forth in Rule 10A-3 under the Exchange. Under the rules of the NYSE MKT, a director will only qualify as an "independent director" if, in the opinion of that company's board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director.
In order to be considered to be independent for purposes of Rule 10A-3, a member of an audit committee of a listed company may not, other than in his or her capacity as a member of the audit committee, the board of directors or any other board committee: (1) accept, directly or indirectly, any consulting, advisory or other compensatory fee from the listed company or any of its subsidiaries; or (2) be an affiliated person of the listed company or any of its subsidiaries.
In 2015, our Board of Directors undertook a review of its composition, the composition of its committees and the independence of each director. Based upon information provided by each director concerning his or her background, employment and affiliations, including family relationships, our Board of Directors has determined that at the time none of Messrs. Coelho, Giles and Stevens, representing three of our five directors, has a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and that each of these directors is "independent" as that term is defined by the NYSE MKT. Our Board of Directors also determined that Messrs. Giles, Coelho and Stevens, who comprise our Audit Committee and our Compensation Committee, and Messrs. Giles and Coelho, who comprise our Nominating and Governance Committee, satisfy the independence standards for those committees established by applicable SEC rules and the NYSE MKT rules. In making this determination, our Board of Directors considered the relationships that each non-employee director has with our Company and all other facts and circumstances our Board of Directors deemed relevant in determining their independence, including the beneficial ownership of our capital stock by each non-employee director. The Board of Directors also has determined that Mr. Giles qualifies as an "audit committee financial expert," as defined in Item 401(h) of Regulation S-K promulgated under the Exchange Act.
| Item 14. | Principal Accountant Fees and Services |
EKS&H LLLP has served as our independent auditors since March 2010 and has been appointed by the Audit Committee of the Board of Directors to continue as our independent auditors for the fiscal year ended December 31, 2015.
The following table presents aggregate fees for professional services rendered by our independent registered public accounting firm, EKS&H LLLP for the audit of our annual consolidated financial statements for the respective periods.
| Year Ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
Audit fees (1) | $ | 179,000 | $ | 161,000 | $ | 139,000 | ||||||
Audit-related fees (2) | 10,000 | 142,000 | 25,000 | |||||||||
Tax fees (3) | 67,000 | 19,000 | 14,000 | |||||||||
|
|
|
|
|
| |||||||
Total fees | $ | 256,000 | $ | 322,000 | $ | 178,000 | ||||||
|
|
|
|
|
| |||||||
| (1) | Audit fees are comprised of annual audit fees and quarterly review fees. |
| (2) | Audit-related fees for fiscal years 2015, 2014 and 2013 are comprised of fees related to registration statements and consultation fees. |
| (3) | Tax fees are comprised of tax compliance, preparation and consultation fees. |
In addition, Aytu, paid EKS&H LLLP approximately $242,000 in total professional fees during 2015.
69
Policy on Audit Committee Pre-Approval of Services of Independent Registered Public Accounting Firm
Our Audit Committee has responsibility for appointing, setting compensation and overseeing the work of the independent registered public accounting firm. In recognition of this responsibility, the Audit Committee has established a policy to pre-approve all audit and permissible non-audit services provided by the independent registered public accounting firm. Prior to engagement of the independent registered public accounting firm for the following year's audit, management will submit to the Audit Committee for approval a description of services expected to be rendered during that year for each of following four categories of services:
Audit services include audit work performed in the audit of the annual financial statements, review of quarterly financial statements, reading of annual, quarterly and current reports, as well as work that generally only the independent auditor can reasonably be expected to provide.
Audit-related services are for assurance and related services that are traditionally performed by the independent auditor, including the provisions of consents and comfort letters in connection with the filing of registration statements, due diligence related to mergers and acquisitions and special procedures required to meet certain regulatory requirements.
Tax services consist principally of assistance with tax compliance and reporting, as well as certain tax planning consultations.
Other services are those associated with services not captured in the other categories. We generally do not request such services from our independent auditor.
Prior to the engagement, the Audit Committee pre-approves these services by category of service. The fees are budgeted, and the Audit Committee requires the independent registered public accounting firm and management to report actual fees versus the budget periodically throughout the year by category of service. During the year, circumstances may arise when it may become necessary to engage the independent registered public accounting firm for additional services not contemplated in the original pre-approval. In those instances, the Audit Committee requires specific pre-approval before engaging the independent registered public accounting firm.
The Audit Committee may delegate pre-approval authority to one or more of its members. The member to whom such authority is delegated must report, for informational purposes only, any pre-approval decisions to the Audit Committee at its next scheduled meeting.
70
PART IV
| Item 15. | Exhibits and Financial Statement Schedules |
(a)(1) Financial Statements
The following documents are filed as part of this Form 10-K, as set forth on the Index to Financial Statements found on page F-1.
| • | Report of Independent Registered Public Accounting Firm |
| • | Consolidated Balance Sheets as of December 31, 2015 and 2014 |
| • | Consolidated Statements of Operations for the years ended December 31, 2015, 2014 and 2013 |
| • | Consolidated Statements of Stockholders' Equity (Deficit) for the years ended December 31, 2015, 2014 and 2013 |
| • | Consolidated Statements of Cash Flows for the years ended December 31, 2015, 2014 and 2013 |
| • | Notes to Consolidated Financial Statements |
(a)(2) Financial Statement Schedules
Not Applicable.
(a)(3) Exhibits
Exhibit number | Exhibit title | |||
| 2.1 | Agreement and Plan of Merger, dated March 2, 2010 (1) | |||
| 2.2 | Securities Put and Guarantee Agreement dated March 2, 2010 (1) | |||
| 2.3 | Agreement and Plan of Merger, dated September 4, 2010 (2) | |||
| 2.4 | Amendment to Agreement and Plan of Merger, effective December 31, 2010 (3) | |||
| 2.5 | Amendment to Agreement and Plan of Merger, dated March 22, 2011 (14) | |||
| 3.1 | Certificate of Incorporation of the Registrant, as currently in effect (4) | |||
| 3.2 | Certificate of Amendment to Certificate of Incorporation(4) | |||
| 3.3 | Plan of Conversion of Chay Enterprises, Inc. to a Delaware corporation(4) | |||
| 3.4 | Bylaws of the Registrant, as currently in effect (4) | |||
| 4.1 | Specimen Common Stock Certificate of the Registrant (11) | |||
| 4.2 | Form of Unsecured Senior Convertible Debenture (5) | |||
| 4.3 | Form of Warrant issued with Unsecured Senior Convertible Debenture (5) | |||
| 4.4 | Form of Senior Unsecured Mandatorily Convertible Debenture (6) | |||
| 4.5 | Form of Warrant issued with Senior Unsecured Mandatorily Convertible Debenture (6) | |||
| 4.6 | Form of Underwriter Warrant (19) | |||
| 10.1 | Form of Director and Executive Officer Indemnification Agreement (7) | |||
| 10.2 | 2010 Stock Incentive Plan and forms of option agreements (7)** | |||
| 10.3 | Employment Agreement, dated April 17, 2009, by and between DMI Life Sciences, Inc. and David Bar-Or, M.D.(7)** | |||
| 10.4 | Employment Agreement, dated April 17, 2009, by and between DMI Life Sciences, Inc. and Bruce G. Miller (7)** | |||
| 10.5 | Employment Agreement, effective August 1, 2010, by and between Ampio Pharmaceuticals, Inc. and Donald B. Wingerter, Jr. (8)** | |||
| 10.6 | Employment Agreement, effective August 1, 2010, by and between Ampio Pharmaceuticals, Inc. and David Bar-Or, M.D.(6)** | |||
| 10.7.1 | Employment Agreement, effective August 1, 2010, by and between Ampio Pharmaceuticals, Inc. and Vaughan Clift, M.D.(12)** | |||
71
Exhibit number | Exhibit title | |
| 10.7.2 | Amendment to Employment Agreement, effective October 1, 2011, by and between Ampio Pharmaceuticals, Inc. and Vaughan Clift, M.D. (12)** | |
| 10.7.3 | Letter Agreement, effective May 31, 2011, by and among Ampio Pharmaceuticals, Inc., on the one hand, and Donald B. Wingerter, Jr. and Vaughan Clift, M.D., on the other hand (16) | |
| 10.8 | Sponsored Research Agreement dated September 1, 2009 (7)*** | |
| 10.9 | Exclusive License Agreement, dated July 11, 2005(7)*** | |
| 10.10 | First Amendment to Exclusive License Agreement, dated April 17, 2009 (7)*** | |
| 10.11 | Exclusive License Agreement, dated February 17, 2009 (7)*** | |
| 10.12 | Extension Agreement for Notes Payable dated May 13, 2010 (9) | |
| 10.13 | Extension Agreement for Notes Payable dated May 13, 2010 (9) | |
| 10.14 | Extension Agreement for Notes Payable effective January 31, 2011(12) | |
| 10.15 | Extension Agreement for Notes Payable effective January 31, 2011 (12) | |
| 10.16 | Note Extension and Subordination Agreement, executed February 15, 2011, by and between Ampio Pharmaceuticals, Inc. and DMI BioSciences, Inc. (12) | |
| 10.17 | Note Extension and Subordination Agreement, executed February 15, 2011, by and between DMI Life Sciences, Inc., a subsidiary of the Company, and DMI BioSciences, Inc. (12) | |
| 10.18 | Note Extension and Subordination Agreement, executed February 15, 2011, by and between DMI Life Sciences, Inc., a subsidiary of the Company, and Michael Macaluso (12) | |
| 10.19 | Promissory Note, dated June 23, 2010 (10) | |
| 10.20 | Irrevocable Instructions to Transfer Agent, dated March 10, 2011 (13) | |
| 10.21 | Lease Agreement by and between Ampio Pharmaceuticals, Inc. and CSHV Denver Tech Center, LLC, dated May 20, 2011 (15) | |
| 10.22 | License, Development and Commercialization Agreement between Ampio Pharmaceuticals, Inc. and Daewoong Pharmaceuticals Co., Ltd, effective as of August 23, 2011 (17) | |
| 10.23 | Asset Purchase Agreement by and between Ampio Pharmaceuticals, Inc. and Valeant International (Barbados) SRL, effective as of December 2, 2011 (23)*** | |
| 10.24 | Employment Agreement, effective January 9, 2012, by and between Ampio Pharmaceuticals, Inc. and Michael Macaluso (20)** | |
| 10.25 | Employment Agreement, effective December 15, 2012, by and between Ampio Pharmaceuticals, Inc. and Joshua R. Disbrow (21)** | |
| 10.26 | Clinical Batch Manufacturing Agreement between Ethypharm S.A. and Ampio Pharmaceuticals, Inc. dated September 10, 2012 (22)*** | |
| 10.27 | Manufacturing and Supply Agreement between Ethypharm S.A. and Ampio Pharmaceuticals, Inc. dated September 10, 2012 (22)*** | |
| 10.28 | Amendment to Employment Agreement between Ampio Pharmaceuticals, Inc. and David Bar-Or, M.D., dated July 15, 2013 (24)** | |
| 10.29 | Amendment to Employment Agreement between Ampio Pharmaceuticals, Inc. and Vaughan Clift, M.D., dated July 15, 2013 (24)** | |
| 10.30 | Securities Purchase Agreement by and among Ampio Pharmaceuticals, Inc. and the Purchasers (as defined therein), dated September 25, 2013 (25) | |
| 10.31 | Amendment to Employment Agreement between Ampio Pharmaceuticals, Inc. and Michael Macaluso, dated October 4, 2013 (26)** | |
| 10.32 | Lease Agreement by and between Ampio Pharmaceuticals, Inc. and NCWP – Inverness Business Park, LLC, dated December 13, 2013 (27) | |
| 10.33 | Amendment of 2010 Stock and Incentive Plan (28)** | |
72
Exhibit number | Exhibit title | |
| 10.34 | Human Serum Albumin Ingredient Purchase and Sale Agreement by and between Ampio Pharmaceuticals, Inc. and Supplier, dated October 10, 2013 (29)*** | |
| 10.35 | Employment Agreement between Ampio Pharmaceuticals, Inc. and Gregory A. Gould, executed June 4, 2014 and effective June 10, 2014 (30)** | |
| 10.36 | Amendment to Employment Agreement between Ampio Pharmaceuticals, Inc. and David Bar-Or, M.D., dated August 11, 2014 (31)** | |
| 10.37 | Amendment to Employment Agreement between Ampio Pharmaceuticals, Inc. and Vaughan Clift, M.D., dated August 11, 2014 (32)** | |
| 10.38 | Amendment to Employment Agreement between Ampio Pharmaceuticals, Inc. and Michael Macaluso, dated December 20, 2014 (33)** | |
| 10.39 | Voting Agreement between Rosewind Corporation and Ampio Pharmaceuticals, Inc., dated April 21, 2015 (34) | |
| 10.40 | Amendment to Employment Agreement between Ampio Pharmaceuticals, Inc. and David Bar-Or, M.D., dated August 3, 2015 (35)** | |
| 10.41 | Amendment to Employment Agreement between Ampio Pharmaceuticals, Inc. and Vaughan Clift, M.D., dated July 31, 2015 (35)** | |
| 10.42 | Amendment to Human Serum Albumin Ingredient Purchase and Sale Agreement among Ampio Pharmaceuticals, Inc., Octapharma USA, Inc. and Nova Biologics, Inc., effective as of October 8, 2015 (36) | |
| 16.1 | Letter Regarding Change in Certifying Accountant, dated March 16, 2010 (7) | |
| 21.1 | List of subsidiaries of the Registrant (18) | |
| 23.1* | Consent of EKS&H LLLP | |
| 31.1* | Certificate of the Chief Executive Officer of Ampio Pharmaceuticals, Inc. pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 31.2* | Certificate of the Chief Financial Officer of Ampio Pharmaceuticals, Inc. pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 32.1* | Certificate of the Chief Executive Officer and the Chief Financial Officer of Ampio Pharmaceuticals, Inc. pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| 101 | XBRL (extensible Business Reporting Language). The following materials from Ampio Pharmaceuticals, Inc.'s Annual Report on Form 10-K for the year ended December 31, 2014 formatted in XBRL: (i) the Consolidated Balance Sheets, (ii) the Consolidated Statements of Operations, (iii) the Consolidated Statements of Stockholders' Equity (Deficit), (iv) the Consolidated Statements of Cash Flows, and (v) the Notes to the Consolidated Financial Statements. | |
| (1) | Incorporated by reference from Registrant's Form 8-K filed March 8, 2010. |
| (2) | Incorporated by reference from Registrant's Amendment No. 1 to Form 8-K filed January 7, 2011. |
| (3) | Incorporated by reference from Registrant's Amendment No. 2 to Form 8-K filed January 7, 2011. |
| (4) | Incorporated by reference from Registrant's Form 8-K filed March 30, 2010. |
| (5) | Incorporated by reference from Registrant's Form 8-K filed August 16, 2010. |
| (6) | Incorporated by reference from Registrant's Form 8-K filed November 12, 2010. |
| (7) | Incorporated by reference from Registrant's Form 8-K/A filed March 17, 2010. |
| (8) | Incorporated by reference from Registrant's Form 8-K/A filed August 17, 2010. |
| (9) | Incorporated by reference from Registrant's Quarterly Report on Form 10-Q for the quarter ended March 31, 2010. |
| (10) | Incorporated by reference from Registrant's Quarterly Report on Form 10-Q for the quarter ended June 30, 2010. |
| (11) | Incorporated by reference from Registrant's Registration Statement on Form S-4 filed January 7, 2011. |
| (12) | Incorporated by reference from Registrant's Form 8-K filed February 15, 2011. |
| (13) | Incorporated by reference from Registrant's Form 8-K filed March 16, 2011. |
| (14) | Incorporated by reference from Registrant's Form 8-K filed March 25, 2011. |
| (15) | Incorporated by reference from Registrant's Registration Statement on Form S-1/A filed May 23, 2011. |
| (16) | Incorporated by reference from Registrant's Form 8-K filed June 8, 2011. |
| (17) | Incorporated by reference from Registrant's Form 8-K/A filed October 5, 2011. |
| (18) | Incorporated by reference from Registrant's Registration Statement on Form S-1 filed November 12, 2010. |
| (19) | Incorporated by reference from Registrant's Form 8-K filed July 13, 2012. |
| (20) | Incorporated by reference from Registrant's Form 8-K filed September 13, 2012. |
| (21) | Incorporated by reference from Registrant's Form 8-K filed December 20, 2012. |
73
| (22) | Incorporated by reference from Registrant's Quarterly Report on Form 10-Q for the quarter ended September 30, 2012. |
| (23) | Incorporated by reference from Registrant's Annual Report on Form 10-K for the year ended December 31, 2011. |
| (24) | Incorporated by reference from Registrant's Form 8-K filed July 19, 2013. |
| (25) | Incorporated by reference from Registrant's Form 8-K filed September 26, 2013. |
| (26) | Incorporated by reference from Registrant's Form 8-K filed October 4, 2013. |
| (27) | Incorporated by reference from Registrant's Form 8-K filed December 19, 2013. |
| (28) | Incorporated by reference from Registrant's Proxy Statement on Form 14A filed November 1, 2013. |
| (29) | Incorporated by reference from Registrant's Form 10-K/A filed May 23, 2014. |
| (30) | Incorporated by reference from Registrant's Form 8-K filed June 10, 2014. |
| (31) | Incorporated by reference from Registrant's Form 8-K filed August 15, 2014. |
| (32) | Incorporated by reference from Registrant's Form 8-K filed August 15, 2014. |
| (33) | Incorporated by reference from Registrant's Form 8-K filed December 29, 2014. |
| (34) | Incorporated by reference from Registrant's Form 8-K filed April 22, 2015. |
| (35) | Incorporated by reference from Registrant's Form 8-K filed August 6, 2015. |
| (36) | Incorporated by reference from Registrant's Form 8-K filed October 20, 2015. |
| * | Filed herewith. |
| ** | This exhibit is a management contract or compensatory plan or arrangement. |
| *** | Confidential treatment has been applied for with respect to certain portions of these exhibits. |
74
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities and Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| AMPIO PHARMACEUTICALS, INC. | ||||||
| Date: February 26, 2016 | By: | /s/ Michael Macaluso | ||||
Michael Macaluso Chief Executive Officer (Principal Executive Officer) | ||||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant in the capacities indicated, on February 26, 2016.
Signature | Title | |||
/s/ Michael Macaluso Michael Macaluso | Chairman of the Board and Chief Executive Officer | |||
/s/ Gregory A. Gould Gregory A. Gould | Chief Financial Officer (Principal Financial and Accounting Officer), Secretary and Treasurer | |||
/s/ David Bar-Or David Bar-Or | Director | |||
/s/ Philip H. Coelho Philip H. Coelho | Director | |||
/s/ Richard B. Giles Richard B. Giles | Director | |||
/s/ David R. Stevens David R. Stevens | Director | |||
75
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
AMPIO PHARMACEUTICALS, INC. AND SUBSIDIARIES
| Page | ||||
Report of Independent Registered Public Accounting Firm | F-2 | |||
Consolidated Balance Sheets | F-3 | |||
Consolidated Statements of Operations | F-4 | |||
Consolidated Statements of Stockholders' Equity (Deficit) | F-5 | |||
Consolidated Statements of Cash Flows | F-6 | |||
Notes to Consolidated Financial Statements | F-7 | |||
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
Ampio Pharmaceuticals, Inc.
Englewood, Colorado
We have audited the accompanying consolidated balance sheets of Ampio Pharmaceuticals, Inc. and subsidiaries (the "Company") as of December 31, 2015 and 2014, and the related consolidated statements of operations, stockholders' equity, and cash flows for each of the years in the three-year period ended December 31, 2015. We also have audited the Company's internal control over financial reporting as of December 31, 2015, based on criteria established in Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company's management is responsible for these consolidated financial statements, for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management's Annual Report on Internal Control over Financial Reporting included in Item 9A. Our responsibility is to express an opinion on these consolidated financial statements and an opinion on the Company's internal control over financial reporting based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Ampio Pharmaceuticals, Inc. and Subsidiaries as of December 31, 2015 and 2014, and the results of their operations and their cash flows for each of the years in the three-year period ended December 31, 2015, in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, Ampio Pharmaceuticals, Inc. and Subsidiaries maintained, in all material respects, effective internal control over financial reporting as of December 31, 2015, based on criteria established in Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
As described in "Management's Annual Report on Internal Control Over Financial Reporting," management has excluded the ProstaScint and Primsol businesses from its assessment of internal control over financial reporting as of December 31, 2015, because those businesses were acquired by the Company during 2015. We have also excluded the ProstaScint and Primsol businesses from our audit of internal control over financial reporting. The total assets and total revenues of these 2015 acquired businesses represent approximately 7.4% and 87.8%, respectively, of the related consolidated financial statement amounts as of and for the year ended December 31, 2015.
| EKS&H LLLP |
February 26, 2016
Denver, Colorado
F-2
AMPIO PHARMACEUTICALS, INC. AND SUBSIDIARIES
Consolidated Balance Sheets
| December 31, 2015 | December 31, 2014 | |||||||
| Assets | ||||||||
Current assets | ||||||||
Cash and cash equivalents | $ | 26,957,938 | $ | 50,320,656 | ||||
Prepaid expenses and other | 2,004,699 | 672,716 | ||||||
Prepaid research and development - related party (Note 11) | 265,785 | 265,785 | ||||||
|
|
|
| |||||
Total current assets | 29,228,422 | 51,259,157 | ||||||
|
|
|
| |||||
Fixed assets, net (Note 2) | 9,331,446 | 9,945,428 | ||||||
In-process research and development | 7,500,000 | 7,500,000 | ||||||
Developed technology, customer contracts and trade names, net | 2,909,583 | - | ||||||
Goodwill | 221,000 | - | ||||||
Patents, net | 593,382 | 664,169 | ||||||
Long-term portion of prepaid research and development - related party (Note 11) | 598,016 | 863,802 | ||||||
Deposits | 36,744 | 35,854 | ||||||
|
|
|
| |||||
| 21,190,171 | 19,009,253 | |||||||
|
|
|
| |||||
Total assets | $ | 50,418,593 | $ | 70,268,410 | ||||
|
|
|
| |||||
| Liabilities and Stockholders' Equity | ||||||||
Current liabilities | ||||||||
Accounts payable | $ | 2,880,662 | $ | 3,299,025 | ||||
Primsol payable | 1,111,057 | - | ||||||
Accrued compensation | 1,378,101 | 235,665 | ||||||
Deferred rent | 59,579 | 59,579 | ||||||
Deferred revenue | 85,714 | 85,714 | ||||||
|
|
|
| |||||
Total current liabilities | 5,515,113 | 3,679,983 | ||||||
Convertible promissory notes, net of unamortized discount of $253,448 (Note 8) | 4,921,552 | - | ||||||
Interest payable | 161,988 | - | ||||||
Contingent consideration | 687,685 | - | ||||||
Long-term deferred rent | 641,262 | 661,160 | ||||||
Long-term deferred revenue | 383,036 | 468,749 | ||||||
Warrant derivative liability | 180,969 | - | ||||||
|
|
|
| |||||
Total liabilities | 12,491,605 | 4,809,892 | ||||||
|
|
|
| |||||
Commitments and contingencies (Note 7) | ||||||||
Stockholders' equity | ||||||||
Preferred Stock, par value $.0001; 10,000,000 shares authorized; none issued | - | - | ||||||
Common Stock, par value $.0001; 100,000,000 shares authorized; shares issued and outstanding - 51,998,306 in 2015 and 51,972,266 in 2014 | 5,200 | 5,197 | ||||||
Additional paid-in capital | 170,999,410 | 168,108,278 | ||||||
Advances to stockholders | (90,640 | ) | (90,640 | ) | ||||
Accumulated deficit | (133,914,812 | ) | (101,904,570 | ) | ||||
|
|
|
| |||||
Total Ampio stockholders' equity | 36,999,158 | 66,118,265 | ||||||
Non-controlling interests | 927,830 | (659,747 | ) | |||||
|
|
|
| |||||
Total stockholders' equity | 37,926,988 | 65,458,518 | ||||||
|
|
|
| |||||
Total liabilities and stockholders' equity | $ | 50,418,593 | $ | 70,268,410 | ||||
|
|
|
| |||||
The accompanying notes are an integral part of these consolidated financial statements.
F-3
AMPIO PHARMACEUTICALS, INC. AND SUBSIDIARIES
Consolidated Statements of Operations
| Years Ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
Revenue | ||||||||||||
Product and service revenue, net | $ | 1,076,749 | $ | - | $ | - | ||||||
License revenue | 85,713 | 76,787 | 50,000 | |||||||||
|
|
|
|
|
| |||||||
Total revenue | 1,162,462 | 76,787 | 50,000 | |||||||||
|
|
|
|
|
| |||||||
Operating expenses | ||||||||||||
Cost of sales | 369,309 | - | - | |||||||||
Research and development | 18,624,752 | 26,618,567 | 16,550,556 | |||||||||
Research and development - related party (Note 11) | 335,793 | 304,421 | 45,921 | |||||||||
Selling, general and administrative | 15,134,727 | 12,224,834 | 7,477,396 | |||||||||
|
|
|
|
|
| |||||||
Total operating expenses | 34,464,581 | 39,147,822 | 24,073,873 | |||||||||
|
|
|
|
|
| |||||||
Loss from operations | (33,302,119 | ) | (39,071,035 | ) | (24,023,873 | ) | ||||||
|
|
|
|
|
| |||||||
Other income (expense) | ||||||||||||
Interest (expense) income | (333,760 | ) | 22,263 | 12,287 | ||||||||
Derivative expense | (78,038 | ) | - | (516,840 | ) | |||||||
|
|
|
|
|
| |||||||
Total other (expense) income | (411,798 | ) | 22,263 | (504,553 | ) | |||||||
|
|
|
|
|
| |||||||
Net loss | (33,713,917 | ) | (39,048,772 | ) | (24,528,426 | ) | ||||||
Net loss applicable to non-controlling interests | 1,703,675 | 923,357 | 519,868 | |||||||||
|
|
|
|
|
| |||||||
Net loss applicable to Ampio | $ | (32,010,242 | ) | $ | (38,125,415 | ) | $ | (24,008,558 | ) | |||
|
|
|
|
|
| |||||||
Weighted average number of Ampio common shares outstanding | 51,992,048 | 50,226,555 | 38,294,259 | |||||||||
|
|
|
|
|
| |||||||
Basic and diluted Ampio net loss per common share | $ | (0.62 | ) | $ | (0.76 | ) | $ | (0.63 | ) | |||
|
|
|
|
|
| |||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-4
AMPIO PHARMACEUTICALS, INC. AND SUBSIDIARIES
Consolidated Statements of Stockholders' Equity
| Common Stock | Additional Paid in | Advances to | Accumulated | Non-controlling | Total Stockholders' | |||||||||||||||||||||||
| Shares | Amount | Capital | Stockholders | Deficit | Interests | Equity | ||||||||||||||||||||||
Balance - December 31, 2012 | 37,009,695 | $ | 3,701 | $ | 63,687,558 | $ | (90,640 | ) | $ | (39,770,597 | ) | $ | - | $ | 23,830,022 | |||||||||||||
Issuance of common stock for services | 22,752 | 2 | 88,048 | - | - | - | 88,050 | |||||||||||||||||||||
Issuance of common stock in exchange for cash in September, net of offering costs of $297,768 | 4,600,319 | 460 | 25,003,526 | - | - | - | 25,003,986 | |||||||||||||||||||||
Issuance of common stock of Luoxis for cash net of offering costs of $985,274 | - | - | 3,340,937 | - | - | 639,353 | 3,980,290 | |||||||||||||||||||||
Issuance of common stock of Luoxis in exchange for patents | - | - | 42,510 | - | - | 7,490 | 50,000 | |||||||||||||||||||||
Non-controlling interests on contributed assets | - | - | (10,739 | ) | - | - | 10,739 | - | ||||||||||||||||||||
Options exercised, net | 238,381 | 24 | 159,858 | - | - | - | 159,882 | |||||||||||||||||||||
Warrants exercised, net | 193,884 | 20 | 1,182,761 | - | - | - | 1,182,781 | |||||||||||||||||||||
Stock-based compensation | - | - | 3,448,285 | - | - | - | 3,448,285 | |||||||||||||||||||||
Net loss | - | - | - | - | (24,008,558 | ) | (519,868 | ) | (24,528,426 | ) | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||
Balance - December 31, 2013 | 42,065,031 | 4,207 | 96,942,744 | (90,640 | ) | (63,779,155 | ) | 137,714 | 33,214,870 | |||||||||||||||||||
Issuance of common stock for services | 4,209 | - | 30,000 | - | - | - | 30,000 | |||||||||||||||||||||
Issuance of common stock in exchange for cash in March 2014, net of offering costs of $4,999,777 | 9,775,000 | 978 | 63,424,244 | - | - | - | 63,425,222 | |||||||||||||||||||||
Non-controlling interests on contributed assets | - | - | (125,896 | ) | - | - | 125,896 | - | ||||||||||||||||||||
Options exercised, net | 120,519 | 12 | (15,480 | ) | - | - | - | (15,468 | ) | |||||||||||||||||||
Warrants exercised, net | 7,507 | - | - | - | - | - | - | |||||||||||||||||||||
Stock-based compensation | - | - | 7,852,666 | - | - | - | 7,852,666 | |||||||||||||||||||||
Net loss | - | - | - | - | (38,125,415 | ) | (923,357 | ) | (39,048,772 | ) | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||
Balance - December 31, 2014 | 51,972,266 | 5,197 | 168,108,278 | (90,640 | ) | (101,904,570 | ) | (659,747 | ) | 65,458,518 | ||||||||||||||||||
Common stock issued for services | 7,998 | 1 | 29,999 | - | - | - | 30,000 | |||||||||||||||||||||
Options exercised, net | 10,416 | 1 | 28,748 | - | - | - | 28,749 | |||||||||||||||||||||
Warrants exercised, net | 7,626 | 1 | - | - | - | - | 1 | |||||||||||||||||||||
Warrant modification | - | - | 422,177 | - | - | - | 422,177 | |||||||||||||||||||||
Stock-based compensation | - | - | 5,748,950 | - | - | - | 5,748,950 | |||||||||||||||||||||
Payments for equity-based transactions | - | - | (47,490 | ) | - | - | - | (47,490 | ) | |||||||||||||||||||
Non-controlling interests on contributed assets | - | - | (3,291,252 | ) | - | - | 3,291,252 | - | ||||||||||||||||||||
Net loss | - | - | - | - | (32,010,242 | ) | (1,703,675 | ) | (33,713,917 | ) | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||
Balance - December 31, 2015 | 51,998,306 | $ | 5,200 | $ | 170,999,410 | $ | (90,640 | ) | $ | (133,914,812 | ) | $ | 927,830 | $ | 37,926,988 | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-5
AMPIO PHARMACEUTICALS, INC. AND SUBSIDIARIES
Consolidated Statements of Cash Flows
| Years Ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
Cash flows from operating activities | ||||||||||||
Net loss | $ | (33,713,917 | ) | $ | (39,048,772 | ) | $ | (24,528,426 | ) | |||
Stock-based compensation and warrant expense | 6,171,127 | 7,852,666 | 3,448,285 | |||||||||
Depreciation, amortization and accretion | 1,136,292 | 439,098 | 137,680 | |||||||||
Loss on disposal of fixed assets | - | 28,685 | - | |||||||||
Amortization of prepaid research and development - related party (Note 11) | 265,786 | 210,413 | - | |||||||||
Amortization of debt issuance costs | 147,805 | - | - | |||||||||
Common stock issued for services | 30,000 | 30,000 | 88,050 | |||||||||
Derivative expense | 78,038 | - | 516,840 | |||||||||
Adjustments to reconcile net loss to net cash used in operating activities | ||||||||||||
(Increase) decrease in prepaid expenses and other | (1,331,983 | ) | (540,730 | ) | 32,904 | |||||||
Increase in prepaid research and development - related party (Note 11) | - | (1,340,000 | ) | - | ||||||||
(Decrease) increase in accounts payable | (426,071 | ) | 1,020,496 | 699,454 | ||||||||
Increase in interest payable | 161,988 | - | - | |||||||||
(Decrease) increase in deferred rent | (19,898 | ) | 720,739 | - | ||||||||
(Decrease) increase in deferred revenue | (85,713 | ) | 173,213 | (50,000 | ) | |||||||
Increase (decrease) in accrued compensation | 1,142,436 | (286,391 | ) | 522,056 | ||||||||
|
|
|
|
|
| |||||||
Net cash used in operating activities | (26,444,110 | ) | (30,740,583 | ) | (19,133,157 | ) | ||||||
|
|
|
|
|
| |||||||
Cash flows used in investing activities | ||||||||||||
Purchase of fixed assets | (235,656 | ) | (8,668,351 | ) | (1,311,383 | ) | ||||||
Purchase of ProstaScint business | (1,000,000 | ) | - | - | ||||||||
Purchase of Primsol business | (540,000 | ) | - | - | ||||||||
Proceeds from sale of fixed assets | - | 2,385 | - | |||||||||
Purchase of patents | - | - | (330,000 | ) | ||||||||
Deposits | (890 | ) | 8,002 | (23,856 | ) | |||||||
|
|
|
|
|
| |||||||
Net cash used in investing activities | (1,776,546 | ) | (8,657,964 | ) | (1,665,239 | ) | ||||||
|
|
|
|
|
| |||||||
Cash flows from financing activities | ||||||||||||
Proceeds from sale of common stock | - | 68,394,051 | 25,301,735 | |||||||||
Costs related to sale of common stock | - | (4,999,777 | ) | (297,768 | ) | |||||||
Proceeds from sale of Luoxis common stock | - | - | 4,652,500 | |||||||||
Costs related to sale of Luoxis common stock | - | - | (672,210 | ) | ||||||||
Proceeds from convertible promissory notes (Note 8) | 5,175,000 | - | - | |||||||||
Debt issuance costs (Note 8) | (298,322 | ) | - | - | ||||||||
Proceeds from option and warrant exercise | 28,750 | 15,480 | 441,071 | |||||||||
Payments for equity-based transactions | (47,490 | ) | - | - | ||||||||
|
|
|
|
|
| |||||||
Net cash provided by financing activities | 4,857,938 | 63,409,754 | 29,425,328 | |||||||||
|
|
|
|
|
| |||||||
Net change in cash and cash equivalents | (23,362,718 | ) | 24,011,207 | 8,626,932 | ||||||||
Cash and cash equivalents at beginning of period | 50,320,656 | 26,309,449 | 17,682,517 | |||||||||
|
|
|
|
|
| |||||||
Cash and cash equivalents at end of period | $ | 26,957,938 | $ | 50,320,656 | $ | 26,309,449 | ||||||
|
|
|
|
|
| |||||||
Non-cash transactions: | ||||||||||||
Issuance of Luoxis stock for patents | $ | - | $ | - | $ | 50,000 | ||||||
Warrant compensation from Luoxis common stock offering costs | $ | - | $ | - | $ | 313,064 | ||||||
Debenture warrant exercise fair value adjustment | $ | - | $ | - | $ | 901,611 | ||||||
Fixed asset purchases included in accounts payable | $ | 7,708 | $ | 377,953 | $ | - | ||||||
Primsol business purchase included in Primsol payable, $1,250,000 less future accretion of $173,000 (Note 1) | $ | 1,077,000 | $ | - | $ | - | ||||||
Warrant derivative liability related to the issuance of the convertible promissory notes (Note 8) | $ | 102,931 | $ | - | $ | - | ||||||
Contingent consideration related to ProstaScint | $ | 664,000 | $ | - | $ | - | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-6
AMPIO PHARMACEUTICALS, INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 1 – Basis of Presentation, Merger and Business Combination
These financial statements represent the consolidated financial statements of Ampio Pharmaceuticals, Inc. ("Ampio" or "the Company") and Aytu BioScience, Inc. ("Aytu"), an 81.5% owned subsidiary (See Note-16 Subsequent Events related to our spin-off of Aytu). We are a biopharmaceutical company focused on primarily developing compounds that decrease inflammation by (i) inhibiting specific pro-inflammatory compounds by affecting specific pathways at the protein expression and at the transcription level; (ii) activating specific phosphatase or depleting available phosphate needed for the inflammation process; and (iii) decreasing vascular permeability. Through Aytu, we are also focused on acquiring, developing and commercializing products focused primarily on the urological disorders market, specifically sexual dysfunction, urological cancer, urinary tract infections and male infertility.
Ampio's activities have been primarily related to research and development and raising capital and have not generated significant revenue to date.
Merger/Subsidiary
Aytu BioScience, Inc.
On April 16, 2015, Luoxis Diagnostics, Inc. ("Luoxis") and Vyrix Pharmaceuticals, Inc. ("Vyrix"), each previously a subsidiary of Ampio, entered into an Agreement and Plan of Merger (the "Merger Agreement") by and among Rosewind Corporation, a Colorado corporation and public company ("Rosewind"), Luoxis, Vyrix, two major stockholders of Rosewind and two subsidiaries of Rosewind created solely for the purposes of the Merger (as defined below), and which did not survive the Merger.
In the first stage of the transaction, each of Luoxis and Vyrix merged with and into one of Rosewind's merger subsidiaries. Luoxis and Vyrix survived these mergers. The outstanding shares of stock of Luoxis and the outstanding shares of stock of Vyrix were converted into the right to receive shares of common stock in Rosewind. The Luoxis stock and the Vyrix stock were each converted at an exchange factor. The exchange factor for each of them was determined upon the basis of a relative value opinion obtained by Ampio prior to the Merger. The outstanding shares of Rosewind's merger subsidiary that merged with Luoxis were converted into shares of Luoxis as the surviving corporation. The outstanding shares of Rosewind's merger subsidiary that merged with Vyrix were converted into shares of Vyrix as the surviving corporation. After completion of the first stage of the transaction, Luoxis and Vyrix were wholly-owned subsidiaries of Rosewind.
In the second stage of the transaction, which occurred on the same day as the first stage of the transaction, each of Luoxis and Vyrix was merged with and into Rosewind, with Rosewind surviving. The first and second stage mergers are referred to collectively as the Merger. Following the consummation of the Merger, Ampio became the holder of 81.5% of the common stock of Rosewind.
Pursuant to the Merger, Rosewind changed its fiscal year end from August 31 to June 30.
On June 1, 2015, the Rosewind shareholders voted to change the state of incorporation from Colorado to Delaware and to change Rosewind's name to Aytu BioScience, Inc., which was effective June 8, 2015. Along with the reincorporation, Aytu now has 300 million authorized shares of common stock with a par value of $0.0001 per share and 50 million authorized shares of preferred stock with a par value of $0.0001 per share. The Aytu shareholders also approved the 2015 Stock Option and Incentive Plan, which provides for the award of stock options, stock appreciation rights, restricted stock and other equity awards for up to an aggregate of 10 million shares of common stock. The shares of common stock underlying any awards that are forfeited, canceled, reacquired by Aytu prior to vesting, satisfied without any issuance of stock, expire or are otherwise terminated (other than by exercise) under the 2015 Plan will be added back to the shares of common stock available for issuance under the 2015 Plan.
On June 1, 2015, the Rosewind shareholders voted and approved a reverse stock split that was in effect on June 8, 2015. The reverse stock split was at a ratio of one new share for every 12.174 shares outstanding.
Business Combination-ProstaScint
In May 2015, Aytu entered into and closed on an Asset Purchase Agreement with Jazz Pharmaceuticals, Inc. (the "Seller"). Pursuant to the agreement, Aytu purchased assets related to the Seller's product known as ProstaScint® (capromab pendetide), including certain intellectual property and contracts, and the product approvals, inventory and work in progress (together, the "ProstaScint Business"), and assumed certain of the Seller's liabilities, including those related to product approvals and the sale and marketing of ProstaScint.
F-7
The purchase price consisted of the upfront payment of $1.0 million. Aytu also paid an additional $500,000 for the ProstaScint-related product inventory and $227,000 that was paid in the fourth quarter of 2015 (which represents a portion of certain FDA fees). Aytu also will pay 8% as contingent consideration on its net sales made after October 31, 2017, payable up to a maximum aggregate payment of an additional $2.5 million. The contingent consideration was valued at $664,000 using a discounted cash flow estimate as of the acquisition date. The total fair value consideration for the purchase was $2.4 million.
The Company's allocation of consideration transferred for ProstaScint as of the purchase date of May 20, 2015 is as follows:
| Fair Value | ||||
Tangible assets | $ | 727,000 | ||
Intangible assets | 1,590,000 | |||
Goodwill | 74,000 | |||
|
| |||
Total assets acquired | $ | 2,391,000 | ||
|
| |||
Included in the intangible assets is developed technology of $790,000, customer contracts of $720,000 and trade names of $80,000 each of which will be amortized over a ten-year period.
As of December 31, 2015, the contingent consideration had increased to $688,000 due to accretion.
Aytu's future amortization from the year ended December 31, 2015 is as follows:
2016 | $ | 159,000 | ||
2017 | 159,000 | |||
2018 | 159,000 | |||
2019 | 159,000 | |||
2020 | 159,000 | |||
Thereafter | 696,000 | |||
|
| |||
| $ | 1,491,000 | |||
|
|
Business Combination-Primsol
In October 2015, Aytu entered into and closed on an Asset Purchase Agreement with FSC Laboratories, Inc. (the "Seller"). Pursuant to the agreement, Aytu purchased assets related to the Seller's product known as Primsol® (trimethoprim solution), including certain intellectual property and contracts, inventory, work in progress and all marketing and sales assets and materials related solely to Primsol (together, the "Primsol Business"), and assumed certain of the Seller's liabilities, including those related to the sale and marketing of Primsol arising after the closing.
Aytu paid $500,000 at closing for the Primsol Business and paid an additional $142,000, of which $102,000 went to inventory and $40,000 towards the Primsol Business, for the transfer of the Primsol-related product inventory. Aytu also agreed to pay an additional (a) $500,000 payable no later than March 31, 2016, (b) $500,000 payable no later than June 30, 2016, and (c) $250,000 payable no later than September 30, 2016 (together, the "Installment Payments").
F-8
The Company's allocation on consideration transferred for Primsol as of the purchase date of October 5, 2015 is as follows:
| Fair Value | ||||
Tangible assets | $ | 182,000 | ||
Intangible assets | 1,470,000 | |||
Goodwill | 147,000 | |||
|
| |||
Total assets acquired | $ | 1,799,000 | ||
|
| |||
Included in tangible assets is $102,000 of inventory and $80,000 of work-in-process inventory. Included in the intangible assets is developed technology of $520,000, customer contracts of $810,000 and trade names of $140,000, each of which will be amortized over a six-year period.
Aytu's future amortization from the year ended December 31, 2015 is as follows:
2016 | $ | 245,000 | ||
2017 | 245,000 | |||
2018 | 245,000 | |||
2019 | 245,000 | |||
2020 | 245,000 | |||
Thereafter | 194,000 | |||
|
| |||
| $ | 1,419,000 | |||
|
|
Note 2 – Summary of Significant Accounting Policies
Principals of Consolidation
These consolidated financial statements include the accounts of Ampio and its majority-owned subsidiary. All material intercompany transactions and balances have been eliminated.
Cash and Cash Equivalents
Ampio considers all highly liquid instruments purchased with an original maturity of three months or less to be cash equivalents. Cash equivalents consist primarily of money market fund investments. Ampio's investment policy is to preserve principal and maintain liquidity. Ampio periodically monitors its positions with, and the credit quality of, the financial institutions with which it invests. Periodically, throughout the year, Ampio has maintained balances in excess of federally insured limits.
Accrued Compensation
The accrued compensation consists of a 2015 employee bonus accrual. As of the filing date of this report, a majority of the bonus had not been paid. The majority of the bonus is contingent upon the Company completing a successful Ampion trial. The Compensation Committee will re-evaluate the payment of the bonus during the first half of 2016.
Revenue Recognition/Deferred Revenue
Product & Service Sales
Ampio recognizes revenue from product and service sales when there is persuasive evidence that an arrangement exists, delivery has occurred or service has been rendered, the price is fixed or determinable and collectability is reasonably assured.
License Agreements and Royalties
Payments received upon signing of license agreements are for the right to use the license and are deferred and amortized over the lesser of the license term or patent life of the licensed drug. Milestone payments relate to obtaining regulatory approval in the territories, cumulative sales targets, and other projected milestones and are recognized at the time the milestone requirements are achieved. Royalties will be recognized as revenue when earned.
F-9
Estimated Sales Returns and Allowances
Ampio records estimated reductions in revenue for potential returns of products by customers. As a result, management must make estimates of potential future product returns and other allowances related to current period product revenue. In making such estimates, management analyzes historical returns, current economic trends and changes in customer demand and acceptance of our products. If management were to make different judgments or utilize different estimates, material differences in the amount of the Company's reported revenue could result.
Fixed Assets
Fixed assets are recorded at cost and after being placed in service, are depreciated using the straight-line method over estimated useful lives. Fixed assets consist of the following:
| Estimated | December 31, | |||||||||
| Useful Lives in years | 2015 | 2014 | ||||||||
Manufacturing Facility/Clean Room - not in service | $ | 2,741,000 | $ | 2,684,000 | ||||||
Leasehold improvements | 10 | 6,093,000 | 6,064,000 | |||||||
Office furniture and equipment | 3 - 10 | 665,000 | 556,000 | |||||||
Lab equipment | 5 -10 | 1,109,000 | 1,060,000 | |||||||
Less accumulated depreciation and accretion | (1,277,000 | ) | (419,000 | ) | ||||||
|
|
|
| |||||||
Fixed assets, net | $ | 9,331,000 | $ | 9,945,000 | ||||||
|
|
|
| |||||||
The Company recorded the following depreciation expense in the respective periods:
| Year Ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
Depreciation Expense | $ | 858,000 | $ | 368,000 | $ | 72,000 | ||||||
In-Process Research and Development
In-process research and development ("IPRD") relates to Aytu's Zertane product and clinical trial data acquired in connection with the 2011 business combination of BioSciences. The $7,500,000 recorded was based on an independent, third party appraisal of the fair value of the assets acquired. IPRD is considered an indefinite-lived intangible asset and its fair value will be assessed annually and written down if impaired. Once the Zertane product obtains regulatory approval and commercial production begins, IPRD will be reclassified to an intangible that will be amortized over its estimated useful life. If Aytu decided to abandon the Zertane product, the IPRD would be expensed.
F-10
Patents
Costs of establishing patents, consisting of legal and filing fees paid to third parties, are expensed as incurred. The fair value of the Zertane patents, determined by an independent, third party appraisal to be $500,000, acquired in connection with the 2011 acquisition of BioSciences is being amortized over the remaining U.S. patent life since our acquisition of approximately 11 years. The fair value of the Luoxis patents was $380,000 when they were acquired in connection with the 2013 formation of Luoxis and is being amortized over the remaining U.S. patent life since our acquisition of approximately 15 years. Patents consist of the following:
| December 31, | ||||||||
| 2015 | 2014 | |||||||
Patents | $ | 880,000 | $ | 880,000 | ||||
Less accumulated amortization | (287,000 | ) | (216,000 | ) | ||||
|
|
|
| |||||
Patents, net | $ | 593,000 | $ | 664,000 | ||||
|
|
|
| |||||
The Company recorded the following amortization expense in the respective periods:
| Year Ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
Amortization Expense | $ | 71,000 | $ | 71,000 | $ | 66,000 | ||||||
Aytu's future amortization is as follows:
2016 | $ | 71,000 | ||
2017 | 71,000 | |||
2018 | 71,000 | |||
2019 | 71,000 | |||
2020 | 71,000 | |||
Thereafter | 238,000 | |||
|
| |||
| $ | 593,000 | |||
|
|
Business Combinations
The Company accounts for its business acquisitions under the acquisition method of accounting as indicated in the Financial Accounting Standards Board's ("FASB") Accounting Standards Codification ("ASC") 805, "Business Combinations", which requires the acquiring entity in a business combination to recognize the fair value of all assets acquired, liabilities assumed, and any non-controlling interest in the acquiree; and establishes the acquisition date as the fair value measurement point. Accordingly, the Company recognizes assets acquired and liabilities assumed in business combinations, including contingent assets and liabilities and non-controlling interest in the acquiree, based on the fair value estimates as of the date of acquisition. In accordance with ASC 805, the Company recognizes and measures goodwill as of the acquisition date, as the excess of the fair value of the consideration paid over the fair value of the identified net assets acquired.
Goodwill
The ProstaScint and Primsol purchase price allocations were based upon an analysis of the fair value of the assets and liabilities acquired. The final purchase price may be adjusted up to one year from the date of the acquisition. Identifying the fair value of the tangible and intangible assets and liabilities acquired required the use of estimates by management, and were based upon currently available data, as noted below.
The Company allocated the excess of purchase price over the identifiable intangible and net tangible assets to goodwill. Such goodwill is not deductible for tax purposes and represents the value placed on entering new markets and expanding market share.
F-11
The Company tests its goodwill for impairment annually, or whenever events or changes in circumstances indicate an impairment may have occurred, by comparing the carrying value to its implied fair value. Impairment may result from, among other things, deterioration in the performance of the acquired business, adverse market conditions, adverse changes in applicable laws or regulations and a variety of other circumstances. If the Company determines that an impairment has occurred, it is required to record a write-down of the carrying value and charge the impairment as an operating expense in the period the determination is made. In evaluating the recoverability of the carrying value of goodwill, the Company must make assumptions regarding estimated future cash flows and other factors to determine the fair value of the acquired assets. Changes in strategy or market conditions could significantly impact those judgments in the future and require an adjustment to the recorded balances. The goodwill was recorded as part of the acquisition of ProstaScint that occurred in May 2015 and the acquisition of Primsol that occurred in October 2015. There was no impairment of goodwill for the year ended December 31, 2015.
Use of Estimates
The preparation of consolidated financial statements in accordance with Generally Accepted Accounting Principles in the United States of America, or GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, disclosures of contingent assets and liabilities as of the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting periods. Significant items subject to such estimates and assumptions include the fair value of warrant derivative liability, hybrid debt instruments, valuation allowances, stock-based compensation, useful lives of fixed assets, estimates of contingent consideration in business combinations and assumptions in evaluating impairment of indefinite lived assets. Actual results could differ from these estimates.
Derivatives
Ampio accounted for hybrid financial instruments (debentures with embedded derivative features – conversion options, down-round protection and mandatory conversion provisions) and related warrants by recording the fair value of each hybrid instrument in its entirety and recording the fair value of the warrant derivative liability. The fair value of the hybrid financial instruments and related warrants was calculated using a binomial-lattice-based valuation model. Ampio recorded a derivative expense at the inception of each instrument reflecting the difference between the fair value and cash received. Changes in the fair value in subsequent periods were recorded as unrealized gain or loss on fair value of debt instruments for the hybrid financial instruments and to derivative income or expense for the warrants. Accounting for hybrid financial instruments and derivatives is discussed more fully in Note 4 – Derivative Financial Instruments. The fair value of warrants issued in connection with the common stock offerings was valued using a Black-Scholes option pricing model. The fair value of the warrants issued in connection with the convertible debt were valued using the Monte Carlo valuation methodology.
Income Taxes
Deferred taxes are provided on an asset and liability method whereby deferred tax assets are recognized for deductible temporary differences and operating loss and tax credit carry forwards and deferred tax liabilities are recognized for taxable temporary differences. Temporary differences are the differences between the reported amounts of assets and liabilities and their tax bases. Deferred tax assets are reduced by a valuation allowance when, in the opinion of management, it is more likely than not that some portion or all of the deferred tax assets will not be realized. Deferred tax assets and liabilities are adjusted for the effects of changes in tax laws and rates on the date of enactment.
Net Loss per Common Share
Basic net loss per share includes no dilution and is computed by dividing the net loss available to common stockholders by the weighted-average number of shares outstanding during the period. Diluted net loss per share reflects the potential of securities that could share in the net loss of Ampio. Basic and diluted loss per share was the same in 2015, 2014 and 2013. Although there were common stock equivalents of 7,814,908, 7,084,577 and 5,662,748 shares outstanding at December 31, 2015, 2014 and 2013, respectively, consisting of stock options and warrants; they were not included in the calculation of the diluted net loss per share because they would have been anti-dilutive.
Stock-Based Compensation
Ampio accounts for share based payments by recognizing compensation expense based upon the estimated fair value of the awards on the date of grant. Ampio determines the estimated grant fair value using the Black-Scholes option pricing model and recognizes compensation costs ratably over the vesting period using the graded method.
F-12
Research and Development
Research and development costs are expensed as incurred with expense recorded in the respective periods as follows:
| Year Ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
Research and development costs | $ | 18,961,000 | $ | 26,923,000 | $ | 16,596,000 | ||||||
Fair Value of Financial Instruments
The carrying amounts of financial instruments, including cash and cash equivalents, accounts receivable, accounts payable and accrued liabilities and convertible notes are carried at cost which approximates fair value due to the short maturity of these instruments. Hybrid financial instruments such as convertible debentures and related warrants were recorded at estimated fair value based on a binomial-lattice-based valuation model , see Note – 4 Derivative Financial Instruments Related to Ampio for additional information . The fair value of warrants issued in connection with the Aytu convertible notes was valued using a Monte Carlo option pricing model. The accounting for financial instruments and the Aytu derivatives is discussed more fully in Note 5 – Fair Value Considerations.
Impairment of Long-Lived Assets
Ampio routinely performs an annual evaluation of the recoverability of the carrying value of its long-lived assets to determine if facts and circumstances indicate that the carrying value of assets or intangible assets may be impaired and if any adjustment is warranted. Based on Ampio's evaluation as of December 31, 2015, no impairment existed for long-lived assets.
F-13
Adoption of Recent Accounting Pronouncements
In September 2015, the FASB issued Accounting Standards Update ("ASU") 2015-16, "Business Combinations (Topic 805): Simplifying the Accounting for Measurement-Period Adjustments," which requires that an acquirer recognize adjustments to estimated amounts that are identified during the measurement period in the reporting period in which the adjustment amounts are determined. The amendments require that the acquirer record, in the same period's financial statements, the effect on earnings of changes in depreciation, amortization, or other income effects, if any, as a result of the change to the estimated amounts, calculated as if the accounting had been completed at the acquisition date. The amendments also require an entity to present separately on the face of the income statement or disclose in the notes the portion of the amount recorded in current-period earnings by line item that would have been recorded in previous reporting periods if the adjustment to the estimated amounts had been recognized as of the acquisition date. The amendment is effective for financial statements issued for fiscal years beginning after December 15, 2015 and early adoption is permitted. As of December 31, 2015, the Company early adopted this standard.
In April 2015, the FASB issued ASU 2015-03, "Interest-Imputation of Interest (Subtopic 835-30): Simplifying the Presentation of Debt Issuance Costs" to simplify the presentation of debt issuance costs. The amendments in the update require that debt issuance costs related to a recognized debt liability be presented in the balance sheet as a direct reduction of the carrying amount of the debt. Recognition and measurement of debt issuance costs were not affected by this amendment. In August 2015, FASB issued ASU 2015-15, "Presentation and Subsequent Measurement of Debt Issuance Costs Associated with Line-of-Credit Arrangements - Amendments to SEC Paragraphs Pursuant to Staff Announcement at June 18, 2015 EITF Meeting" which clarified that the SEC would not object to an entity deferring and presenting debt issuance costs as an asset and subsequently amortizing the deferred debt issuance costs ratably over the term of the line-of-credit arrangement. The amendments are effective for financial statements issued for fiscal years beginning after December 15, 2015. As of December 31, 2015, the Company early adopted this standard and will record any debt issuance costs as a debt discount. There was no impact related to this adoption as the Company did not have any debt issuance costs previously.
In November 2015, the FASB issued ASU 2015-17 regarding ASC Topic 470 "Income Taxes: Balance Sheet Classification of Deferred Taxes." The amendments in ASU 2015-17 eliminate the requirement to bifurcate deferred taxes between current and non-current on the balance sheet and requires that deferred tax liabilities and assets be classified as noncurrent on the balance sheet. The amendments for ASU-2015-17 can be applied retrospectively or prospectively and early adoption is permitted. Ampio early adopted ASU 2015-17 and there was no material impact on its financial statements.
Recent Accounting Pronouncements
In February 2016, the FASB issued Accounting Standards Update 2016-02, "Leases (Topic 842)". The new standard establishes a right-of-use (ROU) model that requires a lessee to record a ROU asset and a lease liability on the balance sheet for all leases with terms longer than 12 months. Leases will be classified as either finance or operating, with classification affecting the pattern of expense recognition in the income statement. The new standard is effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. A modified retrospective transition approach is required for lessees for capital and operating leases existing at, or entered into after, the beginning of the earliest comparative period presented in the financial statements, with certain practical expedients available. We are currently evaluating the impact of our pending adoption of this standard on our financial statements.
In January 2016, the FASB issued ASU 2016-01, "Financial Instruments – Overall (Subtopic 825-10): Recognition Measurement and of Financial Assets and Financial Liabilities," which requires that all equity investments to be measured at fair value with changes in the fair value recognized through net income (other than those accounted for under equity method of accounting or those that result in consolidation of the investee). The amendments in this update also require an entity to present separately in other comprehensive income the portion of the total change in the fair value of a liability resulting from a change in the instrument-specific credit risk when the entity has elected to measure the liability at fair value in accordance with the fair value option for financial instruments. In addition, the amendments in this update eliminate the requirement to disclose the fair value of financial instruments measured at amortized cost for entities that are not public business entities and the requirement for to disclose the method(s) and significant assumptions used to estimate the fair value that is required to be disclosed for financial instruments measured at amortized cost on the balance sheet for public business entities. The amendment is effective for financial statements issued for fiscal years beginning after December 15, 2017. Early adoption is not permitted. The Company is currently evaluating the impact of this standard on its financial statements.
In July 2015, the FASB issued ASU 2015-11, "Simplifying the Measurement of Inventory." ASU 2015-11 clarifies that inventory should be held at the lower of cost or net realizable value. Net realizable value is defined as the estimated selling price, less the estimated costs to complete, dispose and transport such inventory. ASU 2015-11 will be effective for fiscal years and interim periods beginning after December 15, 2016. ASU 2015-11 is required to be applied prospectively and early adoption is permitted. The adoption of ASU 2015-11 is not expected to have a material impact on the Company's financial position or results of operations.
In August 2014, the FASB issued ASU 2014-15, "Presentation of Financial Statements-Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity's Ability to Continue as a Going Concern" ("ASU 2014-15"). ASU 2014-15 is intended to define management's responsibility to evaluate whether there is substantial doubt about an organization's ability to continue as a going concern and to provide related footnote disclosures. The amendments in this ASU are effective for reporting periods ending after December 15, 2016, with early adoption permitted. The Company is evaluating the impact the adoption of ASU 2014-15 will have on its financial statements.
In May 2014, the FASB issued ASU 2014-09 regarding ASC Topic 606, "Revenue from Contracts with Customers". The standard provides principles for recognizing revenue for the transfer of promised goods or services to customers with the consideration to which the entity expects to be entitled in exchange for those goods or services. In August 2015, the FASB issued ASU 2015-14, Revenue from Contracts with Customers: Deferral of the Effective Date, which deferred the effective date of the new revenue standard
F-14
for periods beginning after December 15, 2017, with early adoption permitted. Entities have the option of using either a full retrospective or a modified retrospective approach to adopt this new guidance. We are currently evaluating the impact of our pending adoption of this standard on our financial statements.
Note 3 – Revenue Recognition
The $1,077,000 product and service revenue recognized in 2015 represents sales from Ampio's Aytu segment which includes the ProstaScint and Primsol products and the RedoxSYS System. We did not generate any product and service revenue in 2014 or 2013.
The $86,000, $77,000 and $50,000 license revenue recognized in 2015, 2014 and 2013 respectively, represents the amortization of the upfront payments received from Ampio's license agreements. The initial payment of $500,000 from the license agreement of Zertane with a Korean pharmaceutical company was deferred and is being recognized over ten years. The initial payment of $250,000 from the license agreement of Zertane with a Canadian-based supplier was deferred and is being recognized over seven years.
Note 4 - Derivative Financial Instruments Related to Ampio
The warrants associated with the derivative liability expired in December 2013. All of the warrants were exercised prior to expiration.
Ampio elected to measure the Senior Convertible Debentures issued in 2010 at fair value in their entirety, rather than bifurcating the conversion option. The fair value of the hybrid debt instrument comprises the present value of the principal and coupon enhanced by the conversion option. Both the warrants and the conversion options embedded in the hybrid debt instruments were valued using a binomial-lattice-based valuation model. The lattice-based valuation technique was utilized because it embodies all of the requisite assumptions (including the underlying price, exercise price, term, volatility, and risk-free interest-rate) that are necessary to fair value these instruments. For forward contracts that contingently require net-cash settlement as the principal means of settlement, Ampio projects and discounts future cash flows applying probability-weighting to multiple possible outcomes. Estimating fair values of derivative financial instruments requires the development of significant and subjective estimates that may, and are likely to, change over the duration of the instrument with related changes in internal and external market factors. In addition, option-based techniques are highly volatile and sensitive to changes in the trading market price of Ampio's common stock, which has a high-historical volatility. Since derivative financial instruments are initially and subsequently carried at fair value, Ampio's income has reflected the volatility in these estimate and assumption changes.
The following table summarizes the effects on Ampio's income (expense) associated with changes in the fair value of Ampio's derivative financial instruments by type of financing for the respective periods:
| Year Ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
Warrants (dates correspond to financing) | ||||||||||||
Tranche 1 - August 10, 2010 | $ | - | $ | - | $ | (184,000 | ) | |||||
Tranche 2 - October 22, 2010-October 29, 2010 | - | - | - | |||||||||
Tranche 3 - November 12, 2010-November 29, 2010 | - | - | (254,000 | ) | ||||||||
Tranche 4 - December 13, 2010-December 29, 2010 | - | - | (35,000 | ) | ||||||||
Tranche 5 - January 20, 2011-January 31, 2011 | - | - | (44,000 | ) | ||||||||
|
|
|
|
|
| |||||||
| $ | - | $ | - | $ | (517,000 | ) | ||||||
|
|
|
|
|
| |||||||
Note 5 – Fair Value Considerations
The Company's financial instruments include cash and cash equivalents, accounts receivable, accounts payable and accrued liabilities, convertible notes, warrant derivative liability. Primsol payable and contingent consideration. The carrying amounts of cash and cash equivalents, accounts receivable, accounts payable and accrued liabilities approximate their fair value due to their short maturities. The fair value of the convertible notes is approximately the face value of the notes, $5,175,000 based upon the valuation that the Company had completed of all components of the convertible notes at inception and as of December 31, 2015. Subsequent to December 31, 2015, the majority of the Company's convertible notes converted into common stock, (see Note 16 – Subsequent Events for more information). The fair value of the Primsol payable and contingent consideration is based upon discounted cash flow and sales projections. The valuation policies are determined by the Chief Financial Officer and approved by the Company's Board of Directors. Authoritative guidance defines fair
F-15
value as the price that would be received to sell an asset or paid to transfer a liability (an exit price) in an orderly transaction between market participants at the measurement date. The guidance establishes a hierarchy for inputs used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the most observable inputs be used when available. Observable inputs are inputs that market participants would use in pricing the asset or liability developed based on market data obtained from sources independent of Ampio. Unobservable inputs are inputs that reflect the Company's assumptions of what market participants would use in pricing the asset or liability developed based on the best information available in the circumstances. The hierarchy is broken down into three levels based on reliability of the inputs as follows:
| Level 1: | Inputs that reflect unadjusted quoted prices in active markets that are accessible to Ampio for identical assets or liabilities; | |
| Level 2: | Inputs include quoted prices for similar assets and liabilities in active or inactive markets or that are observable for the asset or liability either directly or indirectly; and | |
| Level 3: | Unobservable inputs that are supported by little or no market activity. | |
Ampio's assets and liabilities which are measured at fair value are classified in their entirety based on the lowest level of input that is significant to their fair value measurement. Ampio's policy is to recognize transfers in and/or out of fair value hierarchy as of the date in which the event or change in circumstances caused the transfer. Ampio has consistently applied the valuation techniques discussed below in all periods presented.
The following table presents Ampio's financial liabilities that were accounted for at fair value on a recurring basis as of December 31, 2015, by level within the fair value hierarchy:
| Fair Value Measurements Using | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
December 31, 2015 | ||||||||||||||||
LIABILITIES | ||||||||||||||||
Warrant derivative liability | $ | - | $ | - | $ | 181,000 | $ | 181,000 | ||||||||
The warrant derivative liability for the warrants was valued using the Monte Carlo valuation methodology because that model embodies all of the relevant assumptions that address the features underlying these instruments. Significant assumptions in valuing the warrant derivative liability based on estimates of the value of Aytu common stock and various factors regarding the warrants were as follows as of December 31, 2015 and at issuance:
| December 31, 2015 | At Issuance | |||
Warrants: | ||||
Exercise price | $0.98 | $1.51 - $1.95 | ||
Volatility | 75.0% | 75.0% | ||
Equivalent term (years) | 4.67 | 5.0 - 5.11 | ||
Risk-free interest rate | 1.57% - 1.60% | 1.54% - 1.74% | ||
Potential number of shares | 126,000 - 215,000 | 139,000 - 224,000 |
The following table sets forth a reconciliation of changes in the fair value of financial liabilities classified as Level 3 in the fair valued hierarchy:
| Derivative Instruments | ||||
Balance as of December 31, 2014 | $ | - | ||
Warrant issuances | 103,000 | |||
Included in earnings | 78,000 | |||
|
| |||
Balance as of December 31, 2015 | $ | 181,000 | ||
|
| |||
F-16
Note 6 – Income Taxes
Income tax benefit resulting from applying statutory rates in jurisdictions in which Ampio is taxed (Federal and State of Colorado) differs from the income tax provision (benefit) in Ampio's consolidated financial statements. The following table reflects the reconciliation for the respective periods:
| Years Ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
Benefit at federal statutory rate | (34.0 | )% | (34.0 | )% | (34.0 | )% | ||||||
State, net of federal income tax impact | (2.9 | )% | (3.1 | )% | (3.1 | )% | ||||||
Stock-based compensation | 1.8 | % | 3.5 | % | 3.7 | % | ||||||
True-up and applicable rate adjustment | - | % | (0.5 | %) | - | % | ||||||
Change in valuation allowance | 35.1 | % | 34.1 | % | 33.4 | % | ||||||
|
|
|
|
|
| |||||||
Effective tax rate | 0.0 | % | 0.0 | % | 0.0 | % | ||||||
|
|
|
|
|
| |||||||
Deferred income taxes arise from temporary differences in the recognition of certain items for income tax and financial reporting purposes. The approximate tax effects of significant temporary differences which comprise the deferred tax assets and liabilities are as follows for the respective periods:
| 2015 | 2014 | 2013 | ||||||||||
Long-term deferred income tax assets (liabilities): | ||||||||||||
Accrued liabilities | $ | 511,000 | $ | 42,000 | $ | - | ||||||
Other reserves and allowances | 73,000 | - | - | |||||||||
Deferred rent | 259,000 | - | - | |||||||||
Net operating loss carryforward | 41,898,000 | 32,273,000 | 20,856,000 | |||||||||
Intangible assets | 477,000 | 466,000 | 499,000 | |||||||||
Deferred revenue license agreement | 174,000 | 206,000 | 142,000 | |||||||||
Share-based compensation expense | 4,271,000 | 2,788,000 | 992,000 | |||||||||
Property and equipment | (182,000 | ) | (43,000 | ) | (65,000 | ) | ||||||
Acquired in-process research and development | (2,780,000 | ) | (2,780,000 | ) | (2,780,000 | ) | ||||||
Warrants | 43,000 | - | - | |||||||||
Less: Valuation allowance | (44,744,000 | ) | (32,952,000 | ) | (19,644,000 | ) | ||||||
|
|
|
|
|
| |||||||
Total long-term deferred income tax assets (liabilities) | $ | - | $ | - | $ | - | ||||||
|
|
|
|
|
| |||||||
During the year ended December 31, 2015, Ampio adopted the guidance issued in ASU 2015-17 on presentation of deferred tax liabilities and assets. The guidance requires that deferred tax liabilities and assets be classified as noncurrent in a classified statement of financial position. Adoption of this new guidance did not have a material impact on the Company's financial statements and adoption served to simplify the presentation of the Company's deferred income taxes while maintaining the usefulness of the information provided. For the years ended December 31, 2015, 2014 and 2013, Ampio's net provision for income taxes was zero for all jurisdictions.
As of December 31, 2015, Ampio has approximately $114.0 million in consolidated net operating loss ("NOL") carryforwards that, subject to limitation, may be available in future tax years to offset taxable income. These net operating loss carryforwards expire in 2019 through 2035. Under the provisions of the Internal Revenue Code, substantial changes in the Company's ownership may result in limitations on the amount of NOL carryforwards that can be utilized in future years. As a result of certain realization requirements of GAAP, the table of deferred tax assets and liabilities shown above does not include certain deferred tax assets as of December 31, 2015, 2014 and 2013 that arose directly from (or the use of which was postponed by) tax deductions related to equity compensation in excess of compensation expense recognized for financial reporting. Those deferred tax assets include approximately $5.0 million of net operating loss deductions. Equity will be increased if and when such deferred tax assets are ultimately realized.
Ampio has provided a full valuation allowance against its deferred tax assets as it has determined that it is not more likely than not that recognition of such deferred tax assets will be utilized in the foreseeable future. The amount of income taxes and related income tax
F-17
positions taken are subject to audits by federal and state tax authorities. Ampio has adopted accounting guidance for uncertain tax positions which provides that in order to recognize an uncertain tax benefit, the taxpayer must be more likely than not of sustaining the position, and the measurement of the benefit is calculated as the largest amount that is more than 50% likely to be realized upon recognition of the benefit. Ampio believes that it has no material uncertain tax positions and has fully reserved against Ampio's future tax benefit with a valuation allowance and do not expect significant changes in the amount of unrecognized tax benefits that occur within the next twelve months. Ampio's policy is to record a liability for the difference between benefits that are both recognized and measured pursuant to GAAP and tax positions taken or expected to be taken on the tax return. Then, to the extent that the assessment of such tax positions changes, the change in estimate is recorded in the period in which the determination is made. Ampio reports tax-related interest and penalties as a component of income tax expense. During the periods reported, management of Ampio has concluded that no significant tax position requires recognition. Ampio files income tax returns in the United States federal and Colorado state jurisdictions. The Company is no longer subject to income tax examinations for federal income taxes before 2011 or for Colorado before 2010. Net operating loss carryforwards are subject to examination in the year they are utilized regardless of whether the tax year in which they are generated has been closed by statute. The amount subject to disallowance is limited to the NOL utilized. Accordingly, the Company may be subject to examination for prior NOLs generated as such NOLs are utilized.
Note 7 – Commitments and Contingencies
The following table summarizes the commitments and contingencies including Aytu (see Note 16 – Subsequent Events) as of December 31, 2015 which are described below:
| Total | 2016 | 2017 | 2018 | 2019 | 2020 | Thereafter | ||||||||||||||||||||||
Ampion supply agreement | $ | 5,100,000 | $ | - | $ | 2,550,000 | $ | 2,550,000 | $ | - | $ | - | $ | - | ||||||||||||||
Clinical research and trial obligations | 4,275,000 | 4,275,000 | - | - | - | - | - | |||||||||||||||||||||
Facility leases | 3,309,000 | 436,000 | 450,000 | 418,000 | 326,000 | 335,000 | 1,344,000 | |||||||||||||||||||||
Sponsored research agreement with related party | 1,406,000 | 395,000 | 395,000 | 395,000 | 151,000 | 70,000 | - | |||||||||||||||||||||
Primsol business | 1,250,000 | 1,250,000 | - | - | - | - | - | |||||||||||||||||||||
Manufacturing agreement | 1,000,000 | 1,000,000 | - | - | - | - | - | |||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||
| $ | 16,340,000 | $ | 7,356,000 | $ | 3,395,000 | $ | 3,363,000 | $ | 477,000 | $ | 405,000 | $ | 1,344,000 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||
Ampion Supply Agreement
In connection with the manufacturing facility/clean room, in October 2013, Ampio entered into a human serum albumin ingredient and purchase sale agreement with a remaining commitment of $5,100,000. Per an amendment to the original agreement, Ampio is not committed to purchases in 2016 and has extended the agreement to 2018.
Clinical Research and Trial Obligations
In connection with current and recent clinical trials, as of December 31, 2015, Ampio has a remaining commitment of $4,263,000 on contracts related to the Ampion study trial expense and $12,000 remaining contract commitments related to the Optina study trial expense.
Facility Leases
In December 2013, Ampio entered into a 125-month non-cancellable operating lease for new office space and the manufacturing facility effective May 1, 2014. The new lease has initial base rent of $23,000 per month, with the total base rent over the term of the lease of approximately $3.3 million and includes rent abatements and leasehold incentives. The Company recognizes rental expense of the facility on a straight-line basis over the term of the lease. Differences between the straight-line net expenses on rent payments are classified as liabilities between current deferred rent and long-term deferred rent.
In June 2015, Aytu entered into a 37-month operating lease for a space in Raleigh, North Carolina. This lease has initial base rent of $2,900 a month, with total base rent over the term of the lease of approximately $112,000. In September 2015, the Company entered into a 37-month operating lease in Englewood, Colorado. This lease has an initial base rent of $8,500 a month with a total base rent over the term of the lease of approximately $318,000. The Company recognizes rental expense of the facilities on a straight-line basis over the term of the lease. Differences between the straight-line net expenses on rent payments are classified as liabilities between current deferred rent and long-term deferred rent.
F-18
Rent expense for the respective periods is as follows:
| Years Ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
Rent expense | $ | 317,000 | $ | 306,000 | $ | 118,000 | ||||||
Sponsored Research Agreement with Related Party
Ampio entered into a Sponsored Research Agreement with TRLLC, a related party, in September 2009. Under the terms of the Sponsored Research Agreement, Ampio is to provide personnel and pay for leased equipment. The Sponsored Research Agreement may be terminated without cause by either party on 180 days' notice. As further noted in Note 11 – Related Party Transactions, in March 2014, the Sponsored Research Agreement was extended through March 2019, including a "no termination" period through March 2017. In a subsequent Addendum, the parties also agreed to increase the equivalent value of the personnel provided by Ampio from $264,000 to $325,000 per year.
Aytu entered into a Sponsored Research Agreement with Trauma Research LLC ("TRLLC"), a related party, in June 2013. Under the terms of the Sponsored Research Agreement, TRLLC agreed to work collaboratively in advancing the RedoxSYS System diagnostic platform through research and development efforts. The Sponsored Research Agreement may be terminated without cause by either party on 30 days' notice.
Primsol Business
In October 2015, Aytu entered into an agreement with FSC Laboratories, Inc. for the purchase of Primsol (see Note 1 – Business Combination - Primsol).
Manufacturing Agreement
In October 2015, Aytu entered into a Master Services Agreement with Biovest International, Inc. ("Biovest"). The agreement provides that Aytu may engage Biovest from time to time to provide services in accordance with mutually agreed upon project addendums and purchase orders. Aytu expects to use the agreement from time to time for manufacturing services, including without limitation, the manufacturing, processing, quality control testing, release or storage of its products for the ProstaScint product. Aytu is obligated to pay Biovest $1.0 million for time and materials as they develop a plan to reproduce the manufacturing process.
Note 8 – Convertible Promissory Notes Pertaining to Aytu
Convertible Promissory Notes
During July and August 2015, Aytu closed on note purchase agreements with institutional and high net worth individual investors for the purchase and sale of convertible promissory notes ("Notes") with an aggregate principal amount of $5.2 million. The sale of the Notes was pursuant to a private placement. Debt issuance costs totaled $401,000 which included $103,000 fair value of the warrants.
The Notes are an unsecured obligation. Unless earlier converted, the Notes will mature 18 months from their respective dates of issuance which will be on January 22, February 11 and February 28, 2017, with an option to extend the maturity date up to six months at Aytu's discretion (provided that in the event Aytu exercises such extension option, the then applicable interest rate shall increase by 2% for such extension period). Aytu does not have the right to prepay the Notes prior to the maturity date. Interest will accrue on the Notes in the following amounts: (i) 8% simple interest per annum for the first six months and (ii) 12% simple interest per annum thereafter if not converted during the first six months. If there had not been a registration statement on Form S-1 filed with the SEC for the registration of the shares of common stock underlying the Notes by the expiration of the first six-month period, then (a) the interest rate would have increased to 14% for the remainder of the period in which the Notes remain outstanding and (b) any Notes held by officers and directors of Aytu would have been subordinated to the remaining Notes. Interest will accrue, is payable with the principal upon maturity, conversion or acceleration of the Notes and may be paid in kind or in cash, in Aytu's sole discretion.
The 4% increase in the interest rate is triggered automatically with the passage of time and is not a contingent feature, thus, there is no initial accounting for this feature. However, the periodic interest cost will be calculated using a constant effective interest over the life of the Notes. As Aytu's management does not intend to utilize the extension option, the expected life of the Notes is 18 months.
F-19
Aytu did not give recognition to the registration rights arrangement as management did not believe at issuance that probable payment under the contingent escalation clause would be required, thus there was no impact on the initial measurement of the Notes. Aytu satisfied the registration rights arrangement in October 2015 upon the effectiveness of a registration statement on Form S-1.
The Notes are convertible at any time at the noteholder's discretion into that number of shares of Aytu common stock equal in an amount equal to 120% of the number of shares of common stock calculated by dividing the then outstanding principal and accrued interest by $4.63. A holder of Notes will be obligated to convert on the terms of Aytu's next public offering of its stock resulting in gross proceeds of at least $5,000,000 (excluding indebtedness converted in such financing) prior to the maturity date of the Notes (a "Qualified Financing"). The principal and accrued interest under the Notes will automatically convert into a number of shares of such equity securities of Aytu sold in the Qualified Financing equal to 120% of the principal and accrued interest under such Note divided by the lesser of (i) the lowest price paid by an investor in the Qualified Financing or (ii) $4.63. In the event that Aytu sells equity securities to investors at any time while the Notes are outstanding in a financing transaction that is not a Qualified Financing, then the noteholders will have the option to convert in whole the outstanding principal and accrued interest as of the closing of such financing into a number of shares of Aytu capital stock in an amount equal to 120% of the number of such shares calculated by dividing the outstanding principal and accrued interest by the lesser of (i) the lowest cash price per share paid by purchasers of shares in such financing, or (ii) $4.63.
Aytu determined that the conversion option is not required to be bifurcated and accounted for as an embedded derivative liability. There was no intrinsic value to the beneficial conversion feature as it was determined that the effective conversion price exceeded the commitment date valuation price.
The Notes contained a purchase premium option in the event of a sale transaction by Aytu as defined in the Notes. A holder of the Notes will be entitled to receive, at the holder's option, (i) repayment of the Note balance plus the amount equal to 25% of the original purchase amount or (ii) the consideration the holder would have received on an as-converted basis. Given that the payment under the purchase premium is contingent upon a sale transaction and involves a substantial premium of 25%, the purchase premium is an embedded derivative that must be bifurcated and accounted for as an embedded derivative. No value was recorded related to this derivative at issuance and December 31, 2015.
Placement agents for the offerings sold the institutional portion of the offering of the Notes. Aytu sold the balance of the Notes to individuals and entities with whom Aytu has an established relationship. For Notes sold by the placement agent, Aytu paid the placement agent 8% of the gross proceeds of Notes sold by the placement agents and is obligated to issue warrants for an amount of shares to be equal to 8% of the gross number of shares of Aytu stock issuable upon conversion of the Notes issued to investors introduced to Aytu by the private placement agents in the private placement, in addition to a previously paid non-refundable retainer fee of $20,000. The placement agent warrants have a term of five years, will have an exercise price equal to the lowest conversion price per share at which the Notes are converted into common stock. Change in fair value is recorded in earnings. Fair value at the grant date was recorded as a debt discount and amortized over the term of the debt. The warrants were recorded at fair value as long-term liabilities on the Balance Sheet. See Note 5 Fair Value Considerations.
Upon our adoption of ASU 2015-3, the costs associated with the Notes were recorded as a long–term liability and are presented in the Balance Sheet as a direct reduction of the carrying amount of the Notes on their inception date.
As of December 31, 2015, the carrying value of the Notes was $4.9 million inclusive of an unamortized debt discount of $253,000.
Note 9 – Common Stock
Capital Stock
At December 31, 2015 and 2014, Ampio had 100.0 million shares of common stock authorized with a par value of $0.0001 per share and 10.0 million shares of preferred stock authorized with a par value of $0.0001 per share.
Shelf Registration
In December 2013, Ampio filed an additional shelf registration statement on Form S-3 with the Securities and Exchange Commission to register Ampio common stock and warrants in an aggregate amount of up to $100.0 million for offering from time to time in the future, as well as 1.5 million shares of common stock available for sale by selling shareholders. The shelf registration was declared effective in January 2014 by the Securities and Exchange Commission. As a result of equity raises, approximately $86.3 million remains available under the Form S-3 filed in December 2013 as of December 31, 2015.
F-20
Underwritten Public Offerings
In March 2014, Ampio completed an underwritten public offering for the sale of 9,775,000 shares of common stock at a price of $7.00 per share. Gross proceeds to the Company were $68,425,000 with net proceeds of $63,425,000 after underwriter fees and cash offering expenses.
Private Placement – Luoxis
In 2013, Ampio completed a private placement for its Luoxis subsidiary. A total of 4,652,500 shares of Luoxis common stock were issued at $1.00 per share resulting in $4,653,000 of gross proceeds. Net proceeds were $3,980,000 after placement agent and legal fees. The placement agent also received 465,250 warrants to purchase Luoxis common stock valued at $313,000 in connection with the closing, which amount has been included in total offering costs in the consolidated statement of changes in stockholders' equity (deficit).
Registered Direct Placement
In September 2013, Ampio closed on the sale of 4,600,319 shares of common stock at $5.50 per share, for a total of $25,302,000 of gross proceeds and $25,004,000 net proceeds after offering costs. The sale of the common stock was made pursuant to the September 2011 Form S-3 Shelf Registration.
Common Stock Issued for Services
Ampio issued 7,998, 7,752 and 4,209 shares valued at $30,000, $30,000 and $30,000 for non-employee directors as part of their director fees in 2015, 2014 and 2013, respectively.
Note 10 – Equity Instruments
Options
In 2010, Ampio shareholders approved the adoption of a stock and option award plan (the "2010 Plan"), under which shares were reserved for future issuance under restricted stock awards, options, and other equity awards. The 2010 Plan permits grants of equity awards to employees, directors and consultants. The shareholders have approved a total of 11.7 million shares reserved for issuance under the 2010 plan.
During 2013, an additional 1,120,000 options were granted at a weighted average exercise price of $6.54 to officers, directors, employees and consultants. Of the options granted, 130,000 options vested immediately while the remaining 990,000 vest over a one to three-year period.
During 2014, we granted 1,645,000 options at a weighted average exercise price of $5.63 to officers, directors, employees and consultants. Of the options granted, 592,500 options vested immediately while the remaining 1,052,500 vest over a one to four-year period.
During 2015, we granted 1,093,000 options at a weighted average exercise price of $3.28 to officers, directors, employees and consultants. Of the options granted, 45,000 options vested immediately while 303,000 vest over a one to three-year period. The remaining 470,000 options are performance-based options based upon the outcome of the ongoing Ampion trial. The granted options during the year ended December 31, 2015 also included 275,000 of modified options held by a former executive. The expense related to this modification was recognized in the period ended June 30, 2015.
F-21
Stock option activity is as follows:
| Number of Options | Weighted Average Exercise Price | Weighted Average Remaining Contractual Life | ||||||||||
Outstanding December 31, 2012 | 4,922,815 | $ | 2.25 | 8.36 | ||||||||
Granted | 1,120,000 | $ | 6.54 | |||||||||
Exercised | (333,176 | ) | $ | 3.23 | ||||||||
Forfeited/Cancelled | (574,581 | ) | $ | 1.89 | ||||||||
|
| |||||||||||
Outstanding December 31, 2013 | 5,135,058 | $ | 3.54 | 8.74 | ||||||||
Granted | 1,645,000 | $ | 5.63 | |||||||||
Exercised | (157,226 | ) | $ | 1.95 | ||||||||
Forfeited/Cancelled | (54,584 | ) | $ | 3.29 | ||||||||
|
| |||||||||||
Outstanding December 31, 2014 | 6,568,248 | $ | 3.82 | 7.66 | ||||||||
Granted | 1,093,000 | $ | 3.28 | |||||||||
Exercised | (10,416 | ) | $ | 2.76 | ||||||||
Forfeited | (275,000 | ) | $ | 4.80 | ||||||||
Expired or Cancelled | (60,000 | ) | $ | 3.53 | ||||||||
|
| |||||||||||
Outstanding December 31, 2015 | 7,315,832 | $ | 3.71 | 6.58 | ||||||||
|
| |||||||||||
Exercisable at December 31, 2015 | 6,104,937 | $ | 3.68 | 6.06 | ||||||||
|
| |||||||||||
Available for grant at December 31, 2015 | 2,989,773 | |||||||||||
|
| |||||||||||
F-22
Stock options outstanding and exercisable at December 31, 2015 are summarized in the table below:
Range of Exercise Prices | Number of Options Outstanding and Exercisable | Weighted Average Exercise Price | Weighted Average Remaining Contractual Lives | |||||||||
$1.03 - $4.00 | 5,140,832 | $ | 2.38 | 6.17 | ||||||||
$4.01 - $7.00 | 1,240,000 | $ | 6.17 | 7.75 | ||||||||
$7.01 - $8.93 | 935,000 | $ | 7.73 | 7.27 | ||||||||
|
| |||||||||||
| 7,315,832 | $ | 3.71 | 6.58 | |||||||||
|
| |||||||||||
| Year Ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
Average fair value per share granted | $ | 0.98 | $ | 4.14 | $ | 3.32 | ||||||
Ampio has computed the fair value of all options granted using the Black-Scholes option pricing model. In order to calculate the fair value of the options, certain assumptions are made regarding components of the model, including the estimated fair value of the underlying common stock, risk-free interest rate, volatility, expected dividend yield and expected option life. Changes to the assumptions could cause significant adjustments to valuation. Ampio calculates its volatility assumption using the actual changes in the market value of our stock. Ampio has estimated a forfeiture rate of 5.0-5.7% based upon historical experience; this is an estimate of options granted that are expected to be forfeited or cancelled before becoming fully vested. Ampio estimates the expected term based on the average of the vesting term and the contractual term of the options. The risk-free interest rate is based on the U.S. Treasury yield in effect at the time of the grant for treasury securities of similar maturity. Accordingly, Ampio has computed the fair value of all options granted during the respective years, using the following assumptions:
| Years Ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
Expected volatility | 104% - 113 | % | 72% - 108 | % | 70% - 89 | % | ||||||
Risk free interest rate | 0.05% - 1.64 | % | 1.51% - 2.27 | % | 0.40% - 2.12 | % | ||||||
Expected term (years) | 1.5 - 6.25 | 5.0 - 7.0 | 3.0 - 6.5 | |||||||||
Dividend yield | 0.0 | % | 0.0 | % | 0.0 | % | ||||||
The Luoxis options that were in the money and all outstanding Vyrix options issued under the respective 2013 Option Plans were accelerated and cancelled in connection with the Merger. Option holders received a cash payment per option share equal to the difference between the consideration payable per share of common stock pursuant to the Merger and the exercise price of the option, if the consideration paid to holders of common stock was less than the exercise price of such options, no amount was paid to the option holder in connection with the cancellation. The cash payment during the period ended June 30, 2015 was $27,000. The Company recognized compensation of $422,000 and $189,000 related to the Luoxis and Vyrix options that had accelerated vesting as of the Merger date.
The Luoxis options that were not paid out were terminated pursuant to the terms of the 2013 Luoxis Option Plan. The Company treated these options as pre-vesting forfeitures and $433,000 of previously recognized compensation was reversed.
On June 1, 2015, Aytu's stockholders approved the 2015 Stock Option and Incentive Plan (the "2015 Plan"), which provides for the award of stock options, stock appreciation rights, restricted stock and other equity awards for up to an aggregate of 10 million shares of common stock. The shares of common stock underlying any awards that are forfeited, canceled, reacquired by Aytu prior to vesting, satisfied without any issuance of stock, expire or are otherwise terminated (other than by exercise) under the 2015 Plan will be added back to the shares of common stock available for issuance under the 2015 Plan.
F-23
The fair value of the options was calculated using the Black-Scholes option pricing model. In order to calculate the fair value of the options, certain assumptions are made regarding components of the model, including the estimated fair value of the underlying common stock, risk-free interest rate, volatility, expected dividend yield and expected option life. Changes to the assumptions could cause significant adjustments to valuation. Aytu estimates the expected term based on the average of the vesting term and the contractual term of the options. The risk-free interest rate is based on the U.S. Treasury yield in effect at the time of the grant for treasury securities of similar maturity. Aytu has computed the fair value of all options granted during the year ended December 31, 2015 using the following assumptions:
Expected volatility | 75.00% | |
Risk free interest rate | 1.08% - 2.08% | |
Expected term (years) | 3.0 - 7.0 | |
Dividend yield | 0% |
Aytu stock option activity is as follows:
| Number of Options | Weighted Average Exercise Price | Weighted Average Remaining Contractual Life in Years | ||||||||||
Outstanding December 31, 2014 | - | $ | - | |||||||||
Granted | 3,695,000 | $ | 1.55 | |||||||||
Exercised | - | $ | - | |||||||||
Forfeited/Cancelled | - | $ | - | |||||||||
|
| |||||||||||
Outstanding December 31, 2015 | 3,695,000 | $ | 1.55 | 9.80 | ||||||||
|
| |||||||||||
Exercisable at December 31, 2015 | 1,120,000 | $ | 1.51 | 9.87 | ||||||||
|
| |||||||||||
Available for grant at December 31, 2015 | 6,305,000 | |||||||||||
|
| |||||||||||
F-24
Stock-based compensation expense related to the fair value of stock options was included in the consolidated statements of operations as research and development expenses and selling, general and administrative expenses as set forth in the table below. Ampio and its subsidiary determined the fair value as of the date of grant using the Black-Scholes option pricing model and expenses the fair value ratably over the vesting period. The following table summarizes stock-based compensation expense for the years ended 2015, 2014 and 2013:
| Years Ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
Research and development expenses | ||||||||||||
Stock options | ||||||||||||
Ampio | $ | 2,866,000 | $ | 4,293,000 | $ | 1,691,000 | ||||||
Aytu | 336,000 | 349,000 | 306,000 | |||||||||
General and administrative expenses | ||||||||||||
Common stock issued for services | 30,000 | 30,000 | 88,000 | |||||||||
Stock options | ||||||||||||
Ampio | 2,092,000 | 2,884,000 | 1,138,000 | |||||||||
Aytu | 455,000 | 327,000 | 313,000 | |||||||||
|
|
|
|
|
| |||||||
| $ | 5,779,000 | $ | 7,883,000 | $ | 3,536,000 | |||||||
|
|
|
|
|
| |||||||
Unrecognized expense at December 31, 2015 | ||||||||||||
Ampio | $ | 1,435,055 | ||||||||||
Aytu | $ | 1,890,000 | ||||||||||
Weighted average remaining years to vest | ||||||||||||
Ampio | 1.49 | |||||||||||
Aytu | 3.16 | |||||||||||
Of the options that Aytu issued during the year ended December 31, 2015, 1,440,000 were to Ampio board members and employees. This was recorded as a return of capital to Ampio and Ampio recorded stock-based compensation expense equal to $1.3 million on their financial statements related to these option grants.
Warrants
Ampio issued warrants in conjunction with its 2011 Senior Convertible Debentures, 2011 Private Placement and an underwritten public offering. A summary of all Ampio warrants is as follows:
| Number of Warrants | Weighted Average Exercise Price | Weighted Average Remaining Contractual Life | ||||||||||
Outstanding December 31, 2012 | 754,371 | $ | 3.00 | 3.01 | ||||||||
Warrants exercised - Debenture holders | (160,679 | ) | $ | 1.75 | ||||||||
Warrants exercised - Private/Registered Direct Placements | (4,504 | ) | $ | 3.13 | ||||||||
Warrants exercised - Private/Registered Direct Placements | (61,498 | ) | $ | 4.06 | ||||||||
|
| |||||||||||
Outstanding December 31, 2013 | 527,690 | $ | 2.93 | 2.44 | ||||||||
Warrants exercised - Private/Registered Direct Placements | (11,361 | ) | $ | 3.13 | ||||||||
|
| |||||||||||
Outstanding December 31, 2014 | 516,329 | $ | 3.26 | 1.44 | ||||||||
Warrants exercised - Private/Registered Direct Placements | (17,253 | ) | $ | 3.94 | ||||||||
|
| |||||||||||
Outstanding December 31, 2015 | 499,076 | $ | 3.24 | 1.19 | ||||||||
|
| |||||||||||
In November 2015, the Company modified select outstanding warrants which extended the expiration for an additional year from March 31, 2016 to March 31, 2017. All of the $422,000 expense related to this modification was recognized in the period ended December 31, 2015.
Luoxis had 465,250 warrants with an exercise price of $1.00 which were converted into Aytu warrants to purchase 102,613 shares of common stock at a price of $4.53. This conversion occurred in April 2015 when the Aytu transaction closed. These warrants have been adjusted to reflect the reverse stock split which occurred in June 2015. All of these warrants remain outstanding with a weighted average remaining contractual life of 2.41 years.
Warrant Obligation related to the Aytu Convertible Promissory Notes
Aytu has the obligation to issue warrants to the private placement agents for the 2015 convertible note financing as part of its fees for the financing. These warrants are classified as a derivative warrant liability due to the fact that the number of shares and exercise price
F-25
have not been set as of December 31, 2015. The number of shares of Aytu stock that these warrants will convert into is equal to 8% of the gross number of shares of the Aytu stock issuable upon conversion of the Notes issued to investors introduced to Aytu by the private placement agents pursuant to the private placement memorandum. The exercise price will be the lower of the lowest conversion price per share at which the Notes are converted into Aytu common stock or $4.63. The warrants have a term of five years from the date the Notes were originally issued in July and August 2015 (see Note 5 - Fair Value Considerations and Note 16 – Subsequent Events).
Note 11 – Related Party Transactions
Ampio entered into a sponsored research agreement with TRLLC, an entity controlled by Ampio's director and Chief Scientific Officer, Dr. Bar-Or, in September 2009, which has been amended six times with the last amendment occurring in January 2015. Under the amended terms of the research agreement, Ampio will provide personnel with an equivalent value of $325,000 per year. With the fifth amendment, Ampio also paid $725,000 in 2014 which is being amortized over the contractual term of 60.5 months and is divided between current and long-term on the balance sheet. In return, TRLLC will assign any intellectual property rights it develops on Ampio's behalf under the research agreement and undertake additional activities to support Ampio's commercial activities and business plan. This agreement is set to expire in March 2019 and cannot be terminated prior to March 2017.
In June 2013, the TRLLC agreement was amended to include Luoxis, which is now a part of Aytu. The agreement, which was amended again in January 2015, provides for Aytu to pay $6,000 per month to TRLLC in consideration for services related to research and development of Aytu's RedoxSYS platform. In March 2014, Luoxis also agreed to pay $615,000 which is being amortized over the contractual term of 60.5 months and is divided between current and long-term on the balance sheet; this amount has been paid in full. This agreement has the same termination and expiration as the agreement between Ampio and TRLLC and was assumed by Aytu.
The Company has advances to one executive and three employees that were used to purchase stock in the Company when it was formed during 2010. These advances are non-interest bearing and due on demand and are classified as a reduction to stockholders' equity. As of December 31, 2015 and 2014, advances of $91,000 to stockholders remained outstanding.
The convertible promissory notes include $275,000 invested by relatives of senior management of Aytu (see Note 8- Convertible Promissory Notes Pertaining to Aytu).
In July 2015, Ampio entered into an agreement with Aytu whereby Aytu agreed to pay Ampio $30,000 per month for shared overhead which includes costs related to the shared facility, corporate staff, and other miscellaneous overhead expenses. These agreements will be in effect until they are terminated in writing by both parties.
F-26
Note 12-Segment Information
Prior to our spin-off of Aytu (see Note 16 – Subsequent Events), we managed our Company and aggregated our operational and financial information in accordance with two reportable segments: Ampio and Aytu. The Ampio segment consists of our core biopharmaceuticals compounds and the clinical trials associated with them. The Aytu segment contains our men's health platform which consists of its diagnostic device platform and sexual dysfunction portfolio. Select financial information for these segments is as follows:
| Years ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
Revenue: | ||||||||||||
Ampio | $ | - | $ | - | $ | 38,000 | ||||||
Aytu | 1,163,000 | 77,000 | 12,000 | |||||||||
|
|
|
|
|
| |||||||
Consolidated revenue | $ | 1,163,000 | $ | 77,000 | $ | 50,000 | ||||||
|
|
|
|
|
| |||||||
Consolidated net loss: | ||||||||||||
Ampio | $ | (24,108,000 | ) | $ | (31,305,000 | ) | $ | (21,504,000 | ) | |||
Aytu | (9,606,000 | ) | (7,743,000 | ) | (3,024,000 | ) | ||||||
|
|
|
|
|
| |||||||
Consolidated net loss | (33,714,000 | ) | (39,048,000 | ) | (24,528,000 | ) | ||||||
Reconciliation of consolidated net loss attributable to Ampio: | ||||||||||||
Net loss applicable to non-controlling interests | 1,704,000 | 923,000 | 519,000 | |||||||||
|
|
|
|
|
| |||||||
Net loss attributable to Ampio | $ | (32,010,000 | ) | $ | (38,125,000 | ) | $ | (24,009,000 | ) | |||
|
|
|
|
|
| |||||||
| December 31, 2015 | December 31, 2014 | |||||||
Total assets | ||||||||
Ampio | $ | 26,009,000 | $ | 61,326,000 | ||||
Aytu | 24,410,000 | 8,942,000 | ||||||
|
|
|
| |||||
Total assets | $ | 50,419,000 | $ | 70,268,000 | ||||
|
|
|
| |||||
The consolidated net loss above includes Ampio interest expense of $22,000 in 2015 and no interest expense in 2014 or 2013. Aytu had interest expense of $416,000 and $121,000 in 2015 and 2014, respectively, and no interest expense in 2013.
Note 13 – Litigation
As previously disclosed, on May 8, 2015 and May 14, 2015, purported stockholders of the Company brought two putative class action lawsuits in the United States District Court in the Central District of California, Napoli v. Ampio Pharmaceuticals, Inc., et al., Case No. 2:15-cv-03474-TJH and Stein v. Ampio Pharmaceuticals, Inc., et al., Case No. 2:15-cv-03640-TJH (the "Securities Class Actions"), alleging that Ampio and certain of its current and former officers violated federal securities laws by misrepresenting and/or omitting information regarding the STEP study. The cases were consolidated, and on February 8, 2016, plaintiffs filed a consolidated amended complaint alleging claims under Sections 10(b) and 20(a) and Rule 10b-5 under the Exchange Act and Sections 11 and 15 under the Securities Act of 1933 on behalf of a putative class of purchasers of common stock from January 13, 2014 through August 21, 2014, including purchasers in the Company's offering on February 28, 2014. The lawsuits seek unspecified damages, pre-judgment and post-judgment interest, and attorneys' fees and costs.
On August 6, 2015 and September 25, 2015, purported stockholders of the Company brought derivative actions in the United States District Court in the Central District of California, Oglina v. Macaluso et al ., Case No. 2:15-cv-05970-TJH-PJW (" Oglina action") and the Colorado state court in Denver, Loyd v. Giles et al ., Case No. 2015CV33429 (" Loyd action"), alleging primarily that the directors and officers of Ampio breached their fiduciary duties because of their alleged misstatements and/or omissions regarding the STEP study. Pursuant to the parties' stipulation, the United States District Court in the Central District of California has stayed the proceedings in the Oglina action at the present time in accordance with the terms of the parties' stipulation. Pursuant to the parties' stipulation, the Colorado state court in Denver has stayed the Loyd action at the present time in accordance with the terms of the parties' stipulation.
The Company believes these claims are without merit and intends to defend these lawsuits vigorously. We currently believe the likelihood of a loss contingency related to these matters is remote and, therefore, no provision for a loss contingency is required.
Note 14 – Employee Benefit Plan
Ampio has a 401(k) plan that allows participants to contribute a portion of their salary, subject to eligibility requirements and annual IRS limits. Ampio does not match employee contributions.
F-27
Note 15 – Selected Quarterly Data (unaudited)
Quarterly results were as follows:
| Quarters Ended | ||||||||||||||||
| March 31, | June 30, | September 30, | December 31, | |||||||||||||
2015 | ||||||||||||||||
Total revenue | $ | 21,429 | $ | 184,435 | $ | 487,385 | $ | 469,213 | ||||||||
|
|
|
|
|
|
|
| |||||||||
Operating expenses | ||||||||||||||||
Cost of sales | - | 87,884 | 37,325 | 244,100 | ||||||||||||
Research and development | 4,660,216 | 3,811,362 | 4,711,350 | 5,777,617 | ||||||||||||
Selling, general and administrative | 2,961,058 | 4,007,195 | 3,410,920 | 4,755,554 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Total operating expenses | 7,621,274 | 7,906,441 | 8,159,595 | 10,777,271 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Other income (expense) | 8,425 | 8,328 | (114,425 | ) | (314,126 | ) | ||||||||||
|
|
|
|
|
|
|
| |||||||||
Net loss | (7,591,420 | ) | (7,713,678 | ) | (7,786,635 | ) | (10,622,184 | ) | ||||||||
Net loss applicable to non-controlling interests | 307,837 | 358,252 | 421,095 | 616,491 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Net loss applicable to Ampio | $ | (7,283,583 | ) | $ | (7,355,426 | ) | $ | (7,365,540 | ) | $ | (10,005,693 | ) | ||||
|
|
|
|
|
|
|
| |||||||||
Weighted average number of Ampio common shares outstanding | 51,981,340 | 51,989,986 | 51,998,306 | 51,998,306 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Basic and diluted Ampio net loss per common share | $ | (0.14 | ) | $ | (0.14 | ) | $ | (0.14 | ) | $ | (0.19 | ) | ||||
|
|
|
|
|
|
|
| |||||||||
| Quarters Ended | ||||||||||||||||
| March 31, | June 30, | September 30, | December 31, | |||||||||||||
2014 | ||||||||||||||||
Total revenue | $ | 12,500 | $ | 21,429 | $ | 21,429 | $ | 21,429 | ||||||||
|
|
|
|
|
|
|
| |||||||||
Operating expenses | ||||||||||||||||
Research and development | 7,828,985 | 5,657,507 | 6,196,097 | 7,240,399 | ||||||||||||
Selling, general and administrative | 2,658,772 | 3,304,123 | 3,322,544 | 2,939,395 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Total operating expenses | 10,487,757 | 8,961,630 | 9,518,641 | 10,179,794 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Other income | 3,495 | 7,505 | 5,547 | 5,716 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Net loss | (10,471,762 | ) | (8,932,696 | ) | (9,491,665 | ) | (10,152,649 | ) | ||||||||
Net loss applicable to non-controlling interests | 229,579 | 241,176 | 211,635 | 240,967 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Net loss applicable to Ampio | $ | (10,242,183 | ) | $ | (8,691,520 | ) | $ | (9,280,030 | ) | $ | (9,911,682 | ) | ||||
|
|
|
|
|
|
|
| |||||||||
Weighted average number of Ampio common shares outstanding | 44,950,267 | 51,917,528 | 51,969,836 | 51,972,266 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Basic and diluted Ampio net loss per common share | $ | (0.23 | ) | $ | (0.17 | ) | $ | (0.18 | ) | $ | (0.19 | ) | ||||
|
|
|
|
|
|
|
| |||||||||
Note 16 – Subsequent Events
On January, 4, 2016, Ampio distributed a majority of its shares of common stock of Aytu to the Ampio shareholders on a pro rata basis. This transaction changed Ampio's ownership from 81.5% down to 8.6% of Aytu's outstanding shares on that date. Ampio believes it continues to have significant influence over Aytu subsequent to the spin-off. Therefore, we plan to account for our remaining investment in Aytu on the equity method of accounting in 2016.
F-28
On January 5, 2016, Aytu accelerated the vesting of 335,000 options to employees of Ampio and Ampio will recognize the expense in the amount of $137,000 related to this modification.
On January 20, 2016, Aytu entered into subscription agreements with Joshua R. Disbrow, Aytu's Chief Executive Officer, and Jarrett T. Disbrow, Aytu's Chief Operating Officer, pursuant to which each officer agreed to purchase 153,846 shares of Aytu's common stock (for an aggregate of 307,692 shares) at a price of $0.65 per share. These sales triggered Aytu's obligation to notify noteholders who held convertible notes issued by Aytu in July and August 2015 of their resulting conversion option under the notes.
Pursuant to the terms of the Aytu notes, the notes (inclusive of accrued but unpaid interest) were convertible, at the option of the noteholder, into shares of Aytu's common stock in an amount equal to 120% of the number of such shares issuable calculated by dividing the outstanding principal and accrued interest by $0.65. On February 10, 2016, the date of the conversion, an aggregate of $4,125,000 of principal and $142,810 of accrued interest on the notes converted into an aggregate of 7,879,096 shares of Aytu's common stock. After giving effect to the conversion, Aytu had 22,446,481 shares of common stock outstanding on February 10, 2016. Convertible notes in the aggregate principal amount of $1,050,000 remain outstanding.
In connection with the conversion of the Aytu notes, Aytu was obligated to issue to the placement agents for the convertible note offering warrants for an amount of shares equal to 8% of the number of shares of Aytu's common stock for the notes sold by the placement agents issued upon conversion of the notes. As a result of the optional note conversion, on February 10, 2016, Aytu issued warrants to the placement agents to purchase an aggregate of 267,073 shares of our common stock at an exercise price of $0.65 per share. These warrants are exercisable for five years from the date of issuance of the related notes in July and August 2015. The warrants have a cashless exercise feature.
In February 2016, Ampio entered into a Controlled Equity Offering SM Sales Agreement (the "Agreement") with a placement agent to implement an "at-the-market" equity program under which Ampio, from time, to time may offer and sell shares of its common stock having an aggregate offering price of up to $25 million through the placement agent.
Disposition of Aytu and Pro Forma (unaudited)
As stated above, and, as previously disclosed, Ampio distributed a majority of its shares of common stock of Aytu to the Ampio shareholders on a pro rata basis. This transaction changed Ampio's ownership from 81.5% down to 8.6% of Aytu's outstanding shares on that date. See below for unaudited pro forma financial information related to this significant disposition.
F-29
Unaudited Pro Forma Condensed Combined Balance Sheet
As of December 31, 2015
| Consolidated December 31, 2015 | Disposition of Aytu December 31, 2015 | Disposition Adjustments | Pro Forma December 31, 2015 | |||||||||||||
Assets | ||||||||||||||||
Cash and cash equivalents | $ | 26,957,938 | $ | 10,959,546 | $ | 15,998,392 | ||||||||||
Other current assets | 2,270,484 | 1,805,108 | 38,451 | 503,827 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Total current assets | 29,228,422 | 12,764,654 | 38,451 | 16,502,219 | ||||||||||||
Long-term assets | 21,190,171 | 11,645,142 | 1,312,254 | 10,857,283 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Total assets | $ | 50,418,593 | $ | 24,409,796 | $ | 1,350,705 | $ | 27,359,502 | ||||||||
|
|
|
|
|
|
|
| |||||||||
Liabilities and Stockholders' Equity | ||||||||||||||||
Current liabilities | ||||||||||||||||
Total current liabilities | $ | 5,515,113 | $ | 2,804,101 | $ | 38,451 | $ | 2,749,463 | ||||||||
Total long-term liabilities | 6,976,492 | 6,346,924 | - | 629,568 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Total liabilities | 12,491,605 | 9,151,025 | 38,451 | 3,379,031 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Commitments and contingencies | ||||||||||||||||
Stockholders' equity | ||||||||||||||||
Common Stock | 5,200 | 1,426 | 1,426 | 5,200 | ||||||||||||
Additional paid-in capital | 170,999,410 | 39,247,254 | 39,247,254 | 170,999,410 | ||||||||||||
Distribution to stockholders | - | - | (13,018,687 | ) | (13,018,687 | ) | ||||||||||
Advances to stockholders | (90,640 | ) | - | (90,640 | ) | |||||||||||
Accumulated deficit | (133,914,812 | ) | (23,989,909 | ) | (23,989,909 | ) | (133,914,812 | ) | ||||||||
|
|
|
|
|
|
|
| |||||||||
Total Ampio stockholders' equity | 36,999,158 | 15,258,771 | 2,240,084 | 23,980,471 | ||||||||||||
Non-controlling interests | 927,830 | - | (927,830 | ) | - | |||||||||||
|
|
|
|
|
|
|
| |||||||||
Total stockholders' equity | 37,926,988 | 15,258,771 | 1,312,254 | 23,980,471 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Total liabilities and stockholders' equity | $ | 50,418,593 | $ | 24,409,796 | $ | 1,350,705 | $ | 27,359,502 | ||||||||
|
|
|
|
|
|
|
| |||||||||
F-30
Unaudited Pro Forma Condensed Combined Statement of Operations
For the year ended December 31, 2015
| Consolidated December 31, 2015 | Disposition of Aytu December 31, 2015 | Disposition Adjustments | Pro Forma December 31, 2015 | |||||||||||||
Total revenue | $ | 1,162,462 | $ | 1,162,462 | $ | - | ||||||||||
|
|
|
|
|
|
|
| |||||||||
Operating expenses | ||||||||||||||||
Cost of sales | 369,309 | 369,309 | - | |||||||||||||
Research and development | 18,960,545 | 3,848,859 | 15,111,686 | |||||||||||||
Selling, general and administrative | 15,134,727 | 6,078,842 | 9,055,885 | |||||||||||||
|
|
|
|
|
|
|
| |||||||||
Total operating expenses | 34,464,581 | 10,297,010 | 24,167,571 | |||||||||||||
|
|
|
|
|
|
|
| |||||||||
Loss from operations | (33,302,119 | ) | (9,134,548 | ) | (24,167,571 | ) | ||||||||||
|
|
|
|
|
|
|
| |||||||||
Total other (expense) income | (411,798 | ) | (471,651 | ) | 59,853 | |||||||||||
Net loss | (33,713,917 | ) | (9,606,199 | ) | (826,133 | ) | (24,933,851 | ) | ||||||||
Net loss applicable to non-controlling interests | 1,703,675 | - | (1,703,675 | ) | - | |||||||||||
|
|
|
|
|
|
|
| |||||||||
Net loss applicable to Ampio | $ | (32,010,242 | ) | $ | (9,606,199 | ) | $ | (2,529,808 | ) | $ | (24,933,851 | ) | ||||
|
|
|
|
|
|
|
| |||||||||
Weighted average number of Ampio common shares outstanding | 51,992,048 | 51,992,048 | ||||||||||||||
|
|
|
| |||||||||||||
Basic and diluted Ampio net loss per common share | $ | (0.62 | ) | $ | (0.48 | ) | ||||||||||
|
|
|
| |||||||||||||
F-31